

Abstract
Cyclic AMP (cAMP) receptor protein, which acts as the sensor of cAMP levels in cells, is a well-studied transcription factor that is best known for allosteric changes effected by the binding of cAMP. Although genetic and biochemical data on the protein are available from several sources, structural information about the cAMP-free protein has been lacking. Therefore, the precise atomic events that take place upon binding of cAMP, leading to conformational changes in the protein and its activation to bind DNA, have been elusive. In this work we solved the cAMP-free crystal structure of the Mycobacterium tuberculosis homolog of cAMP receptor protein at 2.9 Å resolution, and carried out normal-mode analysis to map conformational transitions among its various conformational states. In our structure, the cAMP-binding domain holds onto the DNA-binding domain via strong hydrophobic interactions, thereby freezing the latter in a conformation that is not competent to bind DNA. The two domains release each other in the presence of cAMP, making the DNA-binding domain more flexible and allowing it to bind its cognate DNA via an induced-fit mechanism. The structure of the cAMP-free protein and results of the normal-mode analysis therefore highlight an elegant mechanism of the allosteric changes effected by the binding of cAMP.
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis, and thus is responsible for one of the most dreadful diseases of mankind. In one of the many processes by which M. tuberculosis establishes a successful infection within the host, it modulates the host immune response to its advantage such that it can survive without being adversely affected by the host's immune system. Adenosine 3′:5′-cyclic monophosphate (cAMP) is believed to play a key role in this modulation of the host's immune system (1,2). Moreover, interference in the cellular signaling processes that are mediated by cAMP is believed to be one of the major contributing factors to the attenuation of the widely used vaccine strain, M. bovis BCG (3). Thus, understanding the signaling events in M. tuberculosis mediated by cAMP, and their influence on both M. tuberculosis and the host, may yield useful insights into M. tuberculosis and host interactions (4).
cAMP mediates a large variety of cellular signaling processes, including the well-characterized catabolite repression in prokaryotes (5). The activities of cAMP-mediated signaling are principally exerted via the catabolite activator protein (CAP), which has served as a paradigm for understanding allostery-mediated gene regulation (6). The dimeric transcriptional regulator in Escherichia coli is allosterically activated by binding to cAMP, thereby triggering a chain of events initiated by its binding to the cognate DNA, recruiting RNA polymerase, and activating expression of several genes in E. coli. The >100 genes that have been identified as regulated by this unique protein are involved in a host of cellular processes. Although originally identified as the positive regulator of catabolic gene functions, CAP has also been found to either activate or repress genes involved in functions other than catabolism. It is therefore also known as cAMP receptor protein (CRP) (5). The major common feature among all the CRP proteins in prokaryotes is cAMP-mediated DNA binding. The M. tuberculosis CRP homolog (CRPMt) encoded by the Rv3676 open reading frame has been characterized for its DNA-binding properties and has been predicted to regulate the expression of several genes in M. tuberculosis (7–9).
The crystal structures of various complexes of E. coli CRP (CRPEc) have been a rich source for elucidating the structural aspects of DNA recognition (10), in addition to the wealth of genetic, biochemical, and biophysical data available regarding this protein. The protein itself is composed of three distinct regions of the polypeptide: a large N-terminal domain that binds cAMP, a long α-helix (termed the C-helix) that mediates most of the intermonomer interactions, and a small C-terminal DNA-binding domain. The DNA recognition is mediated by a helix-turn-helix motif composed of E- and F-helices, which position themselves in successive major grooves of a double helical DNA, thereby facilitating the specific recognition of an inverted repeat sequence (10,11). The DNA, when bound to the two helix-turn-helix motifs of the two monomers, is severely kinked. The CRPEc structure has been a useful model for investigating protein-induced, large conformational changes in the DNA structure.
According to the current understanding, cAMP-free CRPEc is unable to bind DNA with high affinity, or, alternately, it binds to nonspecific sequences. Upon binding to one cAMP molecule, it undergoes a conformational transition that is capable of binding DNA (6). It is also capable of binding to two, three, or four cAMP molecules, but the specificity of recognition sequentially diminishes beyond two cAMP molecules bound to CRP. The third and fourth binding sites are also termed secondary binding sites because of their low affinity of binding (12). The physiological consequences of two, three, or four cAMP molecules bound to CRP are currently being debated, as the affinity of CRP for a specific DNA sequence is maximal in the presence of one cAMP molecule. Among the models proposed for different conformational changes, the most commonly accepted one suggests that the two subunits undergo a transformation characterized by a change in their relative orientation upon cAMP binding. Concomitantly, the cAMP-binding domain also undergoes a change in relative orientation with respect to the DNA-binding domain, to correctly juxtapose the DNA-recognition helices. However, despite the rich data available on CRP, the crystal structure of its apoform is as yet unavailable, which severely hampers the visualization of these conformational changes. In this report, we provide the first view, to our knowledge, of the cAMP-free crystal structure of the CRP homolog of M. tuberculosis. Supplemented by a normal-mode analysis, our results allow us to propose a universal mechanism of allostery-mediated DNA recognition of the CRP protein.
Materials and Methods
We previously reported the crystallization and data collection of CRPMt (13). The three-dimensional diffraction data collected at the XRD1 beamline of the Elettra Synchrotron Light Source (Trieste, Italy) yielded 2.9 Å resolution data, with crystals belonging to the orthorhombic space group P212121. All attempts to determine the structure through molecular replacement using the known structures as templates failed, suggesting considerable differences in the structure of CRPMt. Finally, the structure was determined by molecular replacement using AMoRE (14) with the recently deposited coordinates of M. tuberculosis CRP (15) (Protein Data Bank (PDB) ID: 3D0S). The CRPMt structure reported by us differs from 3D0S by three residues at the N-terminal and has considerable conformational differences, as discussed further below. The structure was refined using Refmac (16) and Phenix (17), and intermittent modeling was carried out using COOT (18). During TLS refinement, three groups were defined as follows: group 1, consisting of coordinates 1-116 of the cAMP-binding domain; group 2, consisting of coordinates 117-144 of the C-helix; and group 3, consisting of coordinates 145-214 of the DNA-binding domain. The TLS parameters at the end of refinement were analyzed using TLSANAL. The final refined structure was validated by MolProbity (19) and Procheck (20). Structure analysis was carried out using locally written scripts and, when mentioned, publicly available programs.
CRPEc in the absence of cAMP was modeled based on the refined coordinates of CRPMt, and similarly CRPMt was modeled in complex with cAMP based on the coordinates of CRPEc. The two modeled structures were energy-minimized using GROMACS by steepest descent (21). Normal-mode analysis was carried out using the elastic network model available from the ElNemo server (22). For the normal-mode analysis, end-point apo and holo structures of CRPEc and CRPMt were submitted, in each case with one belonging to the crystal structure and the other one belonging to the energy-minimized model. Low-frequency modes that showed good collectivity and high overlap were further analyzed in an attempt to understand the conformational changes. Dynamic cross-correlation matrices were calculated using GROMACS and MATLAB 7.0 (The MathWorks, Natick, MA). Distance plots were calculated using locally written scripts, and the inter-Cα distances of all the residues in each structure were calculated and stored. Using these values, when mentioned, differences between the inter-Cα distances were calculated for a pair of structures.
Results and Discussion
Structure determination, crystallographic refinement, and overall structure
The final R and Rfree values for the refined structure were 0.223 and 0.296, respectively (see Table S1 in the Supporting Material). The structure contains 3328 protein atoms in two subunits, 38 water molecules, and two sulfate ions (see Fig. S4). The refined structure shows reasonable geometry as judged by Procheck (20) and MolProbity (19). More than 90% of the residues fall within the most allowed regions of the Ramachandran plot; only three residues (Ser-75, Gly-166, and Lys-209 of the B-chain) are in the disallowed regions. Two of these three residues are located in the region that has high temperature factors. Because of the limited resolution of our structure, the geometrical weights in the refinement were restrained to the extent that the geometrical restraints did not exhibit large deviations from ideality.
Two strong densities in difference maps, located at the noncrystallographic symmetry-related positions of both monomers, were interpreted as sulfate ions because the crystallization buffer contained Li2SO4. The sulfate ions were placed in both subunits in an identical environment and were located where the phosphates of the cAMP would be placed in the primary binding site. They were stabilized by helix dipole interactions in a canonical manner (23,24).
The first 26 residues of one subunit are disordered in the structure, and were modeled only in short fragments where some density was observed. Thus, the two subunits possess residues 2–13, 18–23, and 27–224 in one subunit, and 1–214 residues in the other subunit. The overall tertiary structure of both subunits is similar to that of the well-studied CRP structure of E. coli. Hereafter, we retain the same residue numbers used in the sequence of the M. tuberculosis protein, but we adopt the designation of secondary structures from the well-studied E. coli structure (Fig. S4). The M. tuberculosis protein has eight additional residues at the N-terminal, which form a short α-helix. Although the significance of disorder in the first 26 residues of one subunit is not clear, it may be hypothetically possible that these residues act as a gate in controlling cAMP's access to its binding site. However, no evidence for such a gating mechanism exists in the literature.
Comparison between the two monomers
The two monomer structures are very similar except for a difference in the relative orientation between the N-terminal cAMP-binding and C-terminal DNA-binding domains. When the two monomers are superposed, the root mean-square deviation (RMSD) is 1.73 Å for 152 equivalent Cα atoms (Fig. 1 A). The RMSD for individual domain superposition is considerably better, with values of 1.29 Å (108 equivalent Cα atoms) for the cAMP-binding domains (Fig. S1) and 1.1 Å (54 equivalent Cα atoms) for the DNA-binding domains (Fig. S1). The difference in relative orientation between the cAMP-binding and DNA-binding domains arises due to a rotation of 27° around an axis placed close to residues 142–145. These residues thus act as a hinge, around which the DNA-binding domain changes orientation with respect to the cAMP-binding domain.
Figure 1.
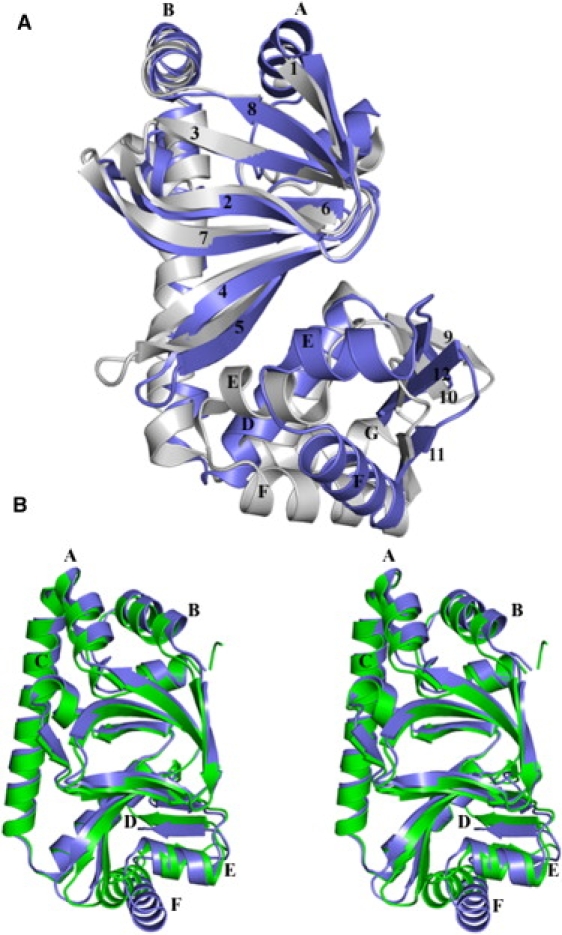
Least-squares superposition of the two subunits of CRPMt (3H3U), and CRPMt (3H3U) and CRPMt (3D0S). The RMSD values for the superposition (see text for details) using all of the residues (A) is considerably higher than those obtained using the coordinates of only the cAMP-binding domain (Fig. S1) or the DNA-binding domain (Fig. S1), reflecting the fact that the two domains are oriented differently in the two subunits. Within the DNA-binding domain, apart from the reorientation of the D-helix, the rest of the domain shows good structural alignment. (B) Superposition of the B-chain of CRPMt (3H3U) and CRPMt (3D0S).
Unlike the recently reported structure of CRPMt, where superposition of the DNA-binding domains of the two chains yields an RMSD of 3.1 Å, the two DNA-binding domains in our structure match well with each other, with an RMSD of 1.1 Å over 54 equivalent Cα positions (15). The only difference in the conformation of the DNA-binding domains is the relative orientation of the D-helix with respect to the rest of the domain. It is therefore likely that the conformational differences in the DNA-binding domains of the two subunits, as observed in the reported CRPMt structure, are due to crystal packing effects rather than inherent conformational heterogeneity (15).
Comparison of our structure with the recently reported cAMP-free CRPMt structure
The recently reported cAMP-free CRPMt structure has significant differences in the conformation of its two DNA-binding domains (15). The asymmetry in the two chains (i.e., the orientations between the two domains), as well as the internal asymmetry within the DNA-binding domains, is reported to be due to the absence of cAMP. On the other hand, our structure shows no internal asymmetry within the individual domains, although the two domains are juxtaposed differently with respect to each other. Since our structure is also devoid of cAMP, it appears unlikely that the absence of cAMP has any effect on the internal asymmetry in the structure. The consistent feature in the two structures is the relative change in orientation between the N- and C-terminal domains, which likely arises due to the absence of cAMP (Fig. 1 B).
The most noticeable difference between the 3D0S structure and ours is in the helix-turn-helix motif of the B-chains. In the 3D0S structure, the DNA-recognition F-helix is drawn toward the D-helix, which makes its conformation significantly different from that of any other reported structures (RMSD = 1.92 Å for 42 equivalent Cα atoms between 3D0S and 1G6N). The conformational difference is spread over the entire DNA-binding domain, as is clearly shown in Fig. 2 A. On the other hand, when the DNA-binding domain of our structure is compared with the cAMP-bound structures of CRPEc, the major conformational change is seen only in the orientation of the D-helix closer to the hinge point (RMSD = 1.71 Å for 54 equivalent Cα atoms of our structure and 1G6N; Fig. 2 B). As a result of the F-helix packing closely with the D-helix in the 3D0S structure, the D-helix is seen to undergo local unwinding toward its N- and C-termini. This unwinding leads to more van der Waals contacts of Phe-198 with other residues (Fig. 3), which are different in our structure (Fig. 3).
Figure 2.
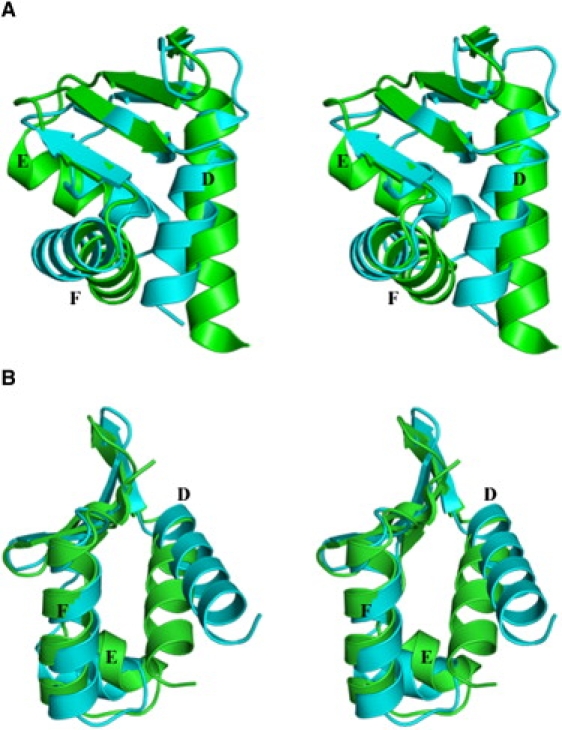
Superposition of the DNA-binding domains of the CRPEc and CRPMt. (A) Superposition of the DNA-binding domain of the cAMP-bound CRPEc (cyan) and cAMP-free CRPMt (green) (PDB ID: 3D0S) shows the change in conformation of the D-helix and helix-turn-helix motif of the cAMP-free CRPMt. (B) Superposition of the DNA-binding domain of the cAMP-bound CRPEc (cyan) and cAMP-free CRPMt (green) (PDB ID: 3H3U) shows the major change in conformation of the D-helix. The helix-turn-helix motif takes a conformation closer to the cAMP-bound conformation of CRPEc (cyan).
Figure 3.
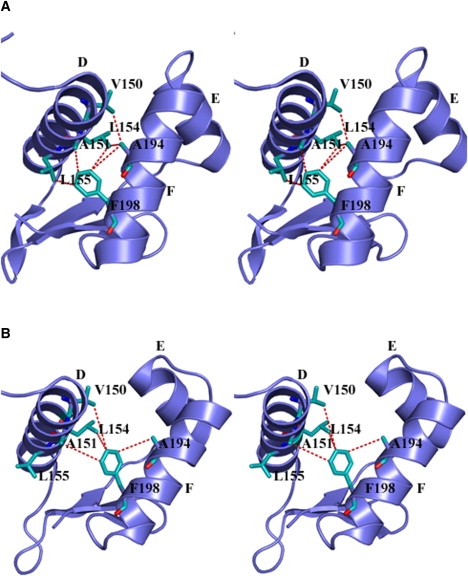
Stereo image of the CRPMt (A) (3D0S) and CRPMt (B) (3H3U) DNA-binding domain, illustrating the protrusion of the F-helix away from the D-helix, and the difference in the interaction of the residues with Phe-198.
The different conformation of the DNA-binding domain in the 3D0S structure may be a consequence of crystal-packing interactions. In the 3D0S structure, this domain is involved in 43 crystal-packing interactions that are closer than 3.5 Å, whereas in our structure it is involved in only nine interactions (Table S2 and Table S3). Moreover, crystal packing brings this domain closer to the cAMP-binding domain of another molecule. Consequently, the first 26 residues, which are disordered in our structure, are in a well-ordered conformation in 3D0S. Thus, the crystal-packing interactions not only affect the conformation of the DNA-binding domain, they also appear to significantly affect the structure of the cAMP-binding domain in 3D0S.
Comparison between cAMP-bound and cAMP-free structures
As predicted by previous genetic studies, one of the interesting differences between the cAMP-bound and cAMP-free structures is the change in relative orientation between the cAMP-binding and DNA-binding domains (25). In the holo CRPEc structure, the two domains are asymmetrically disposed in the two subunits. One is described as “closed”, as observed in the ternary complex of CRPEc, cAMP, and DNA. The “open” conformation is observed in the presence of cAMP and the absence of DNA (26). In our apo structure, this conformation is similar to the “closed” conformation in one of the subunits, and different from both the “open” and “closed” conformations in the other subunit (Fig. 4). Thus, there are likely to be three distinct conformational states of CRP: “open” when bound to cAMP but not to DNA, “closed” when bound to both cAMP and DNA, and “other” in the absence of cAMP and DNA.
Figure 4.
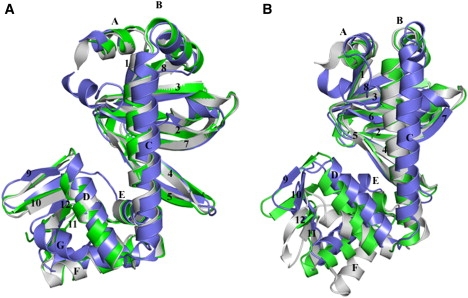
Superposition of CRPEC with CRPMt. (A) Superposition of the A-chains of all three structures—apo (blue), binary complex with cAMP (gray), and ternary complex with cAMP and DNA (green)—shows that the conformations of one subunit in all of the structures are almost identical. (B) Superposition of the B-chains in the three structures, with the same coloring scheme as in panel A. It is clear that the apo structure has a distinct conformation compared to both the binary and ternary complexes.
Quantification of the relative rotation with DynDom (29) revealed that the difference in orientation between the cAMP-binding and DNA-binding domains in the two subunits arises due to two principal motions: a small but significant rotation around an axis placed close to the residues just preceding the C-helix, and a larger rotation at the end of the C-helix. The latter motion corresponds to a rotation of 27° anchored at residues 142–145. Apart from the domain rotations, the C-helices of the two monomers also undergo a relative change in their directions. In the holo structures, these helices are juxtaposed at ∼25° with each other, whereas in the apo structure they are juxtaposed at ∼18° with each other (Table 1). It was previously suggested that the two monomers of CRP may reorient upon binding of cAMP (26). The small but noticeable change in the orientation of the two C-helices as observed in the apo structure confirms this suggestion.
Table 1.
Angles between different helices of two monomers and buried accessible areas (Å2)
| Apo structure of CRPMt (this work) | CRPEc + cAMP complex (1G6N) | CRPEc + cAMP + DNA complex (1O3T) | CRPEc + cAMP + DNA + RNA polymerase C-domain complex (1LB2) | |
|---|---|---|---|---|
| Angle between C-helices of the two monomers (°) | ||||
| 18 | 24 | 24 | 25 | |
| Buried accessible areas | ||||
| Buried accessible surface area of A- and B-chains, respectively (%) | 16 + 18 | 11 + 6 | 12 + 12 | 12 + 12 |
| Area buried by the two chains (Å2) | ||||
| A-chain | 463 | 307 | 347 | 310 |
| B-chain | 550 | 166 | 336 | 310 |
| Angle between E- and F-helices | ||||
| A-chain (°) | 108.0 | 99.0 | 104.0 | |
| B-chain (°) | 91.0 | 102.0 | 104.0 | |
Buried accessible surface areas are between the cAMP-binding and DNA-binding domains among different structures. The PDB code is indicated in parentheses.
The cAMP-binding sites of both of the subunits in our structure are occupied by Arg-130 and Glu-80 (Arg-123 and Glu-72 of the E. coli sequence, respectively). The guanidino group of Arg-130 is located where the adenine moiety of the cAMP would be placed, whereas Glu-80 is located where the ribofuranose ring would be placed. The occupation of this position by Arg appears to be effected by a change in conformation around the χ3 torsion angle of the Arg side chain, and in addition to simply occluding the cAMP-binding site, it has major consequences for many interactions in this region. As illustrated in Fig. 5 A, in all of the structures of CRP where cAMP is present, the side chain of this Arg is involved in ionic interactions with Glu-72 and Asp-68 on either side of the guanidine group (Glu-80 and Asp-76, respectively, in M. tuberculosis). Moreover, it tethers β-strand 6 of the cAMP-binding domain via interaction with the main-chain carbonyl oxygen of residue 69 (77 in M. tuberculosis). The latter interaction is lost in the apo structure due to the change in conformation of Arg-130 (Fig. 5 B). Consequently, β-strand 6 and the entire cAMP-binding domain appear to move further away from the C-helix in the apo form. Thus, the loss of interactions of Arg-130 in the apo structure finally manifest in a rigid-body rotation of the cAMP-binding domain away from the C-helix by almost 30°.
Figure 5.
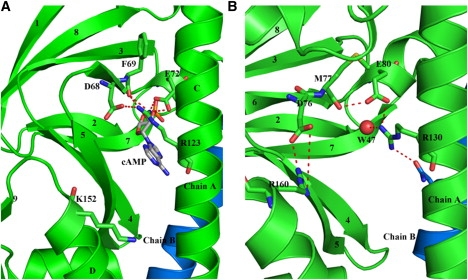
Change in the conformation of Arg-130 and its interactions between the apo and holo structures. (A) Conformation as seen in the E. coli holo structure, where Arg-123 is seen to form ionic interactions with Glu-72 and Asp-68 on either side of its guanidino group. It also interacts with the main carbonyl of residue 69 of β-strand 6. (B) The conformation as seen in the M. tuberculosis apo structure, where the interactions of Arg-130 with Asp-76 and the main-chain carbonyl of residue 77 are now lost.
Reorientation of the cAMP-binding domain as a consequence of ligand binding further leads to several changes in the manner in which this domain interacts with the DNA-binding domain (Fig. 4, A and B). The change in relative reorientation between the two domains is most visible when the cAMP-binding domains of the apo and holo structures are superposed (Fig. 6 B). Whereas the two cAMP-binding domains superpose very well with an RMSD of 1.3 Å over 108 residues, the C-helix appears to move distinctly toward this domain in the holo structure (Fig. 6 B). Consequently, β-strands 4 and 5 of the cAMP-binding domain occupy the position where the E-helix of the DNA-binding domain is present in the apo structure (Fig. 6 A). Occlusion of the E-helix leads to an overall rotation of the DNA-binding domain away from its position. Of interest, β-strands 4 and 5 are juxtaposed in a similar manner in both apo and holo structures with respect to the rest of the cAMP-binding domain, with no apparent conformational change as an effect of cAMP binding. Thus, upon cAMP binding, the DNA-binding domain appears to be forced out, resulting in a rigid body rotation around a hinge centered on residues 142–145.
Figure 6.
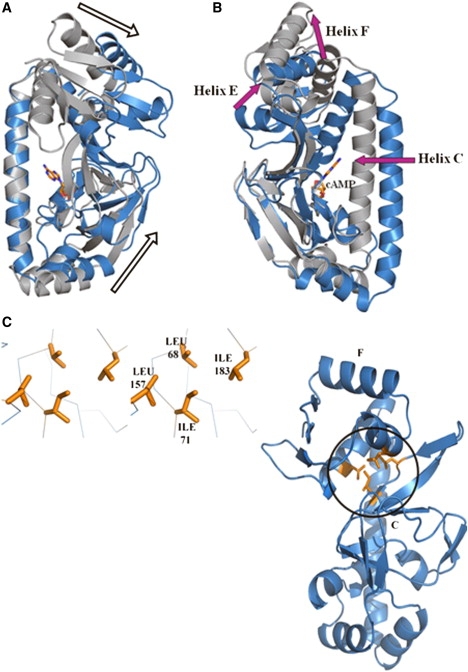
Change in the relative orientation between the cAMP-binding domain and the DNA-binding domain in CRPMt. (A) A stereo view of the superposition of apo (blue) and holo (gray) structures, when the C-helices are aligned, clearly shows that the cAMP- and DNA-binding domains move away from each other in the presence of cAMP. (B) An alternate view of conformational changes between the apo (blue) and holo (gray) structures is seen when the cAMP-binding domains are aligned. In this view, the C-helix is clearly shown to move toward the cAMP-binding domain in the presence of cAMP. (C) Due to the reorientation between the cAMP- and DNA-binding domains, a small hydrophobic core is formed at their interface in the apo structure. Two side chains each from the two domains contribute to the formation of the hydrophobic core. The two domains also now bury a larger accessible surface between them, as shown in Table 2.
The outward orientation of the cAMP-binding domain in the absence of cAMP results in an exposure of several hydrophobic residues at its interface with the DNA-binding domain (Fig. 6 C). This further leads to changes in buried accessible areas between the two domains (Table 1). The apo structure of CRPMt has the highest area buried between the two domains (1013 Å2), which approaches values close to those observed in stable protein/protein complexes (30). Although the A-chains of all of the structures have very similar tertiary structures, the area buried between the two domains is higher in the apo structure by ∼25%. Of interest, the B-subunit of the crystal structure in complex with cAMP (PDB code: 1G6N) has the least amount of area buried between the two domains (473 Å2). Thus, an important step in the cascade of events upon cAMP binding appears to be a weakening of interfacial interactions between the cAMP-binding and DNA-binding domain.
Apart from the changes in buried accessible areas, the formation of many ion pairs between residues of the two domains is also observed in the apo structure. For example, Asp-76 (68 in E. coli), which interacts with Arg-130 (123 in E. coli) in the holo structure, is now placed farther away, severing its interaction with Arg-130. As a result, Asp-76 in both subunits in our structure now occupies a position where it can form a salt bridge with Arg-160 (Lys-152 in E. coli) of the last turn of the D-helix of the DNA-binding domain. The absence of this salt bridge in the holo structure is a direct consequence of cAMP binding, as Arg-130 in the holo structure is well placed to interact with Asp-76. Thus, the overall consequence of binding of cAMP to CRP is a triggering of the reorientation of the cAMP-binding domain with respect to the C-helix and the DNA-binding domain, with Arg-130 (123 in E. coli) playing a pivotal role in initiating this event. The reorientation brings the two domains closer in the absence of cAMP, with the centroids of the two domains closer by ∼4 Å (Table 2).
Table 2.
Distance (in Å) between centroids of DNA-binding and cAMP-binding domains
| Protein | Chain A | Chain B |
|---|---|---|
| 1G6N (CRPEc + cAMP) | 27.5 | 30.3 |
| 1O3T (CRPEc + cAMP + DNA) | 28.4 | 27.7 |
| CRPMt | 24.1 | 23.7 |
Genetic studies have revealed that the hinge residues in CRPEc (residues 138–141) interact closely with the F-helix. These residues therefore affect the distance between the F-helix and the hinge, where bulkier substitutions in these residues lead to CPR∗ mutants (25). In our structure, the corresponding residues (Asp-145, Gly-148, and Ala-151) are also in close proximity to the F-helix, where Ala-151 forms close van der Waals interactions with Phe-198 of the F-helix (Fig. 3). This contact is similar to the Ala-144–Leu-190 van der Waals contact in CRPEc. The presence of Phe in CRPMt at the same place as Leu-190 in CRPEc makes CRPMt resemble CRP∗. It is therefore clear that bulky substitutions in these residues will lead to outward protrusion of the F-helix, as predicted from the genetic studies (25).
The cAMP-free NMR structure of CRPEc has recently become available, which allows us to carry out a comparative analysis between apo CRPEc and CRPMt (26). Of interest, the second half of the C-helix is seen to be highly flexible in the NMR structure, whereas this region has some of the lowest B-factors in the two crystal structures of CRPMt. This region precedes the hinge between the two domains. In the two crystal structures of CRPMt, the hinge has high temperature factors in the B-chain but low temperature factors in the A-chain. The high B-factors in one chain may reflect structural disorder of the hinge. Thus, the NMR and crystal structures have subtle but important differences. The two crystal structures appear to suggest a flexible hinge flanked by rigid domains, giving rise to a fulcrum-like motion. The NMR analysis, on the other hand, suggests a wider flexible region of the structure, starting from the middle of the C-helix.
In the cAMP-free NMR structure, Trp-85 occupies the cAMP-binding pocket (26). This residue is Ser in CRPMt, when sequences of CRPEc and CRPMt are aligned. However, a closely located structurally equivalent Phe-38 in CRPMt appears to function in the same manner as Trp-85. As in the NMR structure, Phe-38 occupies the cAMP-binding pocket. This placement of Phe-38 is further stabilized by Phe-78 and Tyr-48 drawing closer via aromatic interactions. The role of Trp-85 in CRPEc was previously predicted by the results of fluorescence studies (27), which are therefore in agreement with the NMR analysis (26) and the two crystal structures of CRPMt (15) (this work).
Paths of conformational changes
One of the most powerful ways to study conformational changes in proteins is to construct distance maps and then analyze the differences in the distance maps between two conformational states of the protein of interest (31). Such an analysis can yield information regarding the most and least variable regions between two conformational states of the protein. Because CRP exists in three major conformational states, we constructed distance maps for three states of CRP: the apo form (this work), the CRP + cAMP complex (28) (PDB: 1G6N), and the CRP + (cAMP)2 + DNA ternary complex (10) (PDB: 1O3T). The two difference distance maps (one between the first two states, and one between the last two states) clearly show that there are no noticeable changes within the two domains (i.e., the cAMP-binding and DNA-binding domains); rather, most changes occur across the two domains. Of interest, although the entire DNA-binding domain shows large movement away from the cAMP-binding domain, the region spanning the last two turns of the E-helix until the first turn of the F-helix appears invariable when the apo and holo structures are compared (Fig. S2 A). This is also apparent when the binary and ternary complexes are compared (Fig. S2 B). Thus, this region of the polypeptide may act like a fulcrum during the reorientation of the two domains (Fig. 7 A).
Figure 7.
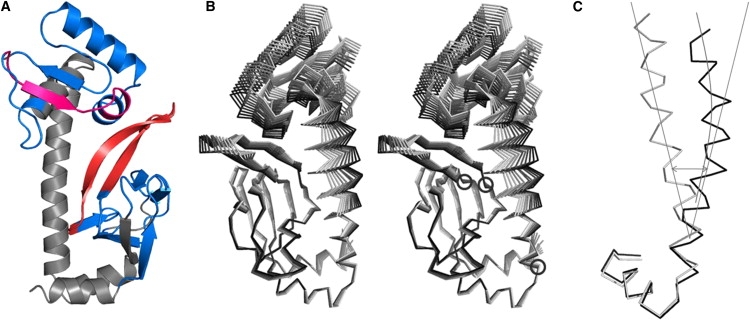
Addressing the dynamic behavior of CRP from normal-mode analysis. (A) Regions of polypeptide that show significant correlated or anticorrelated motions are shown. Those shaded in blue represent polypeptide regions exhibiting anticorrelated motions, whereas those in magenta and red show correlated motions. The structural proximity of residues that exhibit correlated motions (magenta and red) suggests that conformational changes are transmitted through these residues between the two domains. (B) Superposition of intermediate structures for normal mode 13. The intermediate deformed structures were energy-minimized and superposed using their cAMP-binding domains. Apart from the anticorrelated movement between the cAMP- and DNA-binding domains, the spring-like motion of the C-helix is also apparent (see text for details). The hinge regions are shown circled. (C) Normal mode 13 shows significant deformation in the C-helix when the cAMP-binding domains are aligned. The red curve shows the curvature of the helix, and blue lines represent the directions of the two helices. Only the first three turns of the C-helix were considered to calculate its direction vector. This deformation of the C-helix has an overall effect on the reorientation between the cAMP- and DNA-binding domains. Color figures are presented in the online version.
Further, in a comparison of the maps of apoCRP and holo CRP, the residues of the C-helix also show large deviations away from both the cAMP- and DNA-binding domains (Fig. S2 A). These residues of the C-helix do not show as large deviations when the holoCRP is compared with the ternary complex of the CRP structure. It thus appears that there are two conformational transitions in CRP: one when CRP binds cAMP, poising it to bind the cognate DNA, and the other when the cAMP-bound CRP comes in contact with the cognate DNA. In the former, not only is the DNA-binding domain reoriented with respect to the cAMP-binding domain, but the C-helix also shows significant variations in conformation. In the latter, a mere change in the conformation of the DNA-binding domain is sufficient for it to recognize the cognate DNA. These observations are in agreement with our conclusions based on superpositions of the DNA-binding domains as discussed above.
To map the paths of conformational change from the apo structure to the holo structure, we analyzed low-frequency normal modes that showed maximal overlap and collectivity of motions. Since the six lowest-frequency normal modes represent trivial motions, this analysis was carried out for mode 7 onward. Normal-mode analysis of CRPEc based on the cAMP-bound crystal structure of E. coli and the model of its apo structure using CRPMt as a template showed that mode 13 had the maximum overlap (44%) and collectivity (58%). Similarly, normal-mode analysis of CRPMt using the apo crystal structure (this work) and the model of its holo form using CRPEc as a template showed that mode 16 had the maximum overlap (33%) and collectivity (55%). Of interest, the dynamic cross-correlation matrices of these two normal-mode calculations are very similar (Fig. S3, A and B), and therefore might yield useful insights into the paths of conformational changes.
The two normal-mode analyses reveal that the cAMP- and DNA-binding domains exhibit overall anticorrelated motions. This observation agrees well with our crystallographic analyses discussed in the preceding paragraphs. The anticorrelated movements of the two domains are analogous to breathing motions, and may be a direct consequence of the allosteric effects of cAMP binding. Of interest, regions of the polypeptide, including β-strands 4 and 5 of the cAMP-binding domain, and β-strand 10 and the E-helix of the DNA-binding domain, show significant correlated motions. These regions span the two domains but are structurally proximal. It is therefore apparent that the motions of the two domains are communicated through these regions of the polypeptide.
The intermediate deformed structures of the selected normal modes have the ability to reveal paths of conformational changes (32). The normal-mode analyses of CRPMt and CRPEc using apo and holo structures elegantly reveal the conformational changes required to attain one end-point structure starting from another. By superimposing the cAMP-binding domains of all the intermediate structures, we show that not only do the two domains exhibit substantial dynamic behavior, but the C-helix also plays an important role in the change in the conformation of the cAMP binding domain (Fig. 7 B). As noted above, the C-helices were seen to undergo a relative change in orientation by ∼6°. The residues that act like a hinge are also apparent in the superposed structures of intermediate conformations (Fig. 7 B). These hinges appear to be responsible for the motion of β-strands 4 and 5, and the entire region spanning the C-helix and the DNA-binding domain with respect to the cAMP-binding domain.
Apart from a rigid-body rotation between these two helices, they are also seen to exhibit significant flexible motion, with substantial elasticity analogous to that of a long pole (see movies in the Supporting Material). This spring-like movement of the C-helices (Fig. 7 C) has an effect in altering the local interactions toward the C-terminal end of the helix. In the two monomers, these interactions are noticeably different. Thus, the paths of conformational changes between the two domains appear to be communicated in an interplay of bending of the C-helix and hinge-like motion centered around residues 141–145, with the fulcrum being provided by the last two turns of the E-helix and the first turn of the F-helix.
Thus, the overall effect of cAMP binding on CRP begins with reorientation of side chains in the cAMP-binding pocket, followed by drawing of the cAMP-binding domain toward the C-helix and concomitant weakening of interactions between the cAMP- and DNA-binding domains. This leads to enhanced flexibility of the DNA-binding domain as it is released from the rest of the body of the protein. The enhanced flexibility of the DNA-binding domain has further repercussions on the plasticity of the C-helix, with the two together accounting for large swings of the DNA-binding domain in conformational space. The cAMP-induced flexibility of the DNA-binding domain can be elegantly explained by the reported genetic and biochemical data on the protease sensitivity of CRP (6). In the absence of cAMP, CRP is resistant to proteases, but in the presence of micromolar concentrations of cAMP it becomes sensitive to a variety of proteases, generating an N-terminal polypeptide. The apoCRPMt structure clearly shows that the mobility of the DNA-binding domain is restricted in the absence of cAMP due to its close interactions with the cAMP-binding domain, but is more flexible in the presence of cAMP. This increase in flexibility can be correlated with its susceptibility to proteolysis. Similarly, ANS binding to hydrophobic surfaces also can be correlated with the “open” apo structure and the “closed” holo structure. Finally, the release of the DNA-binding domain from the body of the CRP structure in the presence of cAMP, and the concomitant increase in its flexibility, make it adept in binding to the correct DNA sequence by an induced fit.
It is noteworthy that the DNA-recognition F-helices are parallel only in the DNA-bound form of the structures; in the absence of DNA, these helices are not oriented parallel to each other. Parallel juxtaposition of the F-helices is of paramount importance in sequence-specific DNA recognition because these helices are placed in successive major grooves of the DNA. Our proposed mechanism suggests that even though the F-helix of one subunit would be able to place itself in the cognate recognition sequence of the DNA, the F-helix of another subunit would bind DNA only by an induced fit. Thus, the key feature of sequence-specific recognition of DNA lies in the flexibility of the DNA-binding domain only in the presence of the ligand, cAMP.
Acknowledgments
We thank Doriano Lamba, Maurizio Polentarutti, and other staff members of the XRD1 beamline at the Elettra Synchrotron Light Source for help during data collection.
This work was supported by a grant from the Centres of Excellence, Department of Biotechnology, government of India, and a grant from the Department of Science and Technology, government of India, to S.C.M. P.K. is a Senior Research Fellow supported by the University Grants Commission. S.E.H. is a JC Bose National Fellow. We also thank the Department of Science and Technology for travel support for data collection.
Footnotes
Mohd Akif's present address is Department of Biology and Biochemistry, University of Bath, Bath, UK.
Yusuf Akhter's present address is European Molecular Biology Laboratory Outstation, Hamburg, Germany.
Supporting Material
References
- 1.Padh H., Venkitasubramanian T.A. Cyclic adenosine 3′, 5′-monophosphate in mycobacteria. Indian J. Biochem. Biophys. 1976;13:413–414. [PubMed] [Google Scholar]
- 2.Shenoy A.R., Visweswariah S.S. New messages from old messengers: cAMP and mycobacteria. Trends Microbiol. 2005;14:543–550. doi: 10.1016/j.tim.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Spreadbury C.L., Pallen M.J., Cole J.A. Point mutations in the DNA- and cNMP-binding domains of the homologue of the cAMP receptor protein (CRP) in Mycobacterium bovis BCG: implications for the inactivation of a global regulator and strain attenuation. Microbiology. 2005;151:547–556. doi: 10.1099/mic.0.27444-0. [DOI] [PubMed] [Google Scholar]
- 4.Bai G., Schaak D.D., McDonough K.A. cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol. Med. Microbiol. 2009;55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botsford J.L., Harman J.G. Cyclic AMP in prokaryotes. Microbiol. Rev. 1992;56:100–122. doi: 10.1128/mr.56.1.100-122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolb A., Busby S., Adhya S. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 1993;62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- 7.Bai G., McCue L.A., McDonough K.A. Characterization of Mycobacterium tuberculosis Rv3676 (CRPMt), a cyclic AMP receptor protein-like DNA binding protein. J. Bacteriol. 2005;187:7795–7804. doi: 10.1128/JB.187.22.7795-7804.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akhter Y., Tundup S., Hasnain S.E. Novel biochemical properties of a CRP/FNR family transcription factor from Mycobacterium tuberculosis. Int. J. Med. Microbiol. 2007;297:451–457. doi: 10.1016/j.ijmm.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Akhter Y., Yellaboina S., Hasnain S.E. Genome scale portrait of cAMP-receptor protein (CRP) regulons in mycobacteria points to their role in pathogenesis. Gene. 2008;407:148–158. doi: 10.1016/j.gene.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 10.Chen S., Gunasekera A., Berman H.M. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: alteration of DNA binding specificity through alteration of DNA kinking. J. Mol. Biol. 2001;314:75–82. doi: 10.1006/jmbi.2001.5090. [DOI] [PubMed] [Google Scholar]
- 11.Harman J.G. Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta. 2001;1547:1–17. doi: 10.1016/s0167-4838(01)00187-x. [DOI] [PubMed] [Google Scholar]
- 12.Scott S.P., Jarjous S. Proposed structural mechanism of Escherichia coli cAMP receptor protein cAMP dependent proteolytic cleavage protection and selective and nonselective DNA binding. Biochemistry. 2005;44:8730–8748. doi: 10.1021/bi0479609. [DOI] [PubMed] [Google Scholar]
- 13.Akif M., Akhter Y., Mande S.C. Crystallization and preliminary x-ray crystallographic studies of Mycobacterium tuberculosis CRP/FNR family transcription regulator. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:873–875. doi: 10.1107/S1744309106027977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navaza J. AMoRE: an automated package for molecular replacement. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- 15.Gallagher D.T., Smith N., Reddy P.T. Profound asymmetry in the structure of the cAMP-free cAMP receptor protein (CRP) from Mycobacterium tuberculosis. J. Biol. Chem. 2009;284:8228–8232. doi: 10.1074/jbc.C800215200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 17.Adams P.D., Grosse-Kunstleve R.W., Terwilliger T.C. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 18.Emsley P., Cowtan K. COOT: model building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Davis I.W., Leaver-Fay A., Richardson D.C. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laskowski R.A., MacArthur M.W., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 21.Lindahl E., Hess B., van der Spoel R. GROMACS 3.0: a package for molecular simulations and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 22.Suhre K., Sanejouand Y.H. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32(Web Server issue):W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hol W.G.J. The role of the α-helix dipole in protein function and structure. Prog. Biophys. Mol. Biol. 1985;45:149–195. doi: 10.1016/0079-6107(85)90001-x. [DOI] [PubMed] [Google Scholar]
- 24.Chakrabarti P. An assessment of the effect of the helix dipole in protein structures. Protein Eng. 1994;7:471–474. doi: 10.1093/protein/7.4.471. [DOI] [PubMed] [Google Scholar]
- 25.Kim J., Adhya S., Garges S.S. Allosteric changes in the cAMP receptor protein of Escherichia coli: hinge reorientation. Proc. Natl. Acad. Sci. USA. 1992;89:9700–9704. doi: 10.1073/pnas.89.20.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popovych N., Tzeng S.R., Kalodimos C.G. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc. Natl. Acad. Sci. USA. 2009;106:6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wasylewski M., Małecki J., Wasylewski Z. Fluorescence study of Escherichia coli cyclic AMP receptor protein. J. Protein Chem. 1995;14:299–308. doi: 10.1007/BF01886787. [DOI] [PubMed] [Google Scholar]
- 28.Passner J.M., Schultz S.C., Steitz T.A. Modeling the cAMP-induced allosteric transition using the crystal structure of CAP-cAMP at 2.1 Å resolution. J. Mol. Biol. 2000;304:847–859. doi: 10.1006/jmbi.2000.4231. [DOI] [PubMed] [Google Scholar]
- 29.Hayward S., Kitao A., Berendsen H.J.C. Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins. 1997;27:425–437. doi: 10.1002/(sici)1097-0134(199703)27:3<425::aid-prot10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 30.Janin J., Bahadur R.P., Chakrabarti P. Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- 31.Madhusudan, Vijayan M. Rigid and flexible regions in lysozyme and the invariant features in its hydration shell. Curr. Sci. 1991;60:165–170. [Google Scholar]
- 32.Akif M., Suhre K., Mande S.C. Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase: crystal structure and normal-mode analysis. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1603–1611. doi: 10.1107/S0907444905030519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.