

Abstract
Type I interferons (IFN-α/β) are pleitropic cytokines widely used in the treatment of certain malignancies, hepatitis B and C and multiple sclerosis. IFN resistance is a challenging clinical problem to overcome. Hence understanding the molecular mechanism by which IFN immunotherapy ceases to be effective is of translational importance. In this study, we report that continuous IFN-α stimulation of the human Jurkat variant H123 led to resistance to type I IFN-induced apoptosis due to a loss of STAT2 expression. The apoptotic effects of IFN-α were hampered as STAT2 deficient cells were defective in activating the mitochondrial-dependent death pathway and ISGF3-mediated gene activation. Reconstitution of STAT2 restored the apoptotic effects of IFN-α as measured by loss of mitochondrial membrane potential, cytochrome c release from mitochondria, caspase activation, and ultimately cell death. Nuclear localization of STAT2 was a critical event as retention of tyrosine phosphorylated STAT2 in the cytosol was not sufficient to activate apoptosis. Furthermore, silencing STAT2 gene expression in Saos2 and A375S.2 tumor cell lines significantly reduced the apoptotic capacity of IFN-α. Altogether we demonstrate that STAT2 is a critical mediator in the activation of type I IFN-induced apoptosis. More importantly, defects in the expression or nuclear localization of STAT2 could lessen the efficacy of type I IFN immunotherapy.
Keywords: STAT2, Interferon, Mitochondria, Apoptosis, IRF-1
Introduction
Type I interferons (IFN-α/β) are therapeutic cytokines used in the treatment of several forms of cancer (1-3), chronic hepatitis B and C (4, 5) and multiple sclerosis (6). Yet, the beneficial effects of IFNs may be limited as these diseases may already be resistant to type I IFN immunotherapy or patients develop resistance during the course of therapy. This has been reported in patients with chronic myelogenous leukemia (7), melanoma (8, 9) and hepatitis C (10). A clear understanding of the molecular mechanism of IFN resistance remains obscure. A reduced or loss of signal transducers and activators of transcription (STAT)1 (11) and Janus kinase (JAK)1 (12) expression and STAT5 overexpression (13) have been implicated in IFN insensitivity. Other studies report increased free circulating type I IFN receptors in patients with adenocarcinoma (14) and epigenetic silencing of genes that are essential for the biological actions of IFNs (15) as contributors to IFN resistance.
Type I IFNs are a family of pleitropic cytokines that induce their antiproliferative, apoptotic and antiviral effects through activation of receptor associated JAK1 and TYK2 (16). STAT1 and STAT2 are recruited to the IFN-α/β receptor and become tyrosine phosphorylated by JAKs. Activated STATs bind to each other as STAT1 homodimers or STAT1/STAT2 heterodimers that when bound to IRF9 form the IFN-stimulated gene factor-3 (ISGF3) complex. Both STAT complexes translocate to the nucleus and initiate transcription of IFN-stimulated genes (ISGs). STAT1 homodimers recognize and bind to a gamma activated region (GAS) (17) while the ISGF3 complex binds to an IFN-stimulated response element (ISRE) region in the promoter of ISGs (18).
STAT2 is one of seven members of the STAT family of transcription factors (19). In marked contrast to other STATs that can be activated by multiple growth factors and cytokines, only type I IFNs have been found to cause the activation of STAT2 (20). STAT2 is an integral component in the IFN-induced activation of the JAK-STAT signaling pathway. This is supported by the observation that the antiproliferative effects of IFN-α are impaired in STAT2 deficient human fibrosarcoma U6A cells and mouse T cells (21-23). Additionally, studies of STAT2 null mice provide direct evidence to support that STAT2 plays a vital role in type I IFNs signaling and consequently in viral immunity and cancer (24, 25). More importantly, these studies demonstrated that STAT2 is not always required for STAT1 activation. In spite of that, the molecular mechanism of cell growth inhibition and apoptosis mediated by type I IFN and the role STAT2 plays in this response are less studied.
We recently reported on the characterization of a type I IFN-resistant H123 clone that arose after sustained stimulation with IFN-α. Biochemical and genetic analysis revealed this clone had acquired a mutation that changed a conserved proline 630 (P630) to leucine in the SH2 domain of STAT2 and led to impaired IFN-α signaling (26). In contrast, introduction of a tyrosine to phenylalanine mutation, adjacent to P630 enhanced the antiproliferative effects of IFNs due to activation of apoptosis (27). These findings indicated the presence of a regulatory switch in STAT2. In this study, we identify STAT2 as a critical mediator of type I IFN-induced apoptosis. A loss of STAT2 results in impaired activation of the mitochondrial death dependent pathway but not that of STAT1 mediated responses triggered by type I IFNs. Most importantly, nuclear localization of tyrosine phosphorylated STAT2 is a key event for the onset of apoptosis. Altogether our data hint that a loss of STAT2 function may reduce the efficacy of type I IFN immunotherapy.
Results
Development of type I IFN-apoptosis resistance in H123 tumor cells
Several reports have shown that type I IFNs induce apoptosis in some but not all tumor cell lines (28). We previously characterized the human leukemic H123 Jurkat cell line as cells that were isolated for having a defect in T-cell receptor signaling that inexplicably gained sensitivity to the apoptotic effects of IFN-α (29). To begin elucidating the signaling mediators involved in the promotion of apoptosis by IFN-α, we utilized H123 cells to explore this process. H123 cells were maintained under selection pressure in IFN-α for two months. Each week surviving cells were recovered and re-cultured with fresh IFN-α. This approach generated multiple individual clones that survived the apoptotic effects of both type I IFNs, IFN-α and IFN-β. We chose to study one of these IFN resistant clones for further analysis. In response to IFN-α stimulation, parental H123 cells were dramatically growth inhibited as early as 12 h (Fig. 1A, left). In contrast the IFN resistant clone showed no decrease in cell proliferation during the course of 72 h even when high dose IFN-α was used (Fig. 1A, right). Furthermore, while more than 60% of the parental H123 cells underwent apoptosis with IFN-α treatment (Fig. 1B top panel); no such effect took place in the IFN-resistant clone (Fig. 1B, bottom panel). Thus these results demonstrate that we had isolated an H123 variant that is resistant to the antigrowth effects of type IFNs.
Figure 1.
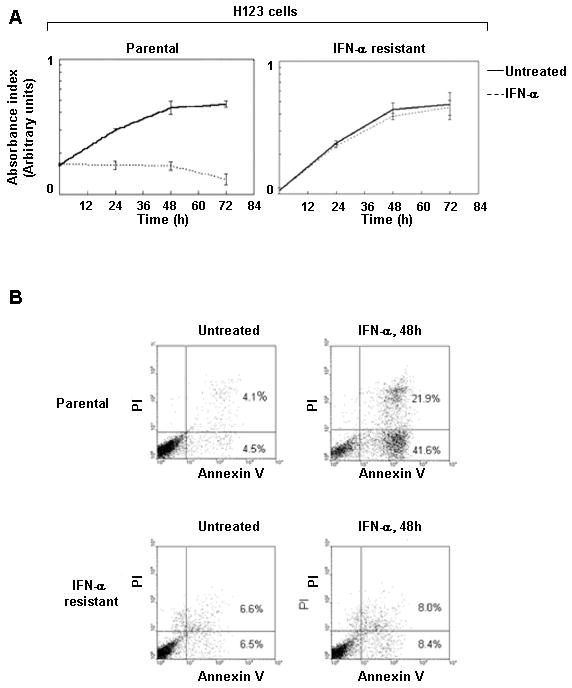
Long-term type I IFN exposure leads to resistance to IFN-induced apoptosis. A) H123 cells (parental and resistant) were left either untreated or treated with 3,000 U/ml IFN-α. Cell proliferation was measured by MTS assay and values are expressed as change in absorbance during the course of 72h. B) Same as A except induction of apoptosis was measured at 48 h by dually labeling with Annexin V (x-axis) and PI (y-axis).
Type I IFN apoptosis resistant H123 does not express STAT2
To help explain why the H123 IFN-resistant line had lost its ability to undergo apoptosis with IFN-α stimulation, we examined the expression of critical signaling components of the JAK/STAT pathway. Western blot analysis revealed that protein levels of JAK1, TYK2, STAT1 and STAT3 remained unchanged between parental and IFN resistant H123 cells (Fig. 2A). However, when STAT2 expression was analyzed, a loss of STAT2 was detected in the IFN resistant H123 clone (Fig. 2B). A loss of STAT2 protein was confirmed by Western blot analysis using two distinct antibodies that recognize the N-terminus (Fig. 2B, left) or C-terminus of STAT2 (Fig. 2B, right). We excluded the possibility that during selection the IFN resistant cells now expressed a truncated form of STAT2 because no other protein bands were detected. This observation was further tested by assaying IFN-α induced formation of the ISGF3 complex in parental and IFN resistant H123 cells. Electrophoretic mobility shift assay (EMSA) using an ISRE oligonucleotide probe demonstrated that binding of ISGF3 to DNA was detected only in parental H123 cells (Fig. 2C). Formation of a different protein complex able to bind the ISRE in the IFN resistant H123 cells was not detected, thereby demonstrating the absence of a truncated version of STAT2. Interestingly, despite a loss of STAT2 protein, RT-PCR and Northern blot analysis showed no discernible changes in the expression of STAT2 mRNA (data not shown). Moreover, STAT2 mRNA was sequenced and although a few single base substitutions were detected, these produced silent mutations. To verify that STAT2 was not targeted for degradation, treatment of IFN resistant H123 cells with two distinct proteosome inhibitors did not restore STAT2 protein expression (Fig. 2D). Thus these results demonstrated that we have isolated a STAT2 deficient clone.
Figure 2.
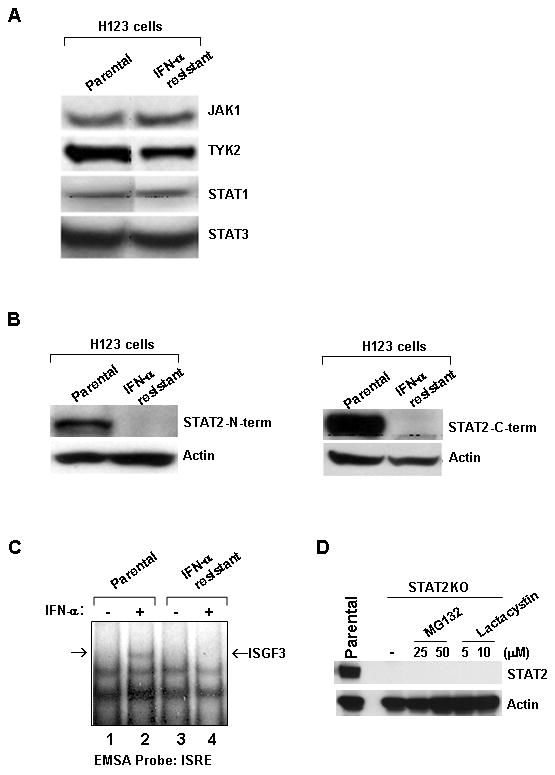
Type I IFN resistant H123 cells show loss of STAT2 protein. Whole cell extracts of parental and IFN resistant H123 cells were prepared and analyzed by Western blot analysis for expression of (A) JAK1, TYK2, STAT1, and STAT3 and B) STAT2 by using antibodies against the N-term or C-term end of STAT2. C) Nuclear extracts from parental and IFN resistant H123 cells were analyzed by EMSA using an ISRE oligonucleotide. D) Cells were pretreated overnight with the indicated doses of proteosome inhibitors and STAT2 protein expression was assessed by Western blot analysis.
STAT2 deficient H123 cells remain responsive to type I FNs and susceptible to Trail-induced apoptosis
Because our IFN-resistant cells now referred to as STAT2 deficient H123 had lost their capacity to undergo apoptosis in response to IFN-α, it was important to establish whether these cells retained any responsiveness to IFN-α. As shown in Fig. 3A, IFN-α stimulation of either parental or STAT2 deficient H123 cells resulted in similar levels of tyrosine phosphorylated STAT1 (Fig. 3A). IFN-α induced responses were evaluated by measuring increases in the expression of IRF1; a STAT1 regulated gene (Fig. 3B). Following 48 h of IFN-α stimulation, IRF1-levels were unchanged in parental and STAT2 deficient H123 cells. Surface expression of MHC class I (A,B and C) molecules was also measured as transactivation of this gene is induced by IFN-α and mediated by STAT1 and IRF-1 (30, 31). Flow cytometric analysis showed that upregulation of MHC class I protein was detected in STAT2 deficient H123 cells (Fig. 3C). However, the level of MHC class I was slightly higher in the parental H123 cells. In response to IFN-α, the fold-change expression levels in parental and STAT2 deficient H123 cells as determined by mean fluorescent intensity (MFI) was 2.1 and 1.7 respectively. Moreover, lack of STAT2 protein did not impair the ability of these cells to respond to other apoptotic stimuli. Indeed, treatment of STAT2 deficient H123 cells with TRAIL induced the activation of apoptosis to similar levels detected with parental H123 cells (Fig. 3D). These results show that, with the exception of induction of apoptosis, other type I IFN-regulated effects are not severely impaired in the absence of STAT2.
Figure 3.
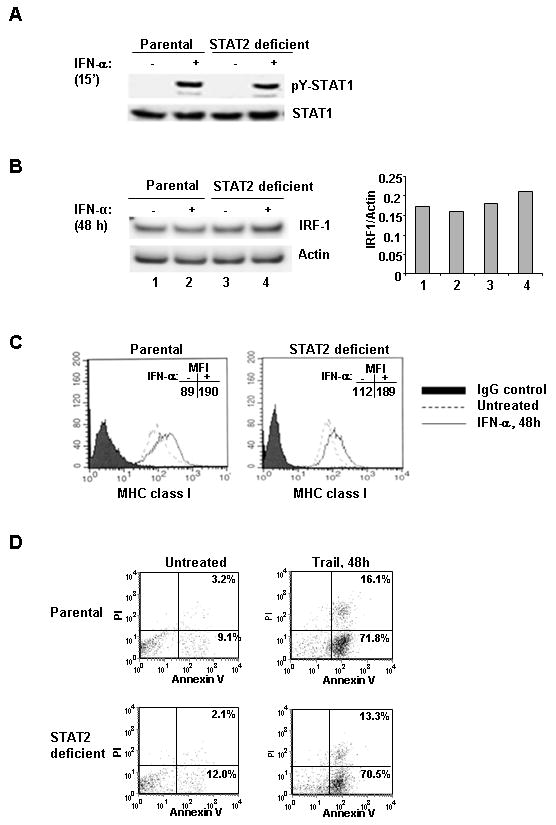
STAT2 deficient H123 cells can respond to type I IFNs and remain susceptible to other death agonists. Parental and IFN resistant H123 cells were left untreated or treated with 3,000 U/ml IFN-α for the indicated times. Whole cell extracts were prepared and analyzed by Western blot analysis for (A) pY (701) STAT1 or (B) IRF-1. Membranes were reprobed for total STAT1 or Actin respectively to assess equal protein loading. The ratio of IRF-1 to Actin was quantified. C) Cells were labeled with isotype control or anti-MHC class-I (A, B and C) FITC labeled antibody and analyzed by flow cytometry. D) Parental and IFN resistant H123 cells were left untreated or treated with 1μg/ml of Trail and dually labeled with Annexin V and PI.
Reconstitution and sufficient levels of STAT2 are required to mediate IFN-α-induced apoptosis
If STAT2 is an integral component in type I IFN-induced apoptosis, re-expression of STAT2 in STAT2 deficient H123 cells should restore this effect. To test this premise, a mammalian expression vector encoding flag-tagged wild type STAT2 was stably expressed in these cells. Individual clones expressing STAT2 levels similar to that of parental H123 cells were selected for analysis. Additionally, clones expressing varied amounts of STAT2 were chosen to determine if the amount of STAT2 was critical in the activation of IFN-α induced apoptosis (Fig. 4A lower panel). STAT2 reconstitution restored IFN-α induced activation of apoptosis (Fig. 4A upper panel). Of interest was the finding that an optimal amount of STAT2 protein was necessary for IFN-α to maximally mediate cell death.
Figure 4.
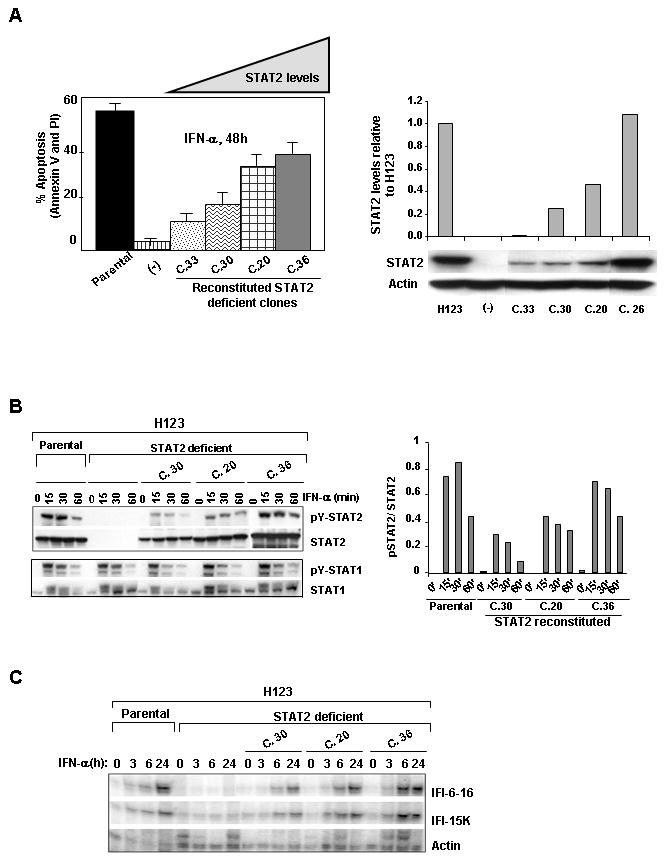
Reconstitution of STAT2 rescues type I IFN-induced apoptosis and ISGF3 driven gene activation. (A) Percent apoptosis was measured by dually staining cells with Annexin V and PI following IFN-α stimulation. STAT2 protein levels in reconstituted clones were assessed by Western blot analysis. Actin served as a control for equal protein loading (right, lower panel). Quantitative analysis of STAT2 levels in reconstituted cell lines relative to H123 cells (right graph). (B) Cells were treated with 1,000 U/ml IFN-α for the indicated times. Western blot analysis was performed with phosphotyrosine specific antibodies against STAT2 and STAT1. Membranes were reprobed with antibodies against total STAT2, STAT1 antibodies to verify for equal protein loading. Levels of pSTAT2 to STAT2 were quantified. C) Total RNA was extracted and expression of IFI-6-16 and IFI-15K RNAs was determined by RNase protection assay. Actin RNA served as an internal control to normalize for equal loading.
The next question we asked was if a deficiency in STAT2 altered the kinetics of IFN-α-induced STAT1 activation and ISGF3 mediated gene expression because based on our results (Fig. 3B) and contrary to earlier published work (19), we found STAT2 not to be required in the sequential activation of STAT1. Western blot analysis showed that STAT1 activation was virtually undistinguishable between parental and STAT2 deficient H123 cells (Fig. 4B). We then measured IFN-α-mediated gene activation by RNase protection assay in the same STAT2 reconstituted clones, with each expressing different amounts of STAT2. Figure 4C shows that in the absence of STAT2, expression of ISGF3-dependent genes, such as IFI-6-16 and IFI-15K was impaired and reintroduction of STAT2 restored transcription of these ISGs. It is important to note that in STAT2 reconstituted clones expressing low levels of STAT2, there was a delay in ISG expression (compare clones 20 and 30 to clone 36 and parental H123 cells). These results indicate that an adequate level of STAT2 protein was required for maximal expression of ISGF3-driven ISGs and activation of type I IFN-induced apoptosis.
STAT2 is essential in IFN-α induced activation of the mitochondrial death pathway
We previously showed that IFN-α induced apoptosis in H123 cells occurred via activation of mitochondrial and caspase-dependent death pathways (29). The hallmark of this signaling process entails (i) loss of mitochondrial membrane potential, (ii) release of cytochrome c from the mitochondria and (iii) activation of caspases. To determine whether STAT2 played a role in the IFN-α induced activation of the mitochondrial-dependent death pathway, first we directly measured changes in mitochondrial membrane potential. Parental H123, STAT2 deficient and STAT2 deficient H123 reconstituted with STAT2 were treated with IFN-α for 24 h or 48 h followed by incubation with the fluorescent dye JC-1 (Fig. 5A). Changes in fluorescence were analyzed by flow cytometry. IFN-α treatment of parental H123 cells and STAT2 deficient H123 cells reconstituted with STAT2 resulted in an increase in JC-1 fluorescence; an indication of loss of mitochondrial membrane potential. In contrast, a change in mitochondrial membrane potential in STAT2 deficient H123 cells was not detected after 48 h of IFN-α treatment. Next, we examined defects in cytochrome c release in the various H123 cell lines after treatment with IFN-α for 48 h. As shown in Figure 5B, in untreated cells, cytochrome c (green) was localized in the mitochondria (red) as demonstrated by the overlay of the two colors (yellow). In contrast, IFN-α treatment resulted in the translocation of cytochrome c to the cytosol away from the mitochondria in parental and STAT2 reconstituted H123 cells, but not in STAT2 deficient H123 cells. Furthermore, measurement of caspase 3 activation after IFN-α treatment of H123 cells also revealed a dependence on STAT2 to drive this event (Fig. 5C). All these results point to STAT2 as an integral mediator in type I IFN-induced activation of the mitochondrial dependent death pathway.
Figure 5.
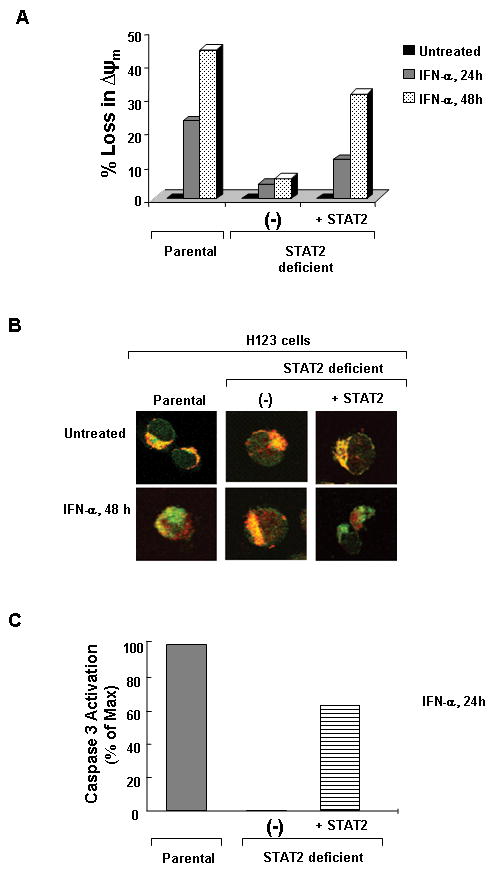
Activation of the mitochondrial dependent death pathway requires STAT2. Cells were left untreated or treated with 3,000 U/ml IFN-α for 24 and 48 h. A) Loss of mitochondrial membrane potential was measured by incubation of cells with the fluorescent cationic dye (JC-1). B) Live cells were incubated with Mitotracker® Red CMXRos then fixed and permeabilized. Cells were then labeled indirectly with anti-cytochrome C (Green). Images were analyzed by confocal microscopy. C) Caspase 3 activity was measured directly in cells after 24 h of IFN-α stimulation. Figures are representative of 3 experiments with similar results.
Requirement of tyrosine phosphorylation and nuclear localization of STAT2 for IFN-α induced apoptosis
We explored the possibility that STAT2 may have other activities in addition to gene transcription as that of an adaptor molecule. To test this hypothesis, we constructed two expression vectors each encoding a distinct STAT2 mutation. Tyrosine (Y)-690 was changed to phenylalanine (Y690F) to produce a transcriptionally inactive protein. Mutation of the tyrosine phosphorylation site prevents STAT2 from dimerizing with STAT1 and their translocation as a complex to the nucleus (32). In contrast, arginine (R)-409 and lysine-(K415) located in the nuclear localization signal (NLS) were changed to alanine (R409A, K415A). This STAT2 mutant can be tyrosine phosphorylated, associate with STAT1, but does not import to the nucleus (33). Both mutants helped us assess whether in response to IFN-α, the transcriptional activity of STAT2 was essential to promote apoptosis. We stably reconstituted STAT2 deficient cells with each STAT2 mutant and evaluated their competence against STAT2 deficient H123 reconstituted wild type STAT2 cells to activate apoptosis. Consistent with published reports, following IFN-α treatment both STAT2 Y690F and STAT2 R409A,K415A (21, 33) failed to localize to the nucleus thus resulting in impaired gene transcription (Fig. 6A and 6B). Interestingly, IFN-α activated STAT2 (STAT2 R409A:K415A) localized in the cytosol was not sufficient to promote apoptosis (Fig. 6C). These results clearly indicate that tyrosine phosphorylation and nuclear localization of STAT2 are two sequential events required for type I IFNs to induce apoptosis. More importantly, detection of activated STAT2 in the cytosol may be wrongly associated with an intact IFN response.
Figure 6.
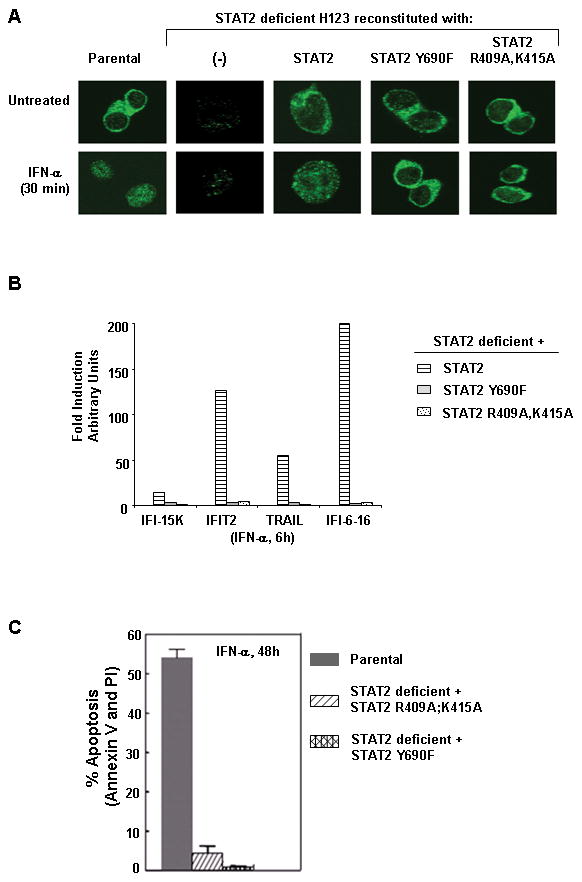
Nuclear localization of activated STAT2 is required for ISGF3-mediated gene expression and induction of type I IFN-induced apoptosis. STAT2 deficient H123 cells reconstituted with various forms of STAT2 were left either untreated or treated with IFN-α (3,000 U/ml) for the indicated times. A) Subcellular distribution of STAT2 was analyzed by confocal immunofluorescence microscopy. B) Gene activation of ISGs was assessed by quantitative RT-PCR. Figure shown is representative of three experiments with similar results. C) Apoptosis was measured by dually staining of cells with Annexin V and PI. Standard error bars were determined from three independent experiments.
STAT2 gene silencing impairs type I IFN-induced apoptosis
To demonstrate a role of STAT2 in the activation of apoptosis, we silenced the expression of STAT2 in other cell lines susceptible to the apoptotic effects of IFN-α. The following tumor cell lines: A375.S2 and SAOS2 were stably transduced with lentiviral vectors expressing mock or specific STAT2 shRNAs. Silencing of STAT2 abrogated type I IFN-induced apoptosis by almost 50 % (Fig 7A). In addition to STAT2, the expression of apoptotic genes driven by STAT1 homodimers may be involved in promoting apoptosis by type I IFNs. Although IFN-α-mediated upregulation of IRF-1 protein was not observed in parental H123 cells; nevertheless, it was important to determine whether IRF1 played a role in promoting apoptosis of H123 cells. Lentiviral silencing of IRF-1 in H123 cells; however did not protect from the killing effects of IFN-α (Fig 7B). This result suggests the existence of other STAT2-regulated apoptotic signaling mediators.
Figure 7.
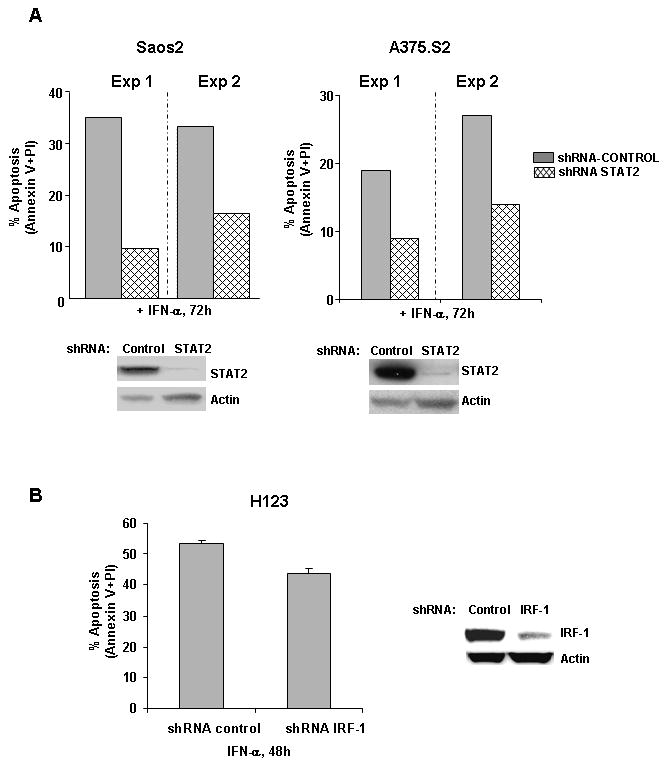
STAT2 gene silencing impairs IFN-α induced apoptosis. A) A375.S2 and SAOS2 were stably infected with shRNA lentivirus targeting STAT2 (ShRNA-STAT2), or non silencing shRNA (ShRNA-control). Percent apoptosis was measured after 72 h of IFN-α treatment by dually staining of cells with Annexin V and PI and flow cytometric analysis. B) H123 cells were infected with shRNA lentivirus targeting IRF-1 and apoptosis was measured after 48 h of IFN-α. Reduction in STAT2 and IRF1 protein levels were analyzed by Western blot analysis. Anti-actin antibody was used to assess equal protein loading. Results are represented as percent apoptosis from two individual experiments.
Discussion
In the present study, we showed that persistent exposure to IFN-α led to the development of a STAT2 deficient cell line that is resistant to type I IFN-induced apoptosis. An unexpected finding was to discover that a lack of STAT2 was not associated with gene methylation, lack of STAT2 mRNA transcripts or mutations that altered its reading frame. Although we do not have an explanation for the lack of STAT2 protein in the presence of mRNA, gene silencing by microRNAs remains a possibility. Another important finding was that STAT2 mutants that do not impair STAT1 activation, but fail to dimerize with STAT1 or localize to the nucleus do not rescue IFN-α-induced apoptosis. Thus our results lead us to speculate that perhaps a subset of patients receiving long-term IFN treatment may experience resistance to type I IFN therapy due to defects in STAT2 function or expression
The role of STAT1 and STAT2 in IFN resistance remains unclear. Work by Chawla-Sarkar et al. showed that defects in STAT1 and STAT2 expression are infrequent in melanoma cell lines and tumor samples and this did not correlate with IFN resistance (35). In a different study, Lesinski et al. reported that the expression levels of STAT1 and STAT2 and their distribution in cytosolic and nuclear compartments in malignant melanoma cells did not always correlate with a favorable response to IFN-α adjuvant therapy (36). However, a small number of melanoma resistant cell lines and tumor samples used in these studies were analyzed for STAT2 defects. In contrast, Zhou et al. reported that IFN-α treatment of patients with carcinoid tumors led to increased levels of STAT1 and STAT2 in their tumors, which correlated with stable disease or objective response (37). While Mischiati et al. showed that STAT2 expression was lost in 67% of 15 metastatic melanoma lesions (38). However in this study, no correlation between IFN-α resistance and loss of STAT2 was made.
Understanding the role of STAT2 in mediating type I IFN-induced apoptosis has been daunting. Before isolation of the STAT2 deficient H123, there were two STAT2 deficient cell lines, in which STAT2 expression was restored. Yet STAT2 reconstitution did not make these cell lines susceptible to IFN-α-induced apoptosis (our unpublished results). In our studies, we demonstrate a requirement for STAT2 in type I IFN-induced apoptosis as reconstitution of STAT2 rescued the apoptotic activity of IFN-α. We observed that a threshold of STAT2 protein was necessary for IFN-α to activate apoptosis. In fact, low expression of STAT2 protein resulted in delayed ISG expression and without or little induction of apoptosis. Furthermore, by silencing STAT2 expression in Saos2 and A375.S2 tumor cells, we show that STAT2 is a critical apoptotic mediator. However, these cells were not fully protected from the lethal effects of IFN-α thus indicating a requirement of additional apoptotic mediators.
IRF-1 is a gene regulated by IFN-activated STAT1 and is known for its role in the activation of IFN-induced apoptosis (39). We found STAT1 activation in H123 cells to be independent of STAT2. Our result supports a previous observation in which STAT1 activation is intact in STAT2 deficient mouse macrophages (40). Next, we found IRF-1 not to be upregulated by IFN-α in parental and STAT2 deficient H123. In addition, IRF-1 gene silencing in H123 cells conferred no significant protection against the killing effects of IFN-α. This observation implies that IRF1 does not always mediate the apoptotic effects of type I IFNs. Recently, a small subset of IFN-inducible, STAT2 dependent apoptotic genes were identified on chromosome 22 (44). Therefore, it is likely that the expression of these genes require transcriptionally competent STAT2 for mediating type I IFN-induced apoptosis.
Consistent with what has been reported about the mode of action of type I IFNs (41), earlier characterization of the H123 cell line demonstrated that IFN-α activated the intrinsic mitochondrial dependent death pathway (29). In this same model, we now show that alterations in mitochondrial membrane potential, cytochome c release from mitochondria to the cytosol, and caspase activation, all of which are characteristics of activation of the intrinsic apoptotic pathway are events regulated by STAT2. In multiple myeloma, members of the Bcl-2 family such as Bax and Bak have been implicated in mediating type I IFN-induced apoptosis (42, 43). However, we did not detect activation of these molecules in H123 cells, possibly due to cell type differences. Collectively, these findings strongly suggest the existence of additional signaling modules that are involved in the activation of apoptosis.
Work by Panaretakis et al. (45) showed an alternative mechanism wherein IFN-α-induced apoptosis is mitochondrial dependent but in a transcriptionally, JAK/STAT signaling independent manner. However, our work seems to contradict these findings because transcriptionally active STAT2 localized to the nucleus was essential for the activation of apoptosis by type IFNs. In addition, tyrosine phosphorylated STAT2 when retained in the cytoplasm is insufficient to mediate apoptosis. This discrepancy could lie on the nature of distinct cell types used for these studies. For instance IFN-α treatment of H123 cells does not activate JNK and pharmacological inhibition of PI3K activity does not protect cells from undergoing apoptosis. Quite the contrary, the presence of these inhibitors in combination with IFN-α accelerate the onset of apoptosis (unpublished results).
In summary, our findings have clinical relevance as a subset of patients who are or become resistant during the course of IFN therapy may be experiencing a loss of STAT2 expression or function. Most importantly, IFN-α responses measured either in the tumor or blood of patients must be interpreted carefully as tyrosine phosphorylated STAT2 may not always be indicative of intact STAT2 function and favorable IFN responses. Further studies are warranted to establish a link between a loss of STAT2 function and type I IFN resistance.
Materials and Methods
Cells and cell culture reagents
H123 cells, a variant of the human leukemic Jurkat cell line, mutant variants derived from H123 (29) and the human osteosarcoma SAOS2 were maintained in RPMI 1640 medium. Human melanoma A375.S2 cells were maintained in DMEM medium. Both media were supplemented with 10% fetal bovine serum containing 2.2 g of GlutaMax™-1, 10 U/ml penicillin G sodium and 10 μg/ml streptomycin sulfate (Invitrogen Corp. Carlsbad CA) at 37°C in a 5% CO2 atmosphere. Stable clones of STAT2 deficient H123 cells reconstituted with wild type or various mutant forms of STAT2 were maintained in G418 at 1 mg/ml. Recombinant human IFN-α-2a, (specific activity 2 × 107 U/ml), was purchased from PeproTech Inc. (Rock Hill NJ). TRAIL was kindly provided by Genentech (San Francisco, CA).
Antibodies and chemical reagents
Antibody against the C-terminus of STAT1 was a gift from Andrew. Larner (VCU, Richmond VA). STAT2 (C-20) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-STAT2 Y690 antibody was obtained from Upstate (Lake Placid, NY) and phospho-STAT3 Y705 was obtained from Cell Signaling (Boston, MA). Antibodies directed against phospho-STAT1 Y701, STAT2, IRF-1, Cytochrome c, JAK1, TYK2, Annexin V-FITC conjugated and mouse IgG isotype FITC conjugated were from BD Biosciences/Pharmingen (Pharmingen, Palo Alto, CA). Antibody against actin was from AbCam (Cambridge MA). Propidium Iodide (PI) was from Calbiochem. Mitotracker Red CMX®Ros and the fluorescent dye JC-1 were purchased from molecular probes, Inc (Eugene, OR). CellTiter 96 Aqueous One Solution Reagent was from Promega (Madison, WI).
Site-directed mutagenesis
Flag-tagged STAT2 construct in pcDNA3, kindly provided by C. Horvath (Northwestern University, Evanston IL), was used as template DNA. STAT2 mutagenesis was performed with the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) using the following primers synthesized by Integrated DNA Technologies (Coralville, IA):
STAT2 Y690F 5′-CAGGAACGGAGGAAATTCCTGAAACACAGGCTC-3′
STAT2 R409A 5′-CTGGAGCAAGCTTCAGGTGGTTCAGCAAAGG-3′
STAT2 K415A 5′- GGTGGTTCAGGAGCGGGCAGCAATAAGG-3″
Mutagenesis was confirmed by sequencing the entire STAT2 sequence.
Transfections
H123 STAT2 deficient cells were transfected with 10 μg of either STAT2-flag tag, STAT2 R409A:K415A or STAT2 Y690F plasmid DNA by using a 2mm cuvette and a BioRad electroporator set at 140 V, 1000 μF Capacitance, ∞ Ω Resistance. Twenty-four hours later, cells were maintained in complete medium containing 1 mg/ml of G418, and two weeks later, individual clones were then selected by limiting dilution.
Apoptosis assay
STAT2 deficient and STAT2 reconstituted H123 cells were left untreated or treated with either 3,000 U/ml IFN-α or TRAIL for the indicated times at 37°C. Cells were harvested, washed in 1× PBS and stained in 50 μl of binding buffer (10mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) containing 2 μl of Annexin V-FITC (Pharmingen, Palo Alto, CA) and 2 μl of propidium iodide (50 μg/ml, Sigma). Cells were then incubated for 10 min at room temperature protected from light followed by the addition of 400 μl of binding buffer and analyzed by FACScan (Becton-Dickinson). Data was collected for 10,000 cells, stored and analyzed using CellQuest™ software (Becton-Dickinson).
Flow Cytometry
Cells were incubated for 30 min on ice in staining buffer (PBS solution containing 0.2% NaN3 and 2% fetal calf serum) containing PE-labeled anti-human MHC class I ABC or isotype matched control antibodies. Surface expression of this marker was analyzed using a FACScan TM cytometer (Becton-Dickinson) and further analyzed using CellQuest software (Becton- Dickinson).
Proliferation Assay
Cells were seeded in triplicate in flat-bottom 96-well plates at a concentration of 5 × 103 cells in 50 μl volume/well. Fifty μl of medium or IFN-α was added to each well and cells were incubated at 37° for 72 hours. Wells without cells containing 100 μl of medium served as background controls. Cell proliferation was assessed by MTS assay, [3-(4,5-dimethythiazol-2-yl)]-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium). Briefly, 20 μl of CellTiter 96-Aqueous One Solution Reagent, was added to each well and the plate incubated at 37° for 1-4 hours. Absorbance was measured at 490 nm using a Victor2™ 1420 multilabel counter (Perkin Elmer, Waltham MA). Background values were first subtracted from each well before proceeding with data analysis.
Preparation of cell extracts
Following treatment, cells were collected, washed in 1× PBS and lysed in ice cold lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 2mM EDTA pH 8.0, 0.5% Triton X-100, 1mM sodium orthovanadate, 200 μM PMSF) supplemented with protease inhibitors (Sigma, St. Louis, MO). After centrifugation at 4°C for 10 min. supernatants were collected and protein concentration measured by standard Bio-Rad Bradford protein assay.
Western blot analysis
Cell extracts were boiled with laemmli buffer and then resolved by 10% SDS-PAGE (Invitrogen). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked for 30 min with blocker™ casein in TBS (Pierce) and then incubated with the specified antibodies. Following washing, immunoblots were incubated with the corresponding IgG isotype secondary antibody (1:10000, Zymed, San Francisco, CA) for 30 minutes. Membranes were washed and developed by chemiluminescence using the ECL Western blotting system (Pierce, Rockford IL) as previously described (Gamero et al. 2004).
RNA Isolation and RNase protection assay
Cells were either left untreated or stimulated with IFN-α for the indicated times. RNA was isolated with RNAzol B (Tel-Test Inc., Friendswood, TX) according to the manufacturer's instructions. RNA concentration was determined by 260/280 ratios. Antisense RNA probes were synthesized by in vitro transcription using T7 or SP6 RNA polymerase (GIBCO-BRL) and [α-32P]-UTP (ICN, Costa Mesa, CA) (29). Ten μg of RNA and 32P-labeled riboprobes were incubated overnight in hybridization buffer containing 80% formamide, 40 mM PIPES pH 6.7, 400 mM NaCl and 1 mM EDTA at 56°C followed by digestion with T1 RNAse (GIBCO-BRL) for 1 h at 37°C, phenol extraction and ethanol precipitation. Protected RNA fragments were solubilized in RNA loading buffer (98% formamide, 10 mM EDTA (pH 8.0), bromophenol blue and xylene cyanole), boiled for 2 min and resolved by electrophoresis on a 4.5% polyacrylamide-urea gel.
Quantitative RT-PCR
Five micrograms of total RNA was reverse transcribed to generate cDNA using Superscript II reverse transcriptase (Invitrogen). qRT-PCR primers were obtained from Applied Biosystems (Foster City, CA). Briefly, cDNA was mixed with Taqman 2× PCR master mix (Applied Biosystems), using primers with FAM reporter dyes, and qPCR reactions were performed using the 7300 Real Time PCR system (Applied Biosystems). Samples were amplified using the following PCR variables: 55°C for 2 minutes (1 cycle), 95°C for 10 minutes (1 cycle), 95°C (40 cycles) for 30 seconds, 60°C for 1 minute. mRNA quantification was normalized by multiplexing with 18S-VIC primers.
Electrophoretic mobility shift assays (EMSA)
Synthetic double-stranded oligonucleotide corresponding to the ISRE of the ISG15 promoter was used as a DNA probe. Probe was end-labeled with [γ-32P]-ATP using T4 polynucleotide kinase (Cell Signaling) as previously described (27). The DNA-protein complexes were subjected to electrophoresis on a 4.7% polyacrylamide gel and visualized by autoradiography.
Mitochondrial membrane potential (ΔΨm)
Loss of mitochondrial membrane integrity was measured by using the fluorescent dye JC-1 (Invitrogen). Cells were left untreated or treated with IFN-α for the indicated times. Cells were stained with 2.5 μM JC-1 resuspended in PBS, incubated for 20 min at 37°C, washed and immediately analyzed by flow cytometry. A loss in mitochrondrial membrane potential was determined by a decrease in green/red double fluorescence to an increase in green single fluorescence.
Confocal microscopy analysis
To measure cytochrome c release, following treatment, cells were incubated with 50 nM Mitotracker® Red CMXRos for 15 min and then washed 3 times with 1× PBS. Samples were cytospun to glass slides at 300 rpm in a Cytospin 2 (Thermo Scientific), fixed in 4% paraformaldehyde for 10 min at room temperature and washed again. Cells were then permeabilized with 0.2% Triton-X 100 for 5 min before being placed in blocking solution (2% goat serum, 2 mg/ml BSA in PBS). Slides were incubated with anti-cytochrome C antibody (1:200 in blocking solution) or anti-Flag antibody (1:200 in blocking solution) overnight at 4°C. The slides were washed with blocking buffer and incubated for 1 h at room temperature with a FITC-labeled mouse IgG isotype (1:200 in blocking solution, Alexis Biochemicals). After several washes with blocking buffer, the slides were mounted with VectaMount (Vector Laboratories, Inc.). To visualize STAT2 localization, permeabilized cells were incubated with anti-STAT2 antibody (1:200 in blocking solution). Confocal images were acquired using a Zeiss LSM510 Meta NLO confocal laser-scanning microscope (Carl Zeiss, Jena, Germany).
Measurement of Caspase 3 activation
Caspase-3 activation was measured using an EnzoLyte™ AMC Caspase 3 Assay Fluorimetric Kit (AnaSpec) following the manufacturer instructions. Briefly, 1 × 105 cells were plated in triplicate in a flat bottom 96-well plate. Cells were stimulated with or without IFN-α. After incubation, caspase 3 substrate was added to each well. Plates were incubated for 30 min at room temperature. Fluorescence intensity was measured in a Victor2TM 1420 multilabel counter (Perkin Elmer), at Ex/Em=354 nm/442 nm.
STAT2 and IRF-1 shRNA constructs and lentiviral infection
STAT2 and IRF-1 shRNA constructs cloned in pLKO.1 puromycin vector were obtained from Open Biosystems (Huntsville AL). Each construct contained a 21-bp sequence targeting specific regions of these genes. Lentivirus production and infections were performed as instructed by the manufacturer. Gene silencing was confirmed by Western blot analysis.
Acknowledgments
We thank Dr. Howard Young for helpful discussions and critical reading of the manuscript.
Grant support: This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health under Contract No. N01-CO-12400 (A. Gamero, A. Romero-Weaver, H-W Wang). This project described was also supported by Award Number K22CA095326 from the National Cancer Institute (A. Gamero).
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the officials views of the National Cancer Institute or the National Institutes of Health
Competing Interest Statement
The authors declare that they have no competing financial interests.
References
- 1.Guilhot F, Roy L, Guilhot J, Millot F. Interferon therapy in chronic myelogenous leukemia. Hematol Oncol Clin North Am. 2004;18:585–603. doi: 10.1016/j.hoc.2004.03.002. viii. [DOI] [PubMed] [Google Scholar]
- 2.Bleumer I, Oosterwijk E, De Mulder P, Mulders PF. Immunotherapy for renal cell carcinoma. Eur Urol. 2003;44:65–75. doi: 10.1016/s0302-2838(03)00191-x. [DOI] [PubMed] [Google Scholar]
- 3.Stoutenburg JP, Schrope B, Kaufman HL. Adjuvant therapy for malignant melanoma. Expert Rev Anticancer Ther. 2004;4:823–35. doi: 10.1586/14737140.4.5.823. [DOI] [PubMed] [Google Scholar]
- 4.Brunetto MR, Bonino F. Treatment of chronic hepatitis B: from research to clinical practice via the consensus conferences. Curr Pharm Des. 2004;10:2063–75. doi: 10.2174/1381612043384277. [DOI] [PubMed] [Google Scholar]
- 5.Picardi A, Gentilucci UV, Zardi EM, D'Avola D, Amoroso A, Afeltra A. The role of ribavirin in the combination therapy of hepatitis C virus infection. Curr Pharm Des. 2004;10:2081–92. doi: 10.2174/1381612043384330. [DOI] [PubMed] [Google Scholar]
- 6.Burks J. Interferon-beta1b for multiple sclerosis. Expert Rev Neurother. 2005;5:153–64. doi: 10.1586/14737175.5.2.153. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian HM, O'Brien S, Anderlini P, Talpaz M. Treatment of myelogenous leukemia: current status and investigational options. Blood. 1996;87:3069–81. [PubMed] [Google Scholar]
- 8.Li Z, Metze D, Nashan D, et al. Expression of SOCS-1, Suppressor of Cytokine Signalling-1, in Human Melanoma. J Investig Dermatol. 2004;123:737–45. doi: 10.1111/j.0022-202X.2004.23408.x. [DOI] [PubMed] [Google Scholar]
- 9.Ruuth K, Berglund A, Munoz V, Lundgren E. Differential resistance of melanoma cells to treatment with recombinant IFN-a2b and leukocyte IFN. Anticancer research. 2007;27:2109–14. [PubMed] [Google Scholar]
- 10.Kemmer Nyingi, N GW. Managing chronic hepatitis C in the difficult-to-treat patient. 2007:1297–310. doi: 10.1111/j.1478-3231.2007.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. The Journal of biological chemistry. 1997;272:28779–85. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 12.Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer research. 2005;65:3447–53. doi: 10.1158/0008-5472.CAN-04-4316. [DOI] [PubMed] [Google Scholar]
- 13.Wellbrock C, Weisser C, Hassel JC, et al. STAT5 Contributes to Interferon Resistance of Melanoma Cells. Curr Biol. 2005;15:1629–39. doi: 10.1016/j.cub.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 14.Ambrus JL, Sr, Dembinski W, Ambrus JL, Jr, et al. Free interferon-alpha/beta receptors in the circulation of patients with adenocarcinoma. Cancer. 2003;98:2730–3. doi: 10.1002/cncr.11843. [DOI] [PubMed] [Google Scholar]
- 15.Reu FJ, Bae SI, Cherkassky L, et al. Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol. 2006;24:3771–9. doi: 10.1200/JCO.2005.03.4074. [DOI] [PubMed] [Google Scholar]
- 16.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 17.Decker T, Lew DJ, Mirkovitch J, Darnell JE., Jr Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. Embo J. 1991;10:927–32. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kessler DS, Veals SA, Fu XY, Levy DS. Interferon-α regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes and Develop. 1990;4:1753–65. doi: 10.1101/gad.4.10.1753. [DOI] [PubMed] [Google Scholar]
- 19.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15:1312–7. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–3. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 21.Qureshi SA, Leung S, Kerr IM, Stark GR, Darnell JE., Jr Function of Stat2 protein in transcriptional activation by alpha interferon. Mol Cell Biol. 1996;16:288–93. doi: 10.1128/mcb.16.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gamero AM, Larner AC. Vanadate facilitates interferon alpha-mediated apoptosis that is dependent on the Jak/Stat pathway. The Journal of biological chemistry. 2001;276:13547–53. doi: 10.1074/jbc.M007948200. [DOI] [PubMed] [Google Scholar]
- 23.Gimeno R, Lee CK, Schindler C, Levy DE. Stat1 and stat2 but not stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T lymphocytes. Mol Cell Biol. 2005;25:5456–65. doi: 10.1128/MCB.25.13.5456-5465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Pham-Mitchell N, Schindler C, Campbell IL. Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J Clin Invest. 2003;112:535–43. doi: 10.1172/JCI18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gamero AM, Sakamoto S, Montenegro J, Larner AC. Identification of a Novel Conserved Motif in the STAT Family That Is Required for Tyrosine Phosphorylation. The Journal of biological chemistry. 2004;279:12379–85. doi: 10.1074/jbc.M310787200. [DOI] [PubMed] [Google Scholar]
- 27.Scarzello AJ, Romero-Weaver AL, Maher SG, et al. A Mutation in the SH2 domain of STAT2 prolongs tyrosine phosphorylation of STAT1 and promotes type I IFN-induced apoptosis. Mol Biol Cell. 2007;18:2455–62. doi: 10.1091/mbc.E06-09-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sangfelt O, Erickson S, Castro J, Heiden T, Einhorn S, Grander D. Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines. Cell Growth Differ. 1997;8:343–52. [PubMed] [Google Scholar]
- 29.Gamero AM, Potla R, Sakamoto S, Baker DP, Abraham R, Larner AC. Type I interferons activate apoptosis in a Jurkat cell variant by caspase-dependent and independent mechanisms. Cellular signalling. 2006;18:1299–308. doi: 10.1016/j.cellsig.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hobart M, Ramassar V, Goes N, Urmson J, Halloran PF. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J Immunol. 1997;158:4260–9. [PubMed] [Google Scholar]
- 31.Massa PT, Whitney LW, Wu C, Ropka SL, Jarosinski KW. A mechanism for selective induction of 2′-5′ oligoadenylate synthetase, anti-viral state, but not MHC class I genes by interferon-beta in neurons. Journal of neurovirology. 1999;5:161–71. doi: 10.3109/13550289909021998. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. The Journal of biological chemistry. 1996;271:5790–4. doi: 10.1074/jbc.271.10.5790. [DOI] [PubMed] [Google Scholar]
- 33.Melen K, Kinnunen L, Julkunen I. Arginine/lysine-rich structural element is involved in interferon-induced nuclear import of STATs. The Journal of biological chemistry. 2001;276:16447–55. doi: 10.1074/jbc.M008821200. [DOI] [PubMed] [Google Scholar]
- 34.Kemmer N, Neff GW. Managing chronic hepatitis C in the difficult-to-treat patient. Liver Int. 2007;27:1297–310. doi: 10.1111/j.1478-3231.2007.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chawla-Sarkar M, Leaman DW, Jacobs BS, et al. Resistance to interferons in melanoma cells does not correlate with the expression or activation of signal transducer and activator of transcription 1 (Stat1) J Interferon Cytokine Res. 2002;22:603–13. doi: 10.1089/10799900252982089. [DOI] [PubMed] [Google Scholar]
- 36.Lesinski GB, Valentino D, Hade EM, et al. Expression of STAT1 and STAT2 in malignant melanoma does not correlate with response to interferon-alpha adjuvant therapy. Cancer Immunol Immunother. 2005 doi: 10.1007/s00262-004-0649-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Wang S, Gobl A, Oberg K. Interferon alpha induction of Stat1 and Stat2 and their prognostic significance in carcinoid tumors. Oncology. 2001;60:330–8. doi: 10.1159/000058529. [DOI] [PubMed] [Google Scholar]
- 38.Mischiati C, Natali PG, Sereni A, et al. cDNA-array profiling of melanomas and paired melanocyte cultures. Journal of cellular physiology. 2006;207:697–705. doi: 10.1002/jcp.20610. [DOI] [PubMed] [Google Scholar]
- 39.Sanceau J, Hiscott J, Delattre O, Wietzerbin J. IFN-beta induces serine phosphorylation of Stat-1 in Ewing's sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene. 2000;19:3372–83. doi: 10.1038/sj.onc.1203670. [DOI] [PubMed] [Google Scholar]
- 40.Zhao W, Cha EN, Lee C, Park CY, Schindler C. Stat2-Dependent Regulation of MHC Class II Expression. J Immunol. 2007;179:463–71. doi: 10.4049/jimmunol.179.1.463. [DOI] [PubMed] [Google Scholar]
- 41.Chawla-Sarkar M, Lindner DJ, Liu YF, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–49. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 42.Panaretakis T, Pokrovskaja K, Shoshan MC, Grander D. Interferon-alpha-induced apoptosis in U266 cells is associated with activation of the proapoptotic Bcl-2 family members Bak and Bax. Oncogene. 2003;22:4543–56. doi: 10.1038/sj.onc.1206503. [DOI] [PubMed] [Google Scholar]
- 43.Thyrell L, Hjortsberg L, Arulampalam V, et al. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. The Journal of biological chemistry. 2004;279:24152–62. doi: 10.1074/jbc.M312219200. [DOI] [PubMed] [Google Scholar]
- 44.Hartman SE, Bertone P, Nath AK, et al. Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev. 2005;19:2953–68. doi: 10.1101/gad.1371305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panaretakis T, Hjortsberg L, Tamm KP, Bjorklund AC, Joseph B, Grander D. Interferon {alpha} Induces Nucleus-independent Apoptosis by Activating Extracellular Signal-regulated Kinase 1/2 and c-Jun NH2-Terminal Kinase Downstream of Phosphatidylinositol 3-Kinase and Mammalian Target of Rapamycin. Mol Biol Cell. 2008;19:41–50. doi: 10.1091/mbc.E07-04-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]