

Abstract
βαβ structural motifs are commonly used building blocks in protein structures containing parallel β-sheets. However, to our knowledge, no stand-alone βαβ structure has been observed in nature to date. Recently, for the first time that we know of, a small protein with an independent βαβ structure (DS119) was successfully designed in our laboratory. To understand the folding mechanism of DS119, in the study described here, we carried out all-atom molecular dynamics and coarse-grained simulations to investigate its folding pathways and energy landscape. From all-atom simulations, we successfully observed the folding event and got a stable folded structure with a minimal root mean-square deviation of 2.6 Å with respect to the NMR structure. The folding process can be described as a fast collapse phase followed by rapid formation of the central helix, and then slow formation of a parallel β-sheet. By using a native-centric Gō-like model, the cooperativity of the system was characterized in terms of the calorimetric criterion, sigmoidal transitions, conformation distribution shifts, and free-energy profiles. DS119 was found to be an incipient downhill folder that folds more cooperatively than a downhill folder, but less cooperatively than a two-state folder. This may reflect the balance between the two structural elements of DS119: the rapidly formed α-helix and the slowly formed parallel β-sheet. Folding times estimated from both the all-atom simulations and the coarse-grained model were at microsecond level, making DS119 another fast folder. Compared to fast folders reported previously, DS119 is, to the best of our knowledge, the first that exhibits a parallel β-sheet.
Introduction
The protein folding problem has been studied using both experimental and theoretical approaches over the years. Despite great advances during the last decades, it is still not fully clarified. With the rapid increase of computational power, all-atom ab initio simulation of protein folding has become feasible. Since the seminal work of Yong Duan and co-workers (1), many folding simulations have been conducted using all-atom models, and most of these have focused on a limited number of fast-folding proteins (2), such as the villin headpiece subdomain (1,3–6), the albumin binding domain (7), protein A (8–11), and Trp-cage (12–14). These model systems have provided valuable information on the protein folding mechanism.
The βαβ motifs, with two parallel β-strands packed against one α-helix, are the building blocks for proteins with α/β structures. Theoretical investigation reveals that the βαβ structure possesses high designability (H. Chen, C. Zeng, and C. Tang, unpublished result). As hypothesized by Li et al., there is a designability principle underlying nature's selection of protein folds (15), and this principle implies that the βαβ fold may exist as an independent structure. However, to our knowledge, the stand-alone form of the βαβ motif has never been observed in nature until now. Recently, a small protein with an individual βαβ motif (DS119) was successfully designed de novo and its monomeric structure was solved by NMR (16). To our knowledge, it is the first reported example of the novel βαβ fold. The small size and the building block nature of DS119 make it an ideal model system for studying the folding of α/β proteins.
De novo protein design has made significant progress in recent years (17), and several novel proteins have been created by either computational or empirical design (18–20). Although well structured and fairly stable, these novel proteins may exhibit folding characteristics that differ from those of natural proteins. For example, most small natural proteins fold cooperatively in a two-state-like manner, whereas the de novo designed protein Top7 (21) was found to fold in a significantly less cooperative way, with severe chevron rollovers and a very rugged corresponding energy landscape. A recent coarse-grained simulation (22) revealed that the noncooperative behavior of Top7 comes from its native topology, which differs from that of natural proteins. Thus, it is of great interest to investigate the folding mechanism of de novo designed proteins, which will be helpful in understanding protein folding mechanism and further improving protein design methods.
Since systematic kinetic/thermodynamic studies of DS119 or similar proteins with parallel β-sheets have not been available, to our knowledge, in this study we performed all-atom molecular dynamics and coarse-grained simulations to investigate the folding pathway and energy landscape of DS119. In the all-atom simulations, the folding event was observed in one of the four trajectories. The folding process could be described as a fast collapse phase followed by a fast formation of the helix, then slow formation of the parallel β-sheet. The major rate-limiting step of DS119 folding was the formation of the parallel β-sheet. From the coarse-grained Gō model simulations, DS119 was shown to be an incipient downhill folder, that is, one that folds more cooperatively than a downhill folder but less cooperatively than a two-state folder. Folding times estimated from all-atom simulations and the coarse-grained model were both at the microsecond level, making DS119 another fast folder with a unique parallel β-sheet that, to the best of our knowledge, has not been observed in other fast folders.
Models and Methods
All-atom folding simulations
The DS119 protein contains 36 residues in the sequence GSGQVRTIWVGGTPEELKKLKEEAKKANIRVTFWGD, as described in our article about its design (16). Simulations were performed using the AMBER (23) package. The FF03 force field (24) was used. The solvation effects were incorporated with the GB/SA implicit solvent model. Starting from a fully extended structure, 500 steps of steepest descent followed by 500 steps of conjugate gradient minimization were carried out. The minimized structure was further heated and equilibrated at 300 K for 50 ps. For the production run, the temperature was controlled at 300 K with a coupling time constant of 2 ps. Ionic strength was set to 0.2 M. The time step used was 2 fs. Bonds involving hydrogen atoms were constrained with SHAKE (25). The nonbonded interaction and GB pairwise summation cutoff was 12 Å. Four trajectories lasting for 1 μs each, with different initial speed assignments, were generated. Among these, the folding event was observed in the third trajectory only, which was then extended to 2 μs; the folding-pathway analysis was based on this extended trajectory.
Among the 20 NMR structures obtained for DS119, the first structure was used as the reference structure in the root mean-square deviation (RMSD) calculation. The RMSD was calculated over the main-chain atoms (Cα, N, C, O) of the helix and sheet residues, i.e., Val5-Val10, Thr13-Ala27, and Ile29-Trp34, because other residues have relatively large fluctuations in the NMR structures. We also calculated the RMSD with the Cα atoms of all residues except the two flexible ends (Gly1-Ser2 and Gly35-Asp36) as the fitting atoms. The value was ∼1 Å larger than the main-chain RMSD. Thus, we reported only the main-chain RMSD here. The solvent-accessible surface area was calculated with an in-house program using Boolean logic and look-up tables (26). The hydrophobic core was defined as I8, V10, L17, L20, A24, I29, V31, and F33.
Coarse-grained Gō model simulation
As an alternative to all-atom simulations, we carried out coarse-grained simulations on DS119 and compared its folding behavior with those of nine other small proteins to study its folding cooperativity. The structure-based continuum Gō-like model (27,28) was used, where the protein conformation is represented by Cα atoms. The interactions were specified to stabilize the native structure. The potential function and parameters used in the model were identical to those of the “without-solvation” model of Liu and Chan (29). The interaction strength was controlled by the parameter ɛ. A pair of residues, i, j, was defined as being in native contact if 1), the residues were separated in the sequence by at least three other residues, that is, ; and 2), two nonhydrogen atoms, one from each residue, were <4.5 Å apart in the Protein Data Bank (PDB) structure. Langevin dynamics was used to perform thermodynamic and kinetic simulations. Histogram techniques with bias potentials were applied to improve the sampling efficiency of equilibrium properties.
The PDB codes and residue truncations of the protein fragments studied in the coarse-grained simulations were summarized in Table S1 in the Supporting Material. For PDB structures determined by NMR, we used the first conformer in the coordinate file to construct the coarse-grained model in all cases except BBL, where only one NMR structure was available. The same choice of conformation was used by Artem et al. in their recent work (30).
Results and Discussion
Folding simulation by all-atom molecular dynamics using the implicit solvation model
We performed simulations with the AMBER (23) package using the generalized Born model (31) and surface area (GB/SA) to account for solvation effects (see Models and Methods for details). After energy minimization and equilibration of the fully extended starting conformation, four trajectories, each lasting for 1 μs, with different initial speed assignments, at 300 K, were generated. The folding event was observed in the third trajectory only, which was then extended to 2 μs, and the following analysis was based on that trajectory.
Folding event
Starting from a fully extended chain, the protein underwent fast collapse within 50 ps (Fig. 1, inset). This phenomenon has been observed in many folding simulation studies at similar timescales (3,8,10,32). In the 300 ns after the collapse, there was a large conformational adjustment during which the RMSD increased from <6 Å to >9 Å (Fig. 1). The RMSD decreased to 4 Å at ∼300 ns, indicating that the structure was near the native state. After that, small structural readjustments lasted for ∼500 ns. At ∼900 ns, the protein reached the native state and stayed in the basin for the rest of the simulation time. The best simulated structure had a minimal main-chain RMSD of 2.6 Å (aligned with the NMR structure in Fig. 2). The simulated structure closely resembled the key features of the NMR structure: the helix was well formed, and the two strands were aligned in the correct direction. Despite the similarity in global fold and secondary structures, small differences in local structure were observed: for the simulated structure, the helix was slightly bent and the N-terminal formed a hairpin due to backbone-backbone hydrogen bonds. These small differences may be caused by the implicit solvation model used in this study and the possible structural fluctuations.
Figure 1.
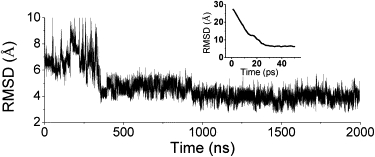
RMSD change of the main-chain atoms in the third trajectory of the all-atom simulation of DS119. (Inset) RMSD of the first 50 ps, illustrating fast collapse of the extended conformation. The structure comparison and RMSD calculation were done with the regular secondary structures assigned in the NMR structure of DS119.
Figure 2.
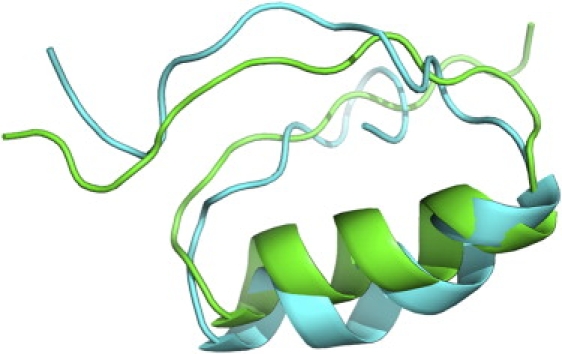
Alignment of the best simulated structure (cyan) and the NMR structure (green, PDB code 2KI0). The main-chain RMSD is 2.6 Å.
Considering that the folding event was observed in one of the four trajectories, the folding time estimated from our simulations was at least at the microsecond level, which is comparable to that of many fast-folding proteins (2). It is noteworthy that most fast-folding proteins are either all-α proteins or proteins containing antiparallel β-sheets only (see Kubelka et al. (2)). A parallel β-sheet generally requires tertiary contacts that have longer sequence distance than an antiparallel β-sheet, which may result in a longer folding time. The fast folding behavior of DS119 observed here may be due to its simple topology, which smoothes the energy landscape and lowers the energy barrier between the folded and unfolded states.
Hydrophobic packing
We also monitored the change of the solvent-accessible surface area (SASA) of the hydrophobic core with time (Fig. S1). After the initial rapid decrease, the SASA increased in the conformation adjustment stage. After the protein had arrived at its near-native state, there were still relatively large fluctuations in SASA, which indicated that the two β-strands were not well formed during the simulation. The minimal SASA was ∼400 Å2, larger than the average SASA of the NMR structures (292 Å2).
When DS119 was designed, a pair of tryptophan residues was introduced to prevent protein aggregation. The alignment of the two indole rings might be face to face according to the nuclear Overhauser effect distances (16). We also monitored the distance between the centers of the two indole rings during the folding process. In the NMR structure, the distance between the two indole rings is 3.6 Å. However, during the simulation, the distance was ∼10 Å after the protein folded (Fig. S2). This result, together with the SASA analysis, indicated that the hydrophobic core of the protein was partially exposed during the simulation, which may stem from the inability of the implicit water model to account for hydrophobic interactions.
Folding pathway
After the initial fast collapse, the protein formed a compact structure that had a size similar to that of the NMR structure. The N-terminal of the helix then formed quickly within 50 ns. The helix further elongated in the subsequent 70 ns. The C-terminal of the helix was formed at 122 ns, accompanied by the formation of a hairpinlike structure of the helical region (Fig. 3). This conformation corresponded to the adjustment phase mentioned above. The helix was fully formed at ∼250 ns. Considering the different scales of simulation time and real time, this is comparable to the 1.7-μs folding time of a naturally occurring helical peptide investigated experimentally (33). Although the helix formed quickly, the two strands were highly flexible. The N-terminal strand bent over initially at ∼355 ns, which caused the sharp RMSD decrease seen in Fig. 1, and it took another 1.3 μs for the other strand to form a compact packing. The folding pathway can be described as an initial collapse followed by fast formation of the central helix and slow formation of the β-sheet. Compared with β-sheet formation, helix formation is easier, because it is determined by local interactions. For the β-sheet, especially the two-stranded parallel β-sheet here, which lacks tight turns between its strands, the two strands are controlled by nonlocal interactions, and constraining the strands together will cause large entropy loss. Thus, the major rate-limiting step of the DS119 folding is the formation of the parallel β-sheet.
Figure 3.
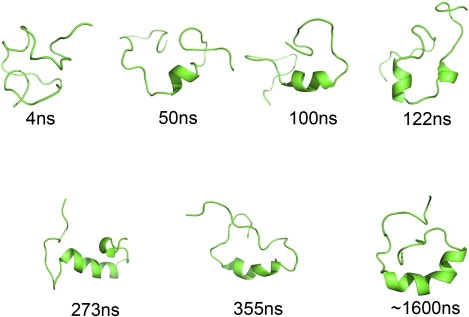
Representative snapshots of the DS119 folding trajectory.
Coarse-grained Gō model simulations reveal that DS119 is an incipient downhill folder
Though all-atom simulations provide valuable information in atomic detail for the folding process, it is often difficult to construct the folding energy landscape with parallel pathways (34,35), as they are computationally expensive even with the implicit solvent models. As an alternative, coarse-grained models are more efficient for obtaining the folding energy landscape. The structure-based continuum Gō-like model (27,36) with coarse-grained chain representation has been widely used in protein-folding problems to facilitate understanding of the topology (37), the cooperativity (38–40), the folding-rate diversity in small proteins (41), and, recently, the coupling between folding and binding (42,43). In this section, we utilized the native-centric Gō model to characterize the folding energy landscape and cooperativity of DS119.
Contact order of DS119 and comparison with proteins of similar size
The overall topology is a significant factor influencing the folding kinetics of a protein. It has been pointed out that for proteins of similar chain length, contact order (CO) from residue contact counts and relative contact order (RCO) from atomic contact counts provide useful information on the folding rates (44,45) and cooperativity (38). We constructed the CO and RCO values for DS119 and other proteins of similar size (Table 1). To make the comparison more reasonable, we focused on proteins with similar chain lengths of ∼35 residues, i.e., NTL9, DS119, Psbd41, HP36, and BBL. Although CO and RCO gave slightly different rank orders, a clear trend emerged: the CO and RCO values of NTL9 and DS119 were much higher than those of Psbd41, HP36 and BBL, suggesting that NTL9 and DS119 were more cooperative than the latter proteins.
Table 1.
Thermodynamic properties and contact order of Cα-chain model proteins
Calorimetric cooperativity criterion
Thermodynamic simulations of the coarse-grained Gō model allowed us to explore the calorimetric cooperativity of DS119 by comparing it with proteins of similar chain length. The heat capacities of six protein fragments differed remarkably in peak height and half-peak width, from the sharp, narrow curve of NTL9 to the flat, broad curve of BBL. DS119 lay in the middle in terms of both sharpness and width (Fig. 4).
Figure 4.
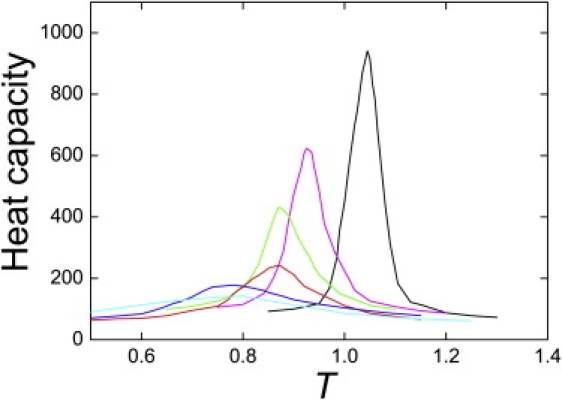
Heat capacities of the coarse-grained protein fragments NTL9 (black), E3BD F166W (magenta), BBL H166W (green), DS119 (red), HP36 (blue), and BBL (cyan). The temperature was measured in units of ɛ/R, where ɛ is the interaction scale used in the model potential.
Although a peak in the heat capacity curve was not a sufficient condition for thermodynamic cooperativity and two-state folding (46), we constructed the calorimetric criterion, , which has been widely used in protein-folding experiments and simulations. In this expression, the calorimetric enthalpy is defined as and the van 't Hoff enthalpy is defined as , where Tm is the transition temperature determined from the peak of the heat capacity (Fig. 4) and Cp(Tm) is the corresponding heat capacity value (47). The ratio was calculated for eight protein models (Table 1). Baseline was constructed by following the protocol proposed by Artem et al. (30). We found that CI2 was the most cooperative and BBL the least cooperative among the protein fragments studied. DS119 showed a moderate value of , 0.53, indicating that it was less cooperative than CI2 but more cooperative than BBL (Table 1). The rank order of cooperativity determined by was CI2 > NTL9 > E3BD F166W > BBL H166W > DS119 > HP36 > Psbd41 > BBL. It has been pointed out (30) that when desolvation effects and/or the many-body effects are taken into consideration, the folding cooperativity of the models increases and the rank order for models with similar values may change. This rank order of folding cooperativity, especially the rank location of DS119, will be further discussed below.
The sigmoidal transitions
Sigmoidal transition of average energy or folding fraction as a function of temperature is a feature of a cooperative two-state transition. We compared the temperature variations of potential energy and the fractional number of native contacts for six proteins: NTL9, E3BD F166W, BBL H166W, DS119, HP36, and BBL (Fig. 5). The normalized potential energy is defined as in Badasyan et al. (30):
| (1) |
where is the average potential energy at temperature T. and are the corresponding average potential energy values for the native state and denatured state, respectively, and are determined from the low- and high-temperature regions far away from the sigmoidal transition. Both and showed that the sigmoidal transition of NTL9 was significantly sharper than that of BBL. DS119 exhibited a moderate sigmoidal transition between NTL9 and BBL. The rank order of sharpness of sigmoidal transition, CI2 > NTL9 > E3BD F166W > BBL H166W > DS119 > HP36 > BBL, was consistent with the rank order of folding cooperativity obtained from the calorimetric criterion.
Figure 5.
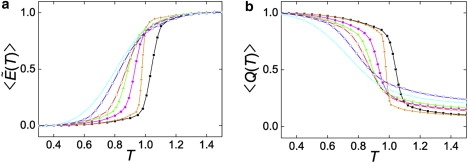
(a) Average normalized potential energy, , and (b) fractional number of native contacts as functions of temperature, T, for CI2 (orange), NTL9 (black), E3BD F166W (magenta), BBL H166W (green), DS119 (red), HP36 (blue), BBL (cyan). Data points were obtained from direct simulations at the given temperature. The average normalized potential energy, , is defined by Eq. 1 in the text.
Conformation distribution shifts with temperature
The population distribution of the fractional number of native contacts, Q, distinguished folding cooperativity among NTL9, DS119, and BBL (Fig. 6). For the most cooperative model, NTL9, the population of the native (or denatured) state decreased (or increased) as temperature increased, whereas the relative location of the native and denatured states remained unchanged (Fig. 6 a). Conversely, for the least cooperative model, BBL, only one populated state was identified, and the location of this state shifted with temperature, i.e., from the denatured-like state to the nativelike state, when temperature decreased (Fig. 6 b), which is consistent with the experimental observation that the folding of BBL is downhill (48). DS119, behaving like NTL9, exhibited two states in the conformation distribution, one identified as the native state (Q ∼ 0.65) and the other as the denatured state (Q ∼ 0.25) (Fig. 6 c). The location of the native and denatured states shifted slightly with temperature (i.e., when simulation temperature increased from 0.75 to 0.85, Q of the folded state decreased from 0.66 to 0.62; when temperature decreased from 0.95 to 0.85, Q of the denatured state increased from 0.21 to 0.30) (Fig. 6 c). Another feature of DS119 was the highly populated states with intermediate Q located between the native and denatured states near the transition temperature. Thus, DS119 was a two-state-like folder with features of a downhill folder.
Figure 6.
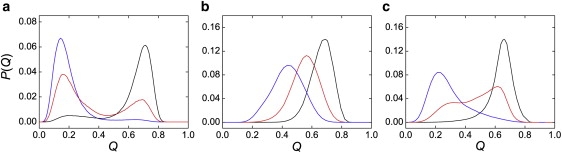
Conformation distribution shifts with temperature change. P(Q) was the normalized population distribution obtained in simulations as a function of Q. The highest temperature, moderate temperature, and lowest temperature (blue, red, and black, respectively) were 1.10, 1.05, and 1.00 for NTL9 (a), 0.80, 0.70, and 0.60 for BBL (b), and 0.95, 0.85, and 0.75 for DS119 (c).
Free-energy profiles
From a comparison of the free-energy profiles of the CI2, DS119, and BBL models under the transition temperatures, it was clear that these three models showed distinct free-energy barrier heights (Fig. 7). CI2 possessed the highest barrier of 3.5 kBT and well-separated folded and unfolded states. BBL, on the other hand, showed no energy barrier along the free-energy profile, with only one free-energy minimum. DS119 showed a broad and flat free-energy landscape with an energy barrier of ∼0.5 kBT, and the locations corresponding to the folded and unfolded states were ambiguous. The heights of the overall free-energy barriers followed the rank order of folding cooperativity, CI2 > DS119 > BBL. It has been suggested that lack of significant free-energy barrier (∼2 kBT) along the folding reaction coordinate results in downhill folding (49). More exactly, it is proposed, folding with a low but nonzero free-energy barrier indicates incipient downhill folding (50), such as the folding behavior of DS119 discussed here. The kinetics and thermodynamics of downhill folders, or incipient downhill folders such as BBL (51), are very sensitive to the environment because of the low barriers involved.
Figure 7.
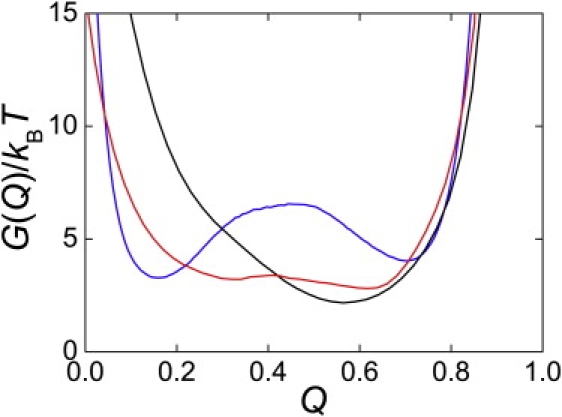
Free-energy profiles for CI2 (blue), DS119 (red), and BBL (black) under transition temperatures. Free energy was calculated using , where Q is the fractional number of native contacts and P(Q) is the normalized population distribution obtained in simulations as a function of Q.
Rank ordering of folding cooperativity
From a different perspective, in the above discussion, the rank order of folding cooperativity was given as CI2 > NTL9 > DS119 > BBL. This rank order, together with the free-energy barrier analysis, unambiguously suggested that the designed DS119 protein was an incipient downhill folder. The high CO value of DS119 was not enough to lead it to fold in a two-state way, which may originate from its distinct topology, consisting of two parallel β-strands and one α-helix. This suggests that when discussing folding cooperativity using only the contact order criterion, care should be taken when proteins with remarkably different topologies are encountered.
Folding kinetics from coarse-grained simulations
We monitored 400 folding trajectories of DS119 under transition temperature (T = 0.85) from 400 randomly chosen unfolded conformations (Q < 0.05) with different initial velocities. A typical trajectory is shown in Fig. S3. Consistent with results from the all-atom simulations, the protein collapsed first and the helix formed quickly and was stable during the entire folding process. Though contacts between the two strands and the helix were partially formed during the folding process, the two strands were rather dynamic and were not correctly packed toward each other, since no native contacts were formed between them. Compared with the C-terminal strand, the N-terminal strand was less dynamic, although fewer contacts existed between this strand and the helix. Taking into consideration all 400 folding trajectories, before reaching the folded state (defined as the minimum with the greater Q from the free-energy profile of Fig. 7), the average contacts within the α-helix, between the α-helix and the N-terminal β-strand, between the α-helix and the C-terminal β-strand, and between the two β-strands were 60%, 30%, 23%, and 5% formed, respectively. Thus, correct packing of the two strands, in particular, packing of the C-terminal strand, was the rate-limiting step. The same conclusion was obtained from the all-atom simulations.
The folding rate prediction for DS119
Computer simulations provide valuable information on the kinetics of protein folding, irrespective of whether an all-atom or coarse-grained model is used. The Gō-like model has been widely used in understanding the folding kinetics of small proteins, and the folding rates from simulations correlate remarkably well with experimentally determined folding rates (41,52). On the basis of this correlation, we predicted the folding rate of DS119, whose kinetic data have not yet been reported. We first constructed a data set of seven small proteins that have been well studied both experimentally (53–58) and theoretically (52,59), then simulated their folding rates under transition midpoints. For the seven proteins we studied here, the simulated folding rates correlated well with the experimentally determined folding rates (Fig. 8). From this correlation, we were able to predict the folding rate of DS119 from the fitting line, and the predicted value was ∼105 s−1. This result was in agreement with the all-atom simulations.
Figure 8.
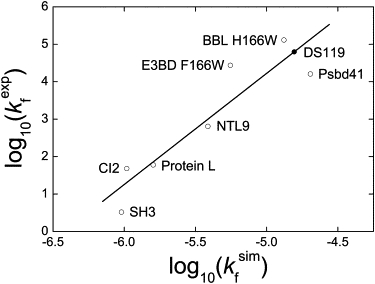
Correlation between simulated folding rates and experimentally determined folding rates. The correlation coefficient was 0.92. Simulated folding rates were estimated by calculating the mean first-passage time for folding transition from 400 randomly chosen unfolded states (Q < 0.05) using the coarse-grained Gō model under transition temperatures, Tm. The experimental folding rates were collected from the literature (53–58). The predicted folding rate for DS119 from the fitting line is denoted by the solid circle.
It is notable that DS119, like many designed proteins, exhibit fast folding behavior. As most protein design methods do not take account of folding dynamics explicitly, the reasons for the fast folding property should be examined. One possible reason is that most designed proteins are relatively small in size and have simple topology, thus requiring fewer distant contacts and less structural rearrangement during folding. For example, the 73-residue three-helix bundle protein α3D folds at a minimal time of 3.2 ± 1.2 μs (60). FSD-1ss, another 29-residue designed protein with one helix and one antiparallel β-sheet, exhibits microsecond folding kinetics near its Tm (61). In fact, several studies have revealed that folding time is positively correlated with protein size (62–65). Conversely, the 93-residue protein Top7 (21), consisting of two helices and five strands, was found to fold in a complex way and its slow folding rate was estimated to approach 1 s−1. The other possible reason might come from currently used design methods, as most computational methods generally result in well-packed hydrophobic cores. As function design is relatively difficult now, most protein design methods focus on designing specific tertiary structures by using protein stability as the objective function. With this stability requirement, the designed proteins—unlike natural proteins, which are also under functional constraints—generally have good hydrophobic packing and stable secondary structures. As the hydrophobic effect is dominant in protein folding, a well-packed core may speed up the folding process. For example, by optimizing the hydrophobic content of the GA module of an albumin-binding domain, Feng Gai and co-workers were able to increase the folding rate up to six folds (66).
Conclusion
In this study, we used all-atom molecular dynamics simulations and a coarse-grained model to study the folding pathway and energy landscape of a de novo designed βαβ protein, DS119. From all-atom simulations, we successfully observed the folding event with a minimal RMSD of 2.6 Å relative to the NMR structure. Different cooperativity studies using the coarse-grained model, i.e., calorimetric criterion, sigmoidal transitions, conformation distribution shifts, and free-energy profiles, all indicate that DS119 is an incipient downhill folder, which folds more cooperatively than a downhill folder such as BBL but less cooperatively than a two-state folder such as CI2. This may reflect a balance between the two structural elements of the βαβ motif: the rapidly formed α helix and the slowly formed parallel β-sheet. Folding time estimated from all-atom simulations and the coarse-grained model were all at the microsecond level. This makes DS119 among the fast folders, which, to the best of our knowledge, is the only one with the unique parallel β-sheet. Further experimental studies are expected to verify this prediction.
Acknowledgments
This work was supported in part by grants from the Ministry of Science of Technology of China (2009CB918500 to Z.L. and L.L.) and the National Natural Science Foundation of China (20228306 and 90103029 to L.L., and 10721403 to L.L. and Z.L.).
Contributor Information
Zhirong Liu, Email: LiuZhiRong@pku.edu.cn.
Luhua Lai, Email: lhlai@pku.edu.cn.
Supporting Material
References
- 1.Duan Y., Kollman P.A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 2.Kubelka J., Hofrichter J., Eaton W.A. The protein folding “speed limit”. Curr. Opin. Struct. Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Lei H., Deng X., Duan Y. The fast-folding HP35 double mutant has a substantially reduced primary folding free energy barrier. J. Chem. Phys. 2008;129:155104. doi: 10.1063/1.2995987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lei H., Duan Y. Two-stage folding of HP-35 from ab initio simulations. J. Mol. Biol. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayachandran G., Vishal V., Pande V.S. Using massively parallel simulation and Markovian models to study protein folding: examining the dynamics of the villin headpiece. J. Chem. Phys. 2006;124:164902. doi: 10.1063/1.2186317. [DOI] [PubMed] [Google Scholar]
- 6.Zagrovic B., Snow C.D., Pande V.S. Simulation of folding of a small α-helical protein in atomistic detail using worldwide-distributed computing. J. Mol. Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 7.Lei H., Duan Y. Ab initio folding of albumin binding domain from all-atom molecular dynamics simulation. J. Phys. Chem. B. 2007;111:5458–5463. doi: 10.1021/jp0704867. [DOI] [PubMed] [Google Scholar]
- 8.Jang S., Kim E., Pak Y. Ab initio folding of helix bundle proteins using molecular dynamics simulations. J. Am. Chem. Soc. 2003;125:14841–14846. doi: 10.1021/ja034701i. [DOI] [PubMed] [Google Scholar]
- 9.Vila J.A., Ripoll D.R., Scheraga H.A. Atomically detailed folding simulation of the B domain of staphylococcal protein A from random structures. Proc. Natl. Acad. Sci. USA. 2003;100:14812–14816. doi: 10.1073/pnas.2436463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei H., Wu C., Duan Y. Folding processes of the B domain of protein A to the native state observed in all-atom ab initio folding simulations. J. Chem. Phys. 2008;128:235105. doi: 10.1063/1.2937135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagielska A., Scheraga H.A. Influence of temperature, friction, and random forces on folding of the B-domain of staphylococcal protein A: all-atom molecular dynamics in implicit solvent. J. Comput. Chem. 2007;28:1068–1082. doi: 10.1002/jcc.20631. [DOI] [PubMed] [Google Scholar]
- 12.Chowdhury S., Lee M.C., Duan Y. Ab initio folding simulation of the Trp-cage mini-protein approaches NMR resolution. J. Mol. Biol. 2003;327:711–717. doi: 10.1016/s0022-2836(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 13.Paschek D., Nymeyer H., García A.E. Replica exchange simulation of reversible folding/unfolding of the Trp-cage miniprotein in explicit solvent: on the structure and possible role of internal water. J. Struct. Biol. 2007;157:524–533. doi: 10.1016/j.jsb.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 14.Zhou R. Trp-cage: folding free energy landscape in explicit water. Proc. Natl. Acad. Sci. USA. 2003;100:13280–13285. doi: 10.1073/pnas.2233312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Helling R., Wingreen N. Emergence of preferred structures in a simple model of protein folding. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 16.Liang H., Chen H., Lai L. De novo design of a βαβ motif. Angew. Chem. Int. Ed. Engl. 2009;48:3301–3303. doi: 10.1002/anie.200805476. [DOI] [PubMed] [Google Scholar]
- 17.Butterfoss G.L., Kuhlman B. Computer-based design of novel protein structures. Annu. Rev. Biophys. Biomol. Struct. 2006;35:49–65. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 18.Kuhlman B., Dantas G., Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 19.Dahiyat B.I., Mayo S.L. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 20.Summa C.M., Rosenblatt M.M., DeGrado W.F. Computational de novo design, and characterization of an A(2)B(2) diiron protein. J. Mol. Biol. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 21.Watters A.L., Deka P., Baker D. The highly cooperative folding of small naturally occurring proteins is likely the result of natural selection. Cell. 2007;128:613–624. doi: 10.1016/j.cell.2006.12.042. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z., Chan H.S. Native topology of the designed protein Top7 is not conducive to cooperative folding. Biophys. J. 2009;96:L25–L27. doi: 10.1016/j.bpj.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Case D.A., Cheatham T.E., 3rd, Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan Y., Wu C., Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 25.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of Cartesian equations of motion of a system with constraints: molecular dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 26.Legrand S.M., Merz K.M. Rapid approximation to molecular surface area via the use of Boolean logic and look-up tables. J. Comput. Chem. 1993;14:349–352. [Google Scholar]
- 27.Go N. Theoretical studies of protein folding. Annu. Rev. Biophys. Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 28.Clementi C., Nymeyer H., Onuchic J.N. Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 29.Liu Z.R., Chan H.S. Solvation and desolvation effects in protein folding: native flexibility, kinetic cooperativity and enthalpic barriers under isostability conditions. Phys. Biol. 2005;2:S75–S85. doi: 10.1088/1478-3975/2/4/S01. [DOI] [PubMed] [Google Scholar]
- 30.Badasyan A., Liu Z., Chan H.S. Interplaying roles of native topology and chain length in marginally cooperative and noncooperative folding of small protein fragments. Int. J. Quantum Chem. 2009;109:3482–3499. [Google Scholar]
- 31.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 32.Lee I.H., Kim S.Y., Lee J. Dynamic folding pathway models of the villin headpiece subdomain (HP-36) structure. J. Comput. Chem. 2009;31:57–65. doi: 10.1002/jcc.21288. [DOI] [PubMed] [Google Scholar]
- 33.Mukherjee S., Chowdhury P., Gai F. Folding kinetics of a naturally occurring helical peptide: implication of the folding speed limit of helical proteins. J. Phys. Chem. B. 2008;112:9146–9150. doi: 10.1021/jp801721p. [DOI] [PubMed] [Google Scholar]
- 34.Dill K.A., Chan H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 35.Bryngelson J.D., Onuchic J.N., Wolynes P.G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 36.Ueda Y., Taketomi H., Go N. Studies on protein folding, unfolding, and fluctuations by computer simulation. 2. 3-Dimensional lattice model of lysozyme. Biopolymers. 1978;17:1531–1548. [Google Scholar]
- 37.Koga N., Takada S. Roles of native topology and chain-length scaling in protein folding: a simulation study with a Go-like model. J. Mol. Biol. 2001;313:171–180. doi: 10.1006/jmbi.2001.5037. [DOI] [PubMed] [Google Scholar]
- 38.Badasyan A., Liu Z., Chan H.S. Probing possible downhill folding: native contact topology likely places a significant constraint on the folding cooperativity of proteins with approximately 40 residues. J. Mol. Biol. 2008;384:512–530. doi: 10.1016/j.jmb.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 39.Kaya H., Chan H.S. Solvation effects and driving forces for protein thermodynamic and kinetic cooperativity: how adequate is native-centric topological modeling? J. Mol. Biol. 2003;326:911–931. doi: 10.1016/s0022-2836(02)01434-1. [DOI] [PubMed] [Google Scholar]
- 40.Cho S.S., Weinkam P., Wolynes P.G. Origins of barriers and barrierless folding in BBL. Proc. Natl. Acad. Sci. USA. 2008;105:118–123. doi: 10.1073/pnas.0709376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferguson A., Liu Z., Chan H.S. Desolvation barrier effects are a likely contributor to the remarkable diversity in the folding rates of small proteins. J. Mol. Biol. 2009;389:619–636. doi: 10.1016/j.jmb.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Turjanski A.G., Gutkind J.S., Hummer G. Binding-induced folding of a natively unstructured transcription factor. PLOS Comput. Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y., Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the ”fly-casting” mechanism. J. Mol. Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- 45.Plaxco K.W., Simons K.T., Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 46.Knott M., Chan H.S. Criteria for downhill protein folding: calorimetry, chevron plot, kinetic relaxation, and single-molecule radius of gyration in chain models with subdued degrees of cooperativity. Proteins. 2006;65:373–391. doi: 10.1002/prot.21066. [DOI] [PubMed] [Google Scholar]
- 47.Kaya H., Chan H.S. Polymer principles of protein calorimetric two-state cooperativity. Proteins. 2000;40:637–661. doi: 10.1002/1097-0134(20000901)40:4<637::aid-prot80>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 48.Sadqi M., Fushman D., Muñoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–321. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 49.Gruebele M. Downhill protein folding: evolution meets physics. C. R. Biol. 2005;328:701–712. doi: 10.1016/j.crvi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Liu F., Du D.G., Gruebele M. An experimental survey of the transition between two-state and downhill protein folding scenarios. Proc. Natl. Acad. Sci. USA. 2008;105:2369–2374. doi: 10.1073/pnas.0711908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Mira M.M., Sadqi M., Muñoz V. Experimental identification of downhill protein folding. Science. 2002;298:2191–2195. doi: 10.1126/science.1077809. [DOI] [PubMed] [Google Scholar]
- 52.Chavez L.L., Onuchic J.N., Clementi C. Quantifying the roughness on the free energy landscape: entropic bottlenecks and protein folding rates. J. Am. Chem. Soc. 2004;126:8426–8432. doi: 10.1021/ja049510+. [DOI] [PubMed] [Google Scholar]
- 53.Jackson S.E., Fersht A.R. Folding of chymotrypsin inhibitor-2. 1. Evidence for a 2-state transition. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 54.Scalley M.L., Baker D. Protein folding kinetics exhibit an Arrhenius temperature dependence when corrected for the temperature dependence of protein stability. Proc. Natl. Acad. Sci. USA. 1997;94:10636–10640. doi: 10.1073/pnas.94.20.10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spector S., Raleigh D.P. Submillisecond folding of the peripheral subunit-binding domain. J. Mol. Biol. 1999;293:763–768. doi: 10.1006/jmbi.1999.3189. [DOI] [PubMed] [Google Scholar]
- 56.Cobos E.S., Filimonov V.V., Martínez J.C. A thermodynamic and kinetic analysis of the folding pathway of an SH3 domain entropically stabilised by a redesigned hydrophobic core. J. Mol. Biol. 2003;328:221–233. doi: 10.1016/s0022-2836(03)00273-0. [DOI] [PubMed] [Google Scholar]
- 57.Horng J.C., Moroz V., Raleigh D.P. Rapid cooperative two-state folding of a miniature α-β protein and design of a thermostable variant. J. Mol. Biol. 2003;326:1261–1270. doi: 10.1016/s0022-2836(03)00028-7. [DOI] [PubMed] [Google Scholar]
- 58.Ferguson N., Sharpe T.D., Fersht A.R. Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J. Mol. Biol. 2005;353:427–446. doi: 10.1016/j.jmb.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 59.Wallin S., Chan H.S. Conformational entropic barriers in topology-dependent protein folding: perspectives from a simple native-centric polymer model. J. Phys. Condens. Matter. 2006;18:S307–S328. [Google Scholar]
- 60.Zhu Y., Alonso D.O., Gai F. Ultrafast folding of α3D: a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. USA. 2003;100:15486–15491. doi: 10.1073/pnas.2136623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sadqi M., de Alba E., Muñoz V. A designed protein as experimental model of primordial folding. Proc. Natl. Acad. Sci. USA. 2009;106:4127–4132. doi: 10.1073/pnas.0812108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gutin A.M., Abkevich V.I., VI, Shakhnovich E.I. Chain length scaling of protein folding time. Phys. Rev. Lett. 1996;77:5433–5436. doi: 10.1103/PhysRevLett.77.5433. [DOI] [PubMed] [Google Scholar]
- 63.Wolynes P.G. Folding funnels and energy landscapes of larger proteins within the capillarity approximation. Proc. Natl. Acad. Sci. USA. 1997;94:6170–6175. doi: 10.1073/pnas.94.12.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li M.S., Klimov D.K., Thirumalai D. Thermal denaturation and folding rates of single domain proteins: size matters. Polymer (Guildf.) 2004;45:573–579. [Google Scholar]
- 65.Naganathan A.N., Muñoz V. Scaling of folding times with protein size. J. Am. Chem. Soc. 2005;127:480–481. doi: 10.1021/ja044449u. [DOI] [PubMed] [Google Scholar]
- 66.Zhu Y., Fu X., Wang T., Tamura A., Takada S. Guiding the search for a protein's maximum rate of folding. Chem. Phys. 2004;307:99–109. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.