

Abstract
A 21-residue peptide segment, LL7-27 (RKSKEKIGKEFKRIVQRIKDF), corresponding to residues 7–27 of the only human cathelicidin antimicrobial peptide, LL37, is shown to exhibit potent activity against microbes (particularly Gram-positive bacteria) but not against erythrocytes. The structure, membrane orientation, and target membrane selectivity of LL7-27 are characterized by differential scanning calorimetry, fluorescence, circular dichroism, and NMR experiments. An anilinonaphthalene-8-sulfonic acid uptake assay reveals two distinct modes of Escherichia coli outer membrane perturbation elicited by LL37 and LL7-27. The circular dichroism results show that conformational transitions are mediated by lipid-specific interactions in the case of LL7-27, unlike LL37. It folds into an α-helical conformation upon binding to anionic (but not zwitterionic) vesicles, and also does not induce dye leakage from zwitterionic lipid vesicles. Differential scanning calorimetry thermograms show that LL7-27 is completely integrated with DMPC/DMPG (3:1) liposomes, but induces peptide-rich and peptide-poor domains in DMPC liposomes. 15N NMR experiments on mechanically aligned lipid bilayers suggest that, like the full-length peptide LL37, the peptide LL7-27 is oriented close to the bilayer surface, indicating a carpet-type mechanism of action for the peptide. 31P NMR spectra obtained from POPC/POPG (3:1) bilayers containing LL7-27 show substantial disruption of the lipid bilayer structure and agree with the peptide's ability to induce dye leakage from POPC/POPG (3:1) vesicles. Cholesterol is shown to suppress peptide-induced disorder in the lipid bilayer structure. These results explain the susceptibility of bacteria and the resistance of erythrocytes to LL7-27, and may have implications for the design of membrane-selective therapeutic agents.
Abbreviations used: AMP, antimicrobial peptide; ANS, anilinonaphthalene-8-sulfonic acid; CD, circular dichroism; CP, cross-polarization; DSC, differential scanning calorimetry; MIC, minimum inhibitory concentration; MLV, multilamellar vesicle; NMR, nuclear magnetic resonance; PBS, phosphate-buffered saline; PISEMA, polarization inversion spin exchange at the magic angle; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol; SUV, small unilamellar vesicle; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine; DMPG, 1,2-dimyristoyl-glycero-3-phosphatidylglycerol
Introduction
Cathelicidins are a family of structurally diverse AMPs that are located at the carboxyl terminus of a 15–18 kDa highly conserved cathepsin-L-inhibitor (cathelin)-like domain (1,2). LL37 is the only human antimicrobial peptide in the cathelicidin family. It is synthesized as an 18 kDa propeptide (hCAP18) consisting of a 13.5 kDa N-terminal cathelin-like domain and the 4.5 kDa active peptide, LL37 (Table 1), consisting of the 37 C-terminal residues. LL37 is active against both Gram-positive and Gram-negative bacteria. Several properties of LL37 make it an excellent candidate for research in this important area and for potential drug development (3,4). LL37 is the only human cathelicidin antimicrobial peptide, and therefore it is of considerable interest to understand how this peptide functions in detail. Unlike many other defensins, LL37 retains a broad-spectrum bactericidal activity at physiological or elevated salt concentrations—a distinct advantage for potential therapeutic uses (5). It is devoid of disulfide bridges, allowing easier and less costly chemical synthesis. Its high rate of microbial killing should provide an advantage for topical applications, since bacterial killing could be achieved before the peptide is mechanically cleared or inactivated. LL37 has also been shown to neutralize lipopolysaccharide activity (6). More details on the multifunctional properties and potential pharmaceutical applications of LL37 can be found elsewhere (3,4).
Table 1.
Amino acid sequences and properties of LL37 and LL7-27
| Peptide | Amino acid sequence | GRAVY30 | Aliphatic index31 |
|---|---|---|---|
| LL37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | −0.724 | 89.46 |
| LL7-27 | RKSKEKIGKEFKRIVQRIKDF | −1.371 | 69.52 |
LL37 appears to form dimeric and trimeric aggregates in solution under experimental conditions (7) as well as in lipid bilayers, as shown by 14N solid-state NMR studies (8), and is significantly protected from proteolytic degradation, whereas other native antimicrobial peptides are highly susceptible to enzymatic degradation. The biological expression and purification of LL37 from Escherichia coli have been reported (9,10). Previous NMR studies on LL37 reported the high-resolution structure in detergent micelles (11,12) and the mechanism of membrane disruption (13,14). Recent biophysics studies have reported membrane interactions (15–18) and fibril formation of LL37 (19).
LL37 and a subset of peptides corresponding to different segments of LL37 have been shown to exhibit potent lytic activities against microbes, cancer cells, and red blood cells (3,20). In our search for peptides with improved antimicrobial activity and cell selectivity, we identified a 21-residue peptide segment corresponding to residues 7–27 of LL37 (which we call LL7-27) that exhibits membrane-selective activity (Table 1). The structures and helical wheel representation of both peptides are given in Fig. 1. In this study, we investigated the hemolytic and antimicrobial activities of LL7-27 and its ability to disrupt the outer membrane of E. coli and to interact with model membranes that mimic bacterial and mammalian membranes. To obtain information on the membrane selectivity, we determined the lipid-induced conformational transitions of the peptide using CD experiments. Specific lipid-peptide interactions were observed by following the phase transition behavior of MLVs formed from a DMPC and also from DMPC/DMPG (3:1) mixture. The peptide's membrane orientation and ability to permeabilize anionic model membranes mimicking the bacterial inner membrane were studied by means of solid-state NMR experiments. Our data from this study suggest that the lipid-induced conformational transitions of the peptide and its integration with anionic membrane form the basis for the observed antimicrobial activity of LL7-27.
Figure 1.
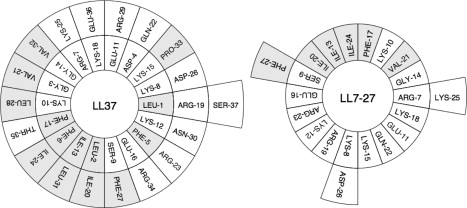
Helical wheel representations of LL37 and LL7-27. The NMR structure of LL37 was used (11).
Materials and Methods
Materials
All phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL). Chloroform and methanol were procured from Aldrich Chemical (Milwaukee, WI). Naphthalene was purchased from Fisher Scientific (Pittsburgh, PA). Buffers were prepared using water obtained from NONApure A filtration system. All protected amino acids, solvents, and reagents were purchased from Bachem (Torrance, CA), Synthetech (Butler, NJ), Aldrich Chemical (Milwaukee, WI), Fisher Scientific (Chicago, IL), and Protein Technologies (Tucson, AZ). All of the chemicals were used without further purification.
Peptide synthesis
Two peptides (LL7-27 and 15N-Val15-LL7-27) were prepared on an automated peptide synthesizer from Protein Technologies using standard solid-phase techniques for N-α-fluorenylmethyloxycarbonyl (Fmoc)-protected amino acids on Rink amide p-methylbenzhydrylamine resin (0.6 mmole/g). A 20% piperidine in N,N-dimethylformamide was used for deprotection. O-(Benzotriazol-1yl)-1,1,3,3-tetramethyluronium hexafluorophosphate was used for coupling. Deprotection and cleavage from the resin were accomplished using 11 mL of a 90% trifluoroacetic acid (TFA)/10% scavenger cocktail (anisole, thioanisole, ethanedithiol, phenol, and water). Crude peptides were purified to homogeneity by preparative reversed-phase high-performance liquid chromatography on a Waters instrument with a Phenomenex C18 column (2.2 × 25.0 cm, 10 mL/min). A linear gradient of 10% acetonitrile (0.1% TFA)/water (0.1% TFA) to 50% acetonitrile (0.1% TFA)/water (0.1% TFA) was employed. The molecular weight of the peptide was confirmed using mass spectrometry.
Bacterial strains and growth
Porphyromonas gingivalis (ATCC 33277) was obtained from the American Type Culture Collection. Enterococcus faecalis cultures were a gift from Dr. Donald B. Clewell (University of Michigan). Bacillus subtilis ATCC 6633, E. coli ATCC 8739, Staphylococcus aureus ATCC 6538, Pseudomonas aeruginosa ATCC 9027, Streptococcus Gordonia, Enterococcus faecalis FA2-2, Enterococcus faecalis OG1X, and Salmonella enteritis Typhimurium ATCC 14028 were obtained from MicroBiologics (St. Cloud, MN). The anaerobic bacteria were maintained by weekly transfer in an anaerobe chamber (Coy Manufacturing, Grass Lake, MI) at 37°C on PRAS Brucella agar plates (Anaerobe Systems, Morgan Hill, CA) in 10% carbon dioxide, 5% hydrogen, and 85% nitrogen atmosphere. Broth cultures were grown in a mixture of 50% Trypticase soy broth, 50% brain heart infusion broth, and 5 g/L yeast extract supplemented with 5 mg/L hemin, 0.01 gm/L sodium bisulfite, and 5 μg/L vitamin K. Aerobic species were maintained by weekly transfer on Trypicase soy agar and broth cultures grown from individual colonies in Trypticase soy broth.
Antimicrobial assay
A doubling dilution series of peptide (100 μg/mL to 0.1 ng/mL) was added to the wells of sterile 384-well microtiter plate (12 replicates per dilution) and dried for ∼12 h. Bacterial suspensions (10 μL, 107/mL) were added to the wells, centrifuged briefly to collect the cells in the bottom of the wells, and incubated at 37°C for 10–40 h depending upon the rate of growth of the bacterial species. MICs were set as the lowest concentration of the peptide at which there was no growth above the inoculated level of bacteria (p < 0.05, n = 12).
Hemolysis assay
The hemolytic activity of the peptides was determined by the released hemoglobin from suspensions of fresh sheep erythrocytes as absorbance at 414 nm. Red blood cells (Colorado Serum, Denver, CO) were centrifuged and washed four times with PBS (0.15 M NaCl, 0.005 M phosphate buffer, pH 7.4). Then 100 μL of red blood cells were added to the wells of a 96-well plate, and 100 μL of the peptide solution (in PBS) were added to each well. The plates were covered with an adhesive plastic sheet, incubated for 1 h at 37°C, and centrifuged at 200 × g for 10 min. Absorbance of the supernatants was measured at 414 nm, and 0% and 100% hemolysis was determined in PBS and 0.1% Triton X-100, respectively.
Outer-membrane disruption assay
The outer-membrane permeabilizing ability was investigated by means of the ANS uptake assay, using E. coli strain BL21 (DE3). Bacterial cells from an overnight culture were inoculated into Luria-Bertani broth medium. Cells from the mid-log phase were centrifuged and washed with Tris buffer (10 mM Tris, 150 mM NaCl, pH 7.4) and then resuspended in Tris buffer to an OD600 of 0.359. A stock solution of ANS was added to 3.0 mL of the cell suspension in a cuvette, to a final concentration of 5.75 μM. The extent of membrane disruption was observed as a function of peptide concentration by the increase in fluorescence intensity at ∼500 nm.
Circular dichroism
SUVs were prepared as described below and used for secondary structure analysis by means of CD experiments. An appropriate amount of lipid was dissolved in chloroform and the clear solution was taken to dryness. Tris buffer (10 mM Tris, 150 mM NaCl, pH 7.4) was added to dry lipid film and subjected to vortex and sonication to obtain a clear dispersion of SUVs. CD spectra were recorded (CDS model 62DS spectropolarimeter; AVIV Biomedical, Lakewood, NJ) at 25°C over a range of 200–250 nm using samples with a peptide/lipid ratio of ∼1:50 in a quartz cuvette (path length = 0.1 cm). Minor contributions from the buffer and SUVs were removed by subtracting the spectra of the corresponding control samples without peptide. The resultant spectra were normalized for path length and concentration.
Dye leakage assay
Carboxyfluorescein dye entrapped SUVs were prepared as described elsewhere (21). Briefly, Tris buffer (10 mM Tris, 100 mM NaCl, pH 7.0) containing 50 mM dye was added to the dry lipid film, vortexed, and sonicated. The dye-containing vesicles were then purified by gel filtration chromatography using a Sephadex G-75 column. Serial concentrations of peptides were added to aliquots of vesicle suspension (50 μM lipid) in Tris buffer, and the fluorescence emission intensity at 520 nm was recorded as a function of time using an excitation wavelength of 490 nm. The maximum leakage was determined by adding 0.1% Triton X-100. The steady-state fluorescence emission spectra of the peptide-vesicle mixtures were measured on a FluoroMax2 spectrofluorimeter (Jobin Yvon-Spex Instruments, Edison, NJ).
Differential scanning calorimetry
Lipid films of DMPC and DMPC/DMPG (3:1) were prepared by mixing the lipid and peptide in a chloroform-methanol mixture in the desired molar ratio, drying the sample under a gentle stream of nitrogen, and then placing it under vacuum overnight to remove any residual solvent. The dried samples were hydrated with Tris buffer (10 mM Tris, 150 mM NaCl, pH 7.4) and then vortexed above the main phase transition temperature to obtain MLVs. Tris buffer (10 mM Tris, 150 mM NaCl, pH 7.4) was added to MLVs to produce a final lipid concentration of 1.0 mg/mL DMPC and DMPC/DMPG (3:1). All buffers and samples were degassed under vacuum for 10 min before being loaded into the Nano-DSC II calorimeter (Calorimetry Sciences, Provo, UT). The scan rate was 1.0°C/min over a temperature range of 5–40°C. The raw data were converted to molar heat capacity using the CPCalc program provided with the calorimeter and taking into account the lipid concentration, molecular weight of each sample, and a partial specific volume of 0.956 mL/g.
Mechanically aligned bilayers
Mechanically aligned POPC and POPC/POPG (3:1) bilayers were prepared as described elsewhere (22). Briefly, 7 mg of lipids and an appropriate amount of peptide were dissolved in a CHCl3/CH3OH (2:1) mixture. The sample was dried under nitrogen gas and dissolved in a CHCl3/CH3OH (2:1) mixture containing equimolar quantities of naphthalene. An aliquot of the solution (∼300 μL) was spread on thin glass plates (Paul Marienfeld GmbH, Bad Mergentheim, Germany). The samples were dried under vacuum at ∼33°C for at least 12 h to effectively remove naphthalene and any residual organic solvents. After drying, the samples were hydrated at 93% relative humidity using saturated NH4H2PO4 solution for 3 days at 37°C, after which ∼2.5 μL of H2O were misted onto the surface of the lipid-peptide film. The glass plates were stacked, wrapped with parafilm, sealed in plastic bags (Plastic Bagmart, Marietta, GA), and then kept at 4°C for 6–24 h.
Solid-state NMR experiments
All NMR spectra of mechanically aligned lipid bilayers were obtained from a Chemagnetics/Varian Infinity 400 MHz solid-state NMR spectrometer operating at resonance frequencies of 40.549 MHz, 161.977 MHz, and 400.1375 MHz for 15N, 31P, and 1H nuclei, respectively, using 1H/31P and 1H/15N in-house-built double-resonance, flat-coil probes. A Chemagnetics temperature controller was used to maintain the sample temperature, and each sample was equilibrated at 37°C for at least 25 min before the experiment was started. The lipid bilayers were positioned in such a way that the bilayer normal was parallel to the external magnetic field. 15N spectra were recorded using a 1.5 ms ramp-CP (with a 10 kHz ramp on the 1H channel) sequence with a 1H π/2 pulse length of 3 μs, a recycle delay of 3 s, 50 kHz CP power, and a 71 kHz TPPM decoupling of protons during acquisition. The spectra were referenced relative to liquid ammonia (0 ppm). 31P spectra were obtained using a spin-echo sequence with a 3 μs 90° pulse width, 35 kHz proton-decoupling radio frequency field, and a 3 s recycle delay. A typical 31P spectrum required the coaddition of ∼1000 transients and was referenced relative to 85% H3PO4 on thin glass plates (0 ppm). Data were processed using Spinsight (Chemagnetics/Varian) software on a Sun Sparc workstation.
Results
Disruption of E. coli membranes
As can be seen from the MIC values given in Table 2, LL7-27 exhibits a broad spectrum of antimicrobial activity against Gram-negative and Gram-positive bacteria at micromolar concentrations. As the antimicrobial activity of LL37 is believed to stem from its ability to permeabilize bacterial membranes, it is likely that LL7-27 also exerts its activity by interacting with the bacterial membranes. Therefore, we compared the abilities of LL37 and LL7-27 to disrupt the bacterial membrane by monitoring the ANS uptake assay. The ANS (5.75 μM) equilibrated with 3 mL E. coli cells showed an emission maximum at ∼519 nm (Fig. 2 A, trace a). The successive addition of aliquots of LL37 to a 3 mL cell suspension resulted in an enhancement in the fluorescence intensity of ANS and a shift in the emission maximum. At 1.85 μM peptide concentration, the observed emission maximum was ∼480 nm. The blue shift in the emission maximum and the enhancement in the fluorescence intensity of ANS indicate that ANS relocates into a relatively less polar environment (presumably the bacterial membrane) as a consequence of outer-membrane disruption by LL37. On the other hand, ANS showed a blue-shifted emission maximum at ∼495 nm when a 3.17 μM concentration of LL7-27 was used against E. coli pretreated with 5.75 μM ANS dye (Fig. 2 B). The enhancement in the fluorescence intensity of ANS at the emission maximum upon addition of the highest concentration of the peptides was ∼1.5-fold (in the case of LL37) and ∼0.25-fold (in the case of LL7-27) higher than the fluorescence intensity of ANS (5.75 μM) in the absence of any peptides. These results suggest that LL7-27 is a weaker membrane-disrupting agent than the lytic peptide LL37, and that these peptides have different modes of interaction with the bacterial membrane.
Table 2.
Antimicrobial activities of LL37 and LL7-27 against different microbes
| Bacteria (OD600 = 0.002) | MIC (μg/mL) LL7-27 | MIC (μg/mL) LL37 |
|---|---|---|
| E. coli ATCC 12014 | 0.4 | 0.1 |
| P. aeruginosa ATCC 10145 | 100 | 12.5 |
| B. subtilus ATCC 11774 | 0.1 | 0.1 |
| S. gordonii | 6.25 | 12.5 |
| S. aureus | 50 | 50 |
| S. enterica ATCC BAA-215 | 50 | 50 |
| E. faecalis Strain FA2-2 | 12.5 | 12.5 |
| E. faecalis Strain OG1X | 50 | 50 |
| P. gingivalis Strain 33277 | 12.5 | 12.5 |
Figure 2.
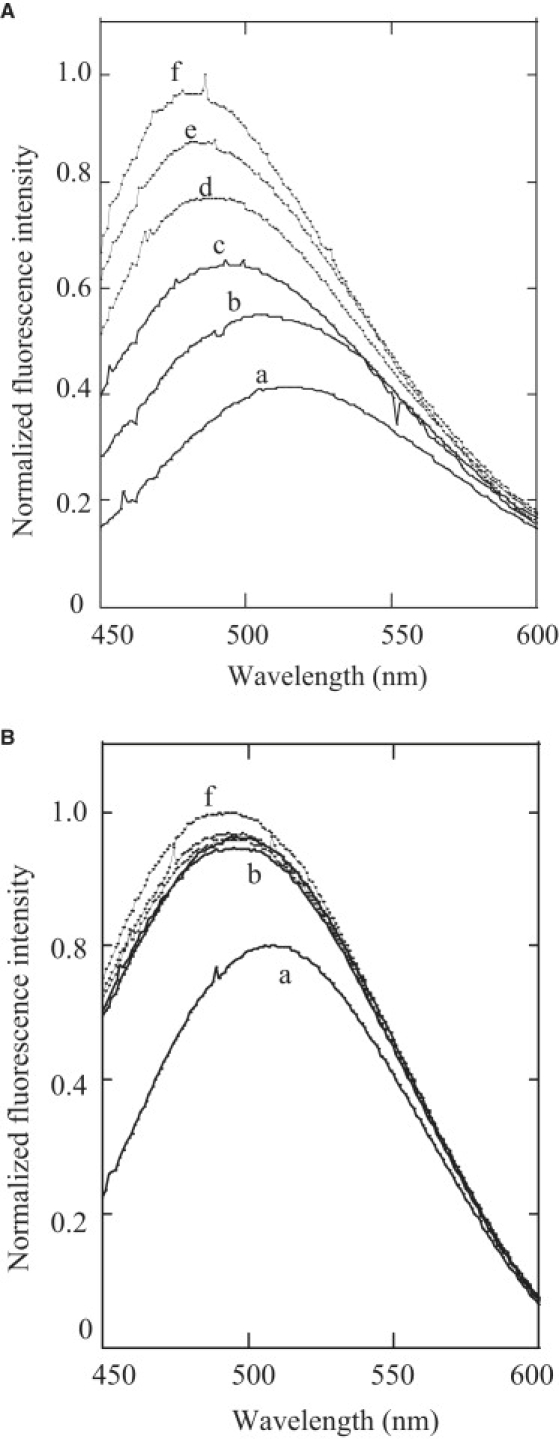
(A) LL37- and (B) LL7-27-induced ANS binding to E. coli membrane. Normalized fluorescence intensities are presented in both figures. The relative fluorescence intensities of ANS (5.75 μM) in the absence of the peptides (trace a in both figures) were comparable in both experiments. (A) LL37 concentrations: trace a (no peptide), b (0.37 μM), c (0.74 μM), d (1.11 μM), e (1.48 μM), and f (1.85 μM). (B) LL7-27 concentrations: trace a (no peptide), b (0.63 μM), c (1.26 μM), d (1.9 μM), e (2.53 μM), and f (3.17 μM).
Membrane bilayer peturbation from dye leakage and 31P NMR experiments
A number of antimicrobial peptides that strongly bind with negatively charged vesicles have been reported to induce leakage of vesicular contents. Because LL7-27 displayed a modest effect in disrupting the bacterial membrane, we studied its ability to induce dye leakage from lipid vesicles using a fluorescence method. Carboxyfluorescein dye-entrapped POPC/POPG (3:1) SUVs (50 μM) were suspended in Tris buffer (pH 7.4), aliquots of peptide solutions were added, and the kinetics of dye leakage was monitored as a function of time. Fig. 3, A and B, show the dye leakage profile upon addition of LL37 (up to 2.97 μM) and LL7-27 (up to 5.07 μM), respectively. As shown in Fig. 3, A and B, LL37 and LL7-27 induced significant dye leakage from POPC/POPG (3:1) vesicles in a concentration-dependent manner. These data clearly show that both LL37 and LL7-27 have the ability to permeabilize negatively charged membrane, and that LL7-27 is a weak membrane-disrupting agent. When aliquots of LL37 and LL7-27 were added to dye-entrapped POPC liposomes, only LL37 induced the release of dye in a concentration-dependent manner (80% dye leakage at P/L = 1:225; Fig. 3 C), and LL7-27 failed to induce any significant dye release even at P/L = 1:8 (Fig. 3 D). Only basal-level fluorescence (∼20%) was observed for POPC MLVs containing LL7-27 (Fig. 3 D), which is typical in dye leakage experiments due to stirring and slight changes in pH, and it was not subtracted in our data.
Figure 3.
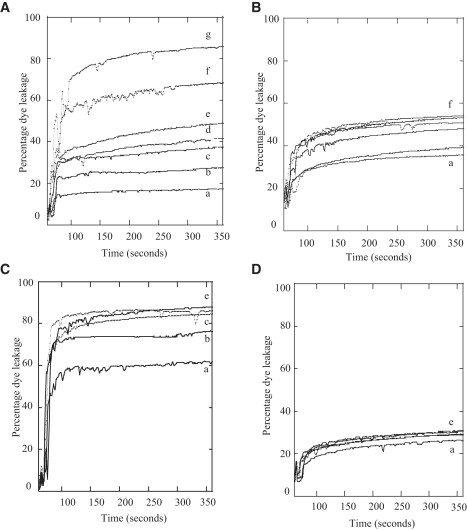
(A) LL37- and (B) LL7-27-induced carboxyfluorescene dye leakage from POPC/POPG (3:1) vesicles (50 μM). (A) LL37 concentrations: trace a (0.15 μM), b (0.37 μM), c (0.74 μM), d (1.11 μM), e (1.48 μM), f (2.26 μM), and g (2.97 μM). (B) LL7-27 concentrations: trace a (0.253 μM), b (0.633 μM), c (1.26 μM), d (2.53 μM), e (3.8 μM), and f (5.07 μM). (C) LL37- and (D) LL7-27-induced carboxyfluorescene dye leakage from POPC vesicles (50 μM). (C) LL37 concentrations: trace a (0.074 μM), b (0.148 μM), c (0.222 μM), d (0.329 μM), and e (0.37 μM). (D) LL7-27 concentrations: trace a (0.63 μM), b (2.52μM), c (3.78 μM), d (5.04 μM), and e (6.3 μM).
Previous solid-state NMR studies reported the LL37 structure and its interaction with various types of membrane (13,14). In this study, to further understand LL7-27's ability to disrupt the bacterial inner membrane, we performed 31P NMR experiments on mechanically aligned POPC and POPC/POPG (3:1) bilayers. A single narrow line at the higher frequency (or the parallel) edge of the 31P chemical shift powder pattern spectrum was observed, as reported in our earlier publications (see Fig. 4 A; data not shown for pure POPC) (13). No significant changes were observed for POPC bilayers containing up to 5 mol of LL7-27, which is consistent with its inability to induce dye leakage from POPC liposomes. On the other hand, when POPC/POPG (3:1) bilayers were incorporated with 3 mol % LL7-27, the linewidth of the peak increased significantly (Fig. 4 B), suggesting that the peptide binding to lipid bilayers affects the conformation of the lipid headgroups, which is in good agreement with the dye leakage from POPC/POPG (3:1) vesicles. Of interest, the inclusion of 3 mol % LL7-27 in POPC/POPG (3:1) bilayers containing 15 mol % cholesterol resulted in the spectrum shown in Fig. 4 C, which indicates that cholesterol inhibits the peptide-induced structural disorder in lipid bilayers.
Figure 4.
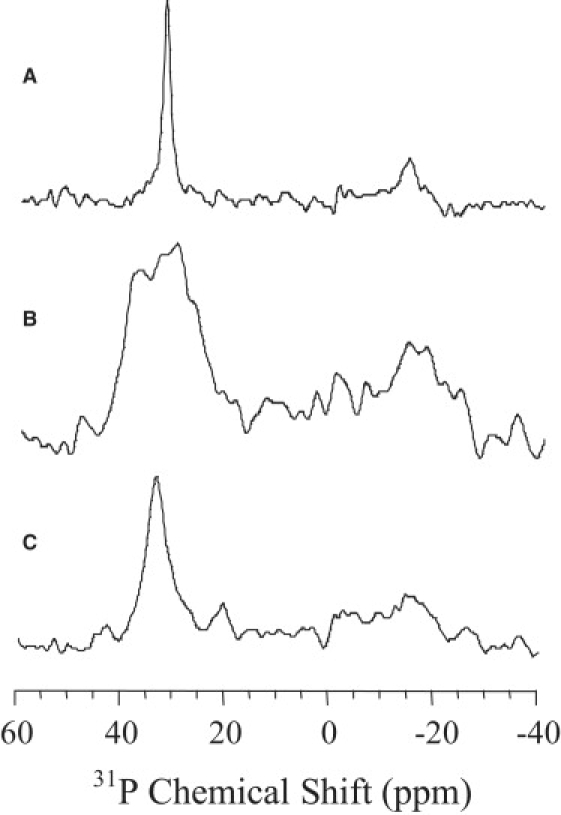
Phosphorus-31 chemical-shift spectra of mechanically aligned lipid bilayers at 37°C: (A) POPC/POPG (3:1), (B) POPC/POPG (3:1) containing 3 mol % of LL7-27, and (C) POPC/POPG (3:1) containing 3 mol % of LL7-27 and 15 mol % of cholesterol.
Secondary structure
Since the modes of interaction of LL7-27 with POPC and POPC/POPG (3:1) membranes are distinct, we studied the secondary structure of LL37 and LL7-27 in the presence of POPC and POPC/POPG (3:1) liposomes. The CD spectrum of LL37 in aqueous buffer (pH 7.4) displayed two broad minima at ∼206 and ∼222 nm, indicating the existence of a helical structure (Fig. 5 A), in good agreement with a previous NMR study of LL37 in solution. Upon the addition of POPC or POPC/POPG (3:1) liposomes, LL37 showed relatively sharp negative minima at 209 and 222 nm, suggesting a conformational transition into a regular α-helical structure. The calculated fractional helicity (∼40%) is in good agreement with the reported NMR structures from detergent micelles or lipid bilayers (11). On the other hand, the CD spectrum of LL7-27 in aqueous buffer exhibited a negative minimum at ∼203 nm, suggesting a random coil conformation (Fig. 5 B). In the presence of POPC/POPG (3:1) SUVs, the peptide showed a negative minimum at ∼208 nm and another minimum at ∼222 nm, indicating the formation of α-helical conformation; the fractional helicity of the peptide was calculated to be ∼25%. Of interest, the addition of POPC liposomes to the LL7-27 solution did not induce any observable transition in the conformation of the peptide; the CD profiles of LL7-27 in buffer and in the presence of POPC vesicles were comparable. This might suggest that the peptide does not bind to the zwitterionic POPC membrane, or it binds weakly and the membrane-induced structural changes in the peptide are minimal. From the CD data, it is clear that LL7-27, unlike its parent peptide LL37, interacts distinctly with anionic and neutral lipid membranes.
Figure 5.
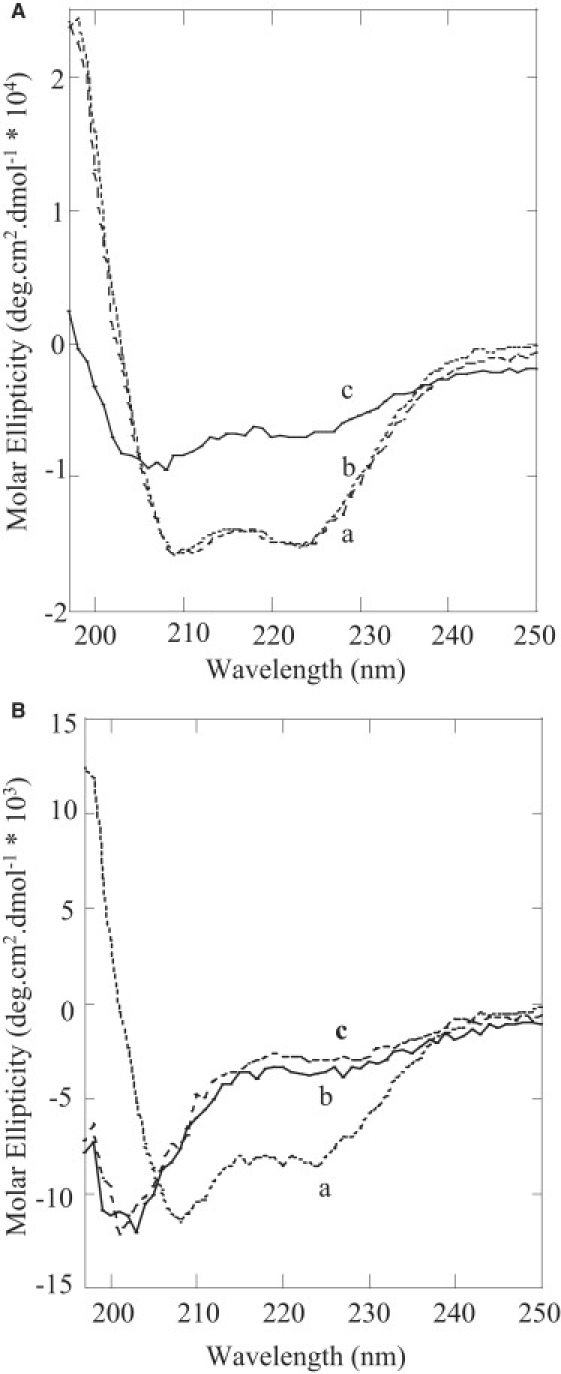
CD spectra of LL37 (A) and LL7-27 (B) in Tris buffer (solid line), POPC SUVs (dashed line), and POPC/POPG (3:1) SUVs (dotted line).
Effect of lipid-LL7-27 interactions on the main phase transition from DSC experiments
To further understand LL7-27-membrane interactions, we investigated the effect of the peptide on the thermotrophic phase transition behavior of multilamellar lipid vesicles. DSC themograms were obtained from pure DMPC and DMPC/DMPG (3:1) MLVs, and in the presence of different amounts of LL7-27. The effect of LL7-27 on the phase transition of negatively charged DMPC/DMPG (3:1) liposomes is shown in Fig. 6 A. The DMPC/DMPG (3:1) MLVs showed a pretransition at 16°C and the main transition at 24.8°C. However, when 0.258 mol % of LL7-27 was incorporated, a reduction in enthalpy of both the pretransition and main transition was observed. The incorporation of increasing quantities of LL7-27 produced a broadening of the phase transition curves as well as a reduction in the main phase transition enthalpy, suggesting a homogeneous intergration of LL7-27 with DMPC/DMPG (3:1) liposomes.
Figure 6.
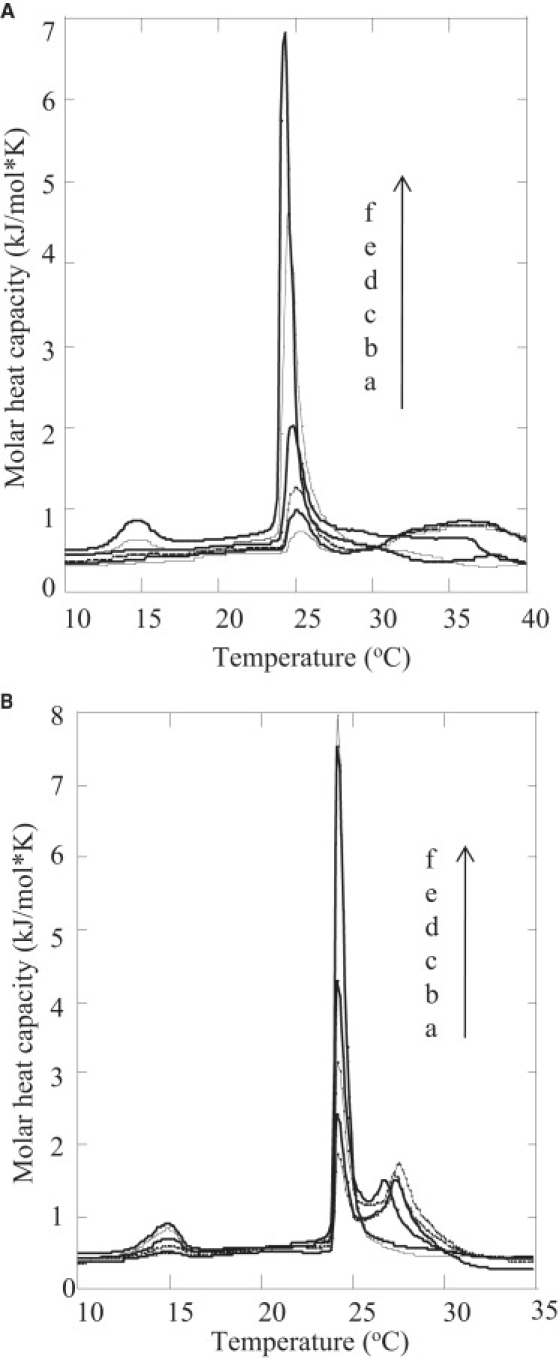
DSC thermograms showing the effects of LL7-27 on the phase transition of (A) DMPC/DMPG (3:1) (MLVs, 1 mg/mL) and (B) DMPC (MLVs, 1 mg/mL) consisting of various peptide concentrations: a (4.136 mol %; the bottom most trace); b (2.068 mol %); c (1.034 mol %); d (0.517 mol %); e (0.258 mol %); f (no peptide).
The enthalpies calculated for pure DMPC and DMPC incorporated with 0.258 mol % of LL7-27 are comparable, as the main phase transition behavior of DMPC MLVs was only marginally influenced by the presence of LL7-27 (Fig. 6 B). These data suggest that there is very little effect on the gel-to-liquid-crystalline (Lα) phase transition temperature at ∼24°C. Similarly, the pretransition arising from the lamellar gel (Lβ′) to lamellar rippled gel (Pβ′) phase is also not significantly affected. The few changes in the transition temperature (Tm) indicate that the peptide does not alter the packing of the hydrocarbon chains of DMPC in the gel and liquid-crystalline states. Consequently, the incorporation of progressive amounts of LL7-27 resulted in the abolition of the pretransition and the emergence of a two-component main transition. The relative contributions of the high-temperature component of the main transition also became more prominent at higher peptide concentrations. The sharp component at 24°C is attributed to the hydrocarbon chain melting of the peptide-poor lipid domain, whereas the broad high-temperature component is believed to arise from the peptide-rich lipid domain. The consistent decrease in the Tm and enthalpy of the sharp low-temperature component may be attributed to a decreasing size of the peptide-poor domain as the concentration of peptide is increased. In summary, these results indicate that LL7-27 does not integrate homogeneously with DMPC membrane and is located at the membrane-water interface, as revealed by the 15N NMR experiments described below.
Membrane orientation of LL7-27 from solid-state NMR experiments
It is important to determine the membrane orientation of LL7-27 to understand the mechanism of membrane disruption by the peptide (23). Solid-state NMR spectroscopy of aligned samples is a unique approach to determine the membrane orientation of the peptide, as demonstrated in previous studies on membrane-associated peptides and proteins (24–27). A two-dimensional PISEMA (28,29) spectrum correlating the 15N chemical shift and 15N-1H dipolar coupling frequencies associated with the amide site of the mechanically aligned POPC/POPG (3:1) bilayers containing 2 mol % 15N-Val15-LL7-27 is given in Fig. 7. The presence of a single peak corresponding to a 85 ppm 15N chemical shift and a 4.0 kHz 15N-1H dipolar coupling suggests that the LL7-27 peptide is oriented with its helix axis perpendicular to the bilayer normal. Since the peptide forms an amphipathic helical structure in a membrane environment and perturbs the lipid headgroup region, as revealed by 13P NMR results (Fig. 4), it is very likely that it is located close to the lipid bilayer surface and near the phosphate headgroup region of lipids. A model developed based on the NMR results presented in this study and also using the NMR structure of LL37 peptide is shown in Fig. 8.
Figure 7.
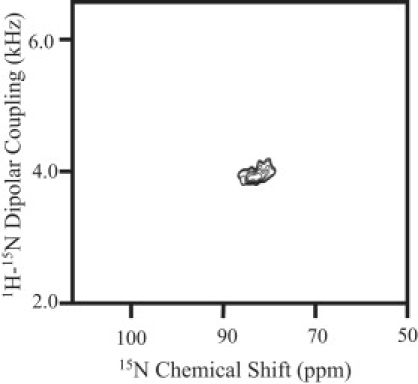
Nitrogen-15 chemical-shift spectrum of mechanically aligned POPC/POPG (3:1) lipid bilayers containing 3 mol % of LL7-27 at 37°C. The bilayer normal of the sample was set along the external magnetic field direction.
Figure 8.
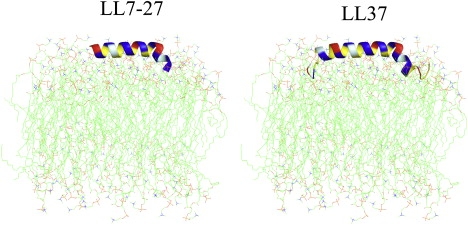
Orientation of LL37 and LL7-27 in phospholipid bilayers depicting a carpet-type mechanism of activity. The model was constructed from solid-state NMR experimental results.
Discussion
The broad spectrum of antimicrobial properties exhibited by the only human cathelicidin antimicrobial peptide, LL37, make it an attractive candidate for the design of potent peptide antibiotic compounds to potentially overcome the problem of increasing bacterial resistance. Previous NMR studies have reported its high-resolution three-dimensional structure (11), membrane orientation (13), and modes of membrane permeation from model membranes to elucidate the antimicrobial and other biological properties of this intriguing molecule (3,14). Since the formation of a helical structure in solution as well as in membrane has been thought to play an important role in the functions of LL37, it is imperative to investigate the property of a peptide segment that comprises a part of the highly helical region (residues 15–31) of LL37. The deletion of N- and C-terminal segments of LL37 would leave LL7-27 with a reduced hydrophobicity and α-helical propensity as compared to LL37. The altered hydrophobicity and helix propensity might minimize or eliminate the undesirable hemolytic activity observed in the case of LL37. With a net charge of +8 at neutral pH, and an aliphatic index of 69.52 (see Table 1) (30,31), LL7-27 can be expected to retain the broad spectrum of antimicrobial property of the parent peptide LL37. These 21 residues with a moderate α-helical propensity can fold, in principle, into ∼6 helical turns, providing a ∼31.5 Å long α-helix that is sufficient to span the membrane bilayer. In this study, we investigated the antimicrobial and membrane permeation properties of LL7-27 and compared them with those of the parent peptide LL37.
LL7-27 exhibits a broad spectrum of antimicrobial activities, but is more effective against Gram-positive bacteria
The MIC values reported in Table 2 indicate that LL7-27 inhibits the growth of several of the bacterial strains examined in this study and is slightly less potent than LL37 against Gram-negative bacteria. The pronounced activity of LL37 observed in ANS uptake by E. Coli cells (Fig. 2 A) and in dye leakage from POPC/POPG (3:1) liposomes (Fig. 3 B) mimicking the bacterial inner membrane suggest that LL37 is potent in disrupting both the inner and outer membranes of bacteria. This property is well correlated with the nonselective cell-lytic activities of LL37 given in Table 2. On the other hand, LL7-27 exhibits a strong disruption of the bacterial inner membrane (Fig. 3 A) but a weak disruption of the outer membrane of E. coli (Fig. 2 B). These membrane-disrupting properties are in good agreement with low MIC values for both peptides against Gram-positive bacteria and high MIC values for LL7-27 in the case of Gram-negative bacteria. For example, the MIC values against Gram-positive bacteria B. subtilus, S. aureus, and E. faecalis are the same for both peptides under the conditions used in this study. However, the MIC values for LL37 against Gram-negative bacteria E. coli and P. aeruginosa are considerably smaller than those of LL7-27. These results suggest that residues 1–6 and 28–37 of LL37 may have a role in disrupting the outer membrane of bacteria, and perhaps explain the nonselective cell-lytic activities of LL37. It is interesting to note that under the experimental conditions used to observe the antimicrobial properties, only LL37 (and not LL7-27) showed hemolytic activity. The selectivity displayed by LL7-27 against bacteria and not against human erythrocytes is intriguing and suggests that the mechanism of cell-peptide interactions should be explored at the molecular level.
Structured LL7-27 selectively permeates anionic membranes
The difference in the membrane composition of bacterial and mammalian cell membranes has been thought to play a major role in the selectivity of antimicrobial peptides. For example, the presence of cholesterol in human erythrocyte membrane imparts rigidity to the membrane bilayer and confers resistance to peptide-induced membrane permeation, as shown by deuterium NMR studies on pardaxin (32) and LL37 (14). Conversely, the presence of anionic lipids in bacterial membrane enables selective interaction with cationic AMPs (33,34). In this study, we determined the role of anionic lipids in the membrane permeating effects of LL7-27 using DSC, CD, and dye leakage experiments on zwitterionic POPC as a model mammalian membrane, and anionic POPC/POPG (3:1) as a model bacterial membrane. The selective interaction of the peptide with anionic membrane was confirmed by the complete integration of LL7-27 into DMPC/DMPG (3:1) membrane, as revealed by DSC thermograms (Fig. 6 A). The formation of peptide-rich domains (Fig. 6 B) in DMPC MLVs can only be explained by a lipid-induced peptide aggregation process. Because the homogeneous integration of the peptide in anionic lipid bilayers and the peptide aggregation in zwitterionic lipid bilayers are two distinct physical processes, the lipid-induced conformational changes in the peptide should also be distinctly different. This hypothesis is corroborated by the random coil to α-helical transition of LL7-27 in the presence of anionic POPC/POPG (3:1) lipids (Fig. 5 B). This hypothesis gains support from the 31P NMR data that reveal the peptide-induced disorder only in anionic lipid bilayers (Fig. 4 B). This proposition also explains our observation of peptide-induced leakage from anionic liposomes and not from zwitterionic liposomes (Fig. 3, B and D). Therefore, it appears that the electrostatic interactions between the cationic LL7-27 and anionic lipids lead to the segregation of cationic and hydrophobic residues along the nascent helix. Such a helix would be amphipathic and exert a profound effect on anionic membranes as compared to the weak interactions of LL7-27 with zwitterionic membranes.
Carpet-type mechanism of membrane disruption by LL7-27
The presence of a single peak in the low-frequency region (85 ppm) in the 15N chemical-shift frequency scale of the two-dimensional PISEMA spectrum with a 4 kHz dipolar coupling value suggests that the helix axis of the peptide is nearly perpendicular to the bilayer normal (Fig. 7). Since these NMR parameters are similar to values previously reported for LL37 (13), the membrane orientations of these two peptides are likely to be similar, at least in the helical regions of these peptides. The peptide-induced disorder measured by 31P chemical-shift spectra (Fig. 4 B) suggests that the cationic peptide should have strong interactions with anionic lipids in the lipid headgroup region of the bilayer. These results rule out the possibility of a transmembrane orientation and any barrel-stave-like, pore-forming mechanism of membrane disruption within the concentrations investigated in this study. Therefore, the peptide is likely to exert its activity via a carpet-type mechanism, as in the case of the parent peptide LL37, and at higher peptide concentrations may lead to either toroidal-pore formation and/or micellization of the membrane, as has been reported for the parent peptide LL37. Solid-state NMR and other studies have revealed the oligomeric nature of LL37 in both solution and membranes owing to the high aliphatic index (89.46) and α-helical propensity of the peptide. On the other hand, the unstructured LL7-27 could be a monomer in solution. Its low aliphatic index (69.52) and lack of ability to oligomerize in solution may form the basis for the peptide's inability to permeabilize zwitterionic POPC and human erythrocyte membranes.
In conclusion, we have identified a 21-residue nonhemolytic peptide fragment of LL37 that retains a broad spectrum of antimicrobial activity and exhibits significantly more potent activities against Gram-positive compared to Gram-negative bacteria. Using liposome assays and CD spectroscopy, we have shown that lipid-induced secondary structure formation is one of the initial steps involved in membrane-selective binding and permeabilization. The results of 15N NMR experiments on mechanically aligned lipid bilayers containing a 15N-labeled LL7-27 provide evidence for the membrane surface orientation of the peptide, which may suggest a “carpet-type mechanism” of membrane permeabilization. Our results also suggest that residues 1–6 and 28–37 in the parent peptide LL37 may have a role in the nonselective lytic activity of LL37, as well as in lipopolysaccharide detoxification. Further efforts to improve membrane selectivity and binding affinity could yield peptides with higher therapeutic indices. We also believe that the results reported in this study, which demonstrate several similarities and differences between LL7-27 and LL37, may provide insights into the in vivo activities of LL37.
Acknowledgments
We thank Ravi Nanga for preparing the structures of the peptides, Jeffrey Brender for valuable discussions, Amy Kruckemeyer and Dr. Vishnu Dhople for help with measuring the MIC values, and Drs. Dong-Kuk Lee and Ulrich Dürr for help with the NMR experiments.
This study was supported by research funds from the National Institutes of Health (AI054515 to A.R.).
References
- 1.Zanetti M., Gennaro R., Circo R. Cathelicidin peptides as candidates for a novel class of antimicrobials. Curr. Pharm. Des. 2002;8:779–793. doi: 10.2174/1381612023395457. [DOI] [PubMed] [Google Scholar]
- 2.Scott M.G., Davidson D.J., Hancock R.E. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002;169:3883–3891. doi: 10.4049/jimmunol.169.7.3883. [DOI] [PubMed] [Google Scholar]
- 3.Dürr U.H.N., Sudheendra U.S., Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta. 2006;1758:1408–1425. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 4.Nijnik A., Hancock R.E.W. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 2009;16:41–47. doi: 10.1097/moh.0b013e32831ac517. [DOI] [PubMed] [Google Scholar]
- 5.Mookherjee N., Hancock R.E.W. Cationic host defence peptides: innate immune regulatory peptides as a novel approach for treating infections. Cell. Mol. Life Sci. 2007;64:922–933. doi: 10.1007/s00018-007-6475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenfeld Y., Papo N., Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 2005;281:1636–1643. doi: 10.1074/jbc.M504327200. [DOI] [PubMed] [Google Scholar]
- 7.Oren Z., Lerman J.C., Shai Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999;341:501–513. [PMC free article] [PubMed] [Google Scholar]
- 8.Ramamoorthy A., Lee D.K., Henzler-Wildman K.A. Nitrogen-14 solid-state NMR spectroscopy of aligned phospholipid bilayers to probe peptide-lipid interaction and oligomerization of membrane associated peptides. J. Am. Chem. Soc. 2008;130:11023–11029. doi: 10.1021/ja802210u. [DOI] [PubMed] [Google Scholar]
- 9.Moon J.Y., Henzler-Wildman K.A., Ramamoorthy A. Expression and purification of a recombinant LL-37 from Escherichia coli. Biochim. Biophys. Acta. 2006;1758:1351–1358. doi: 10.1016/j.bbamem.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Li, Y., X. Li, H. Li, O. Lockridge, and Wang, G. A novel method for purifying recombinant human host defense cathelicidin LL-37 by utilizing its inherent property of aggregation. Protein Expr. Purif. 54:157–165. [DOI] [PubMed]
- 11.Porcelli F., Verardi R., Veglia G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry. 2008;47:5565–5572. doi: 10.1021/bi702036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 13.Henzler Wildman K.A., Lee D.K., Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 2003;42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 14.Henzler-Wildman K.A., Martinez G.V., Ramamoorthy A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry. 2004;43:8459–8469. doi: 10.1021/bi036284s. [DOI] [PubMed] [Google Scholar]
- 15.Neville F., Cahuzac M., Gidalevitz D. The interaction of antimicrobial peptide LL-37 with artificial biomembranes: epifluorescence and impedance spectroscopy approach. J. Phys. Condens. Matter. 2004;16:S2413–S2420. [Google Scholar]
- 16.Neville F., Cahuzac M., Gidalevitz D. Lipid headgroup discrimination by antimicrobial peptide LL-37: insight into mechanism of action. Biophys. J. 2006;90:1275–1287. doi: 10.1529/biophysj.105.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sevcsik E., Pabst G., Lohner K. Interaction of LL-37 with model membrane systems of different complexity: influence of the lipid matrix. Biophys. J. 2008;94:4688–4699. doi: 10.1529/biophysj.107.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sood R., Kinnunen P.K. Cholesterol, lanosterol, and ergosterol attenuate the membrane association of LL-37(W27F) and temporin L. Biochim. Biophys. Acta. 2008;1778:1460–1466. doi: 10.1016/j.bbamem.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 19.Sood R., Domanov Y., Kinnunen P.K. Binding of LL-37 to model biomembranes: insight into target vs host cell recognition. Biochim. Biophys. Acta. 2008;1778:983–996. doi: 10.1016/j.bbamem.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Li X., Li Y., Wang G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- 21.Ramamoorthy A., Thennarasu S., Krensky A.M. Cell selectivity correlates with membrane-specific interactions: a case study on the antimicrobial peptide G15 derived from granulysin. Biochim. Biophys. Acta. 2006;1758:154–163. doi: 10.1016/j.bbamem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Hallock K.J., Henzler Wildman K.A., Ramamoorthy A. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys. J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramamoorthy A. Beyond NMR spectra of antimicrobial peptides: dynamical images at atomic resolution and functional insights. Solid State Nucl. Magn. Reson. 2009;35:201–207. doi: 10.1016/j.ssnmr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bechinger B., Zasloff M., Opella S.J. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state nuclear magnetic resonance spectroscopy. Protein Sci. 1993;2:2077–2084. doi: 10.1002/pro.5560021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Page R.C., Li C., Cross T.A. Lipid bilayers: an essential environment for the understanding of membrane proteins. Magn. Reson. Chem. 2007;45(S1):S2–S11. doi: 10.1002/mrc.2077. [DOI] [PubMed] [Google Scholar]
- 26.Ramamoorthy A., Thennarasu S., Maloy L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bechinger B. Detergent-like properties of magainin antibiotic peptides: a 31P solid-state NMR spectroscopy study. Biochim. Biophys. Acta. 2005;1712:101–108. doi: 10.1016/j.bbamem.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Wu C.H., Ramamoorthy A., Opella S.J. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J. Magn. Reson. A. 1994;109:270–272. [Google Scholar]
- 29.Ramamoorthy A., Wei Y., Lee D.K. PISEMA solid-state NMR spectroscopy. Ann. Rep. NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 30.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 31.Ikai A.J. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88:1895–1898. [PubMed] [Google Scholar]
- 32.Porcelli F., Buck B., Veglia G. Structure and orientation of pardaxin determined by NMR experiments in model membranes. J. Biol. Chem. 2004;279:45815–45823. doi: 10.1074/jbc.M405454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallock K.J., Lee D.K., Ramamoorthy A. Membrane composition determines pardaxin's mechanism of lipid bilayer disruption. Biophys. J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Epand R.M., Epand R.F. Domains in bacterial membranes and the action of antimicrobial agents. Mol. Biosyst. 2009;5:580–587. doi: 10.1039/b900278m. [DOI] [PubMed] [Google Scholar]