

Abstract
We investigate the effect of macromolecular crowding on protein folding, using purely repulsive crowding particles and a self-organizing polymer model of protein folding. We find that the variation in folding stability with crowder size for typical α-, β-, and α/β-proteins is well described by an adaptation of the scaled particle theory. The native state, the transition state, and the unfolded protein are treated as effective hard spheres, with the folded and transition state radii independent of the size and concentration of the crowders. Remarkably, we find that, as the effective unfolded state radius is very weakly dependent on the crowder concentration, it can also be approximated by a single size. The same model predicts the effect of crowding on the folding barrier and therefore refolding rates with no adjustable parameters. A simple extension of the scaled-particle theory model, assuming additivity, can also describe the behavior of mixtures of crowding particles.
Introduction
In contrast with conventional laboratory experiments conducted under dilute conditions, protein folding in a cell occurs in a dense environment consisting of various other macromolecules, commonly referred to as crowders (1). Although the detailed interactions of the protein with the crowders may be quite complex (e.g., direct protein-crowder attractions, water-mediated interactions (2)), the primary physical effect of macromolecular crowding on the protein folding reaction is to reduce the volume available to the protein by that occupied by the crowders (3–10). A complete theoretical understanding of the excluded volume effects will greatly enhance our ability to interpret experiments, as well as all-atom simulations, and to develop coarse-grained models (11,12), of concentrated protein solutions.
Several theories have been put forward to predict the effect of excluded volume crowders on the folding free energy. However, these theories provide strongly contrasting predictions for even the qualitative effects of crowding. We focus here on two theories which provide convenient closed-form expressions suitable for fitting to theory or simulation, although we note that other theories requiring numerical solution have been put forward (13). By treating the folded and unfolded proteins as effective hard spheres, Minton utilized scaled particle theory (SPT) to estimate the change in folding free energy as the difference between the insertion free energy for the folded and the unfolded states. The SPT free energy of inserting a hard sphere of radius R in a hard-sphere fluid of particle radius Rc is (14)
where β = 1/kBT, T is the temperature, kB is the Boltzmann's constant, y = R/Rc, ρ = ϕc/(1 − ϕc), and ϕc is the fluid volume fraction. This theory predicts a strong effect of macromolecular crowding on the folding free energy (13), with a monotonic increase in stability with increasing crowder packing fraction ϕc, in qualitative agreement with previous molecular simulation results (4). We note that the insertion free energy of any given protein conformation in a hard sphere solvent can also be calculated directly from its geometric properties using, e.g., a recently proposed morphometric approach (15). In this work, we concentrate on theories that do not require enumeration of the protein configurations, which cannot be done experimentally.
An alternative theory by Zhou (16) also uses SPT for the effect of crowders on the folded protein, but the free energy of the unfolded protein is calculated using an elegant model of the unfolded chain as a random walk in the presence of a spherical trap. For a Gaussian chain of radius of gyration Rg, with z = Rg/Rc, the change in free energy is (17)
This model predicts a much weaker effect of crowding on the unfolded state free energy, as the unfolded polypeptide can access the voids between the crowders. In addition, a stability maximum is predicted as the crowder packing fraction is increased. The extension of this theory to binary mixtures of different size crowders leads to an intriguing conclusion that there will be an optimum mixing ratio of the two components to achieve maximum protein stability (18). One may expect that different crowding theories will work better under certain conditions of crowder packing fraction and size. However, the boundaries for the validity of these theories in parameter space are relatively unknown.
Model and Methods
In this article, we investigate the quantitative theoretical description of purely repulsive crowders on folding, using molecular simulations of a coarse-grained folding model in a bath of crowders. We consider two-state proteins from the three main protein structural classes: all-α (prb (19)), all-β (TNfn3 (20)) and α/β (protein G (21)), which are described using a self-organizing polymer model (or Gō-like model) (22,23). In our model, each residue is represented by a single particle and a standard procedure is used to build the potential from the experimental native-state structure (24). Interactions between the contacts present in the native state are treated as attractive and all others as repulsive, an approximation motivated by the funneled nature of the folding free energy landscape (25). Previous studies have shown that this type of simplified model can indeed capture relevant features of protein folding (26), such as mechanism (27) and kinetics (28,29). Repulsive protein-crowder and crowder-crowder interactions are given by the pair potential
where r is the distance between particle centers, ɛ = 1 kcal/mol sets the energy scale, σ is the hard core overlap distance, and σref = 6 Å is a reference diameter: for σ = σref, V(r) reduces to a more familiar form. For the pair (i, j), we define σ = Ri + Rj, where Ri, Rj are the radii of crowder particles Rc, or of the various protein residues Rp. We use Langevin dynamics simulations with a time step of 10 fs and a friction coefficient of 0.2 ps−1 using the BBK integrator (30) in the CHARMM simulation package (31). Cubic periodic boundary conditions with a primary cell size of 100 Å were employed. To speed up equilibration at a given temperature, we use replica exchange moves every 30 ps between 12 replicas which are each biased using an umbrella potential of the form
where Q is the fraction of native contacts, 0 ≤ Qi ≤ 1 is the target Q value for replica i, and κ = 300 kcal/mol is the force constant. We define
where the sum runs over Nij native contact pairs (i, j), which are separated by distances in the native structure and by rij in configuration R; β = 5 Å−1; λ = 1.2. The required thermodynamic information is extracted from simulations with different umbrella potentials and temperatures using the weighted histogram method (32,33).
Results and Discussion
For the model proteins considered here, Q is nearly an optimal coordinate for identifying transition states and capturing the dynamics of protein folding with a diffusive Markovian model (29,34,35). Here, we project folding in our systems onto Q, and use it to identify transition states as well as unfolded and folded free energy minima. To estimate the crowding-induced changes on the folding free energy surface, we construct the potential of mean force along Q, defined as
where P(Q) is the equilibrium probability of observing configurations between Q and Q + ΔQ. Fig. 1 shows representative free energy profiles in bulk (ϕc = 0) and under crowded conditions (ϕc > 0) for protein G. The destabilization due to the presence of crowders is clearly visible from an upward shift in the curves near the unfolded basin (Q ≈ 0.1), qualitatively consistent with earlier predictions (3). This is due to the greater cost of inserting the larger unfolded state into a bath of crowders. A secondary effect is that the average size of the unfolded state is reduced with increasing confinement, because the more expanded conformers are disfavored. At the highest packing fractions, we find a reduction of 15% in the average radius of gyration of the unfolded state. This compaction is qualitatively consistent with neutron scattering measurements on random-coil polymers, which have suggested a compaction of up to 30% (36). The quantitative difference from the experimental data may be due to differences between the compact unfolded state of the Gō model, due to residual nativelike interactions, and random-coil chains. For high crowding packing fractions (ϕc > 0.10), there is also a slight destabilization of the transition state (Q ≈ 0.5) with respect to the folded basin (Q ≈ 0.9). The increase in folding temperature Tf (the maximum in heat capacity Cv; see Fig. 1 inset) with increasing ϕc, demonstrates the stabilizing effect of the crowders as predicted by approximate theoretical models (3) and observed in molecular simulations previously (4).
Figure 1.
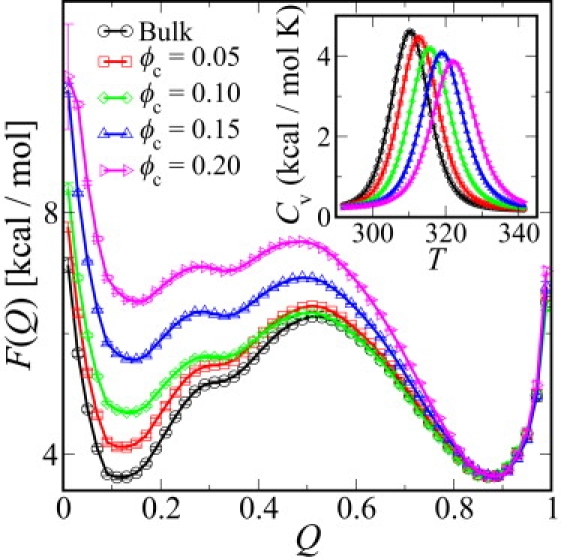
Effect of crowding on folding free energy surface of protein G. The potential of mean force along the coordinate Q, the fraction of native contacts, is shown for bulk conditions and with different crowder packing fractions ϕc for crowder size Rc = 8 Å at the bulk folding temperature Tfbulk = 310 K. The curves have been shifted by an arbitrary constant to match the free energy of the folded state. (Inset) Heat capacity Cv(T) from weighted histogram method analysis.
Fig. 2, A–C, shows the effect of crowding on the free energy of folding,
for each protein, where
is the difference between the native FN and unfolded FU free energy and Q‡ is the location of the transition state along Q. We find a monotonic increase in stability with the packing fraction ϕc for all proteins and crowder sizes. For a given ϕc, smaller crowders are more strongly stabilizing because there are effectively fewer voids of the size of the protein. For a given size and concentration of crowders, we observe more stabilization for a longer protein chain because the size ratio between the unfolded and the folded state increases with chain length (35,37). Chain lengths and are 47 (1.67), 56 (1.89), and 90 (2.2) for prb, protein G, and TNfn3, respectively. We compute radii of gyration and , for the folded and unfolded proteins, respectively, from the simulations and define the folded protein radius aN = (Fig. 2 legend). The Zhou theory is in excellent agreement with the simulation data in the limit ϕc → 0 (Fig. 2). For smaller crowders and higher packing fractions, however, the theory predicts too small a stabilization—most likely due to the neglect of excluded volume in the unfolded chain.
Figure 2.
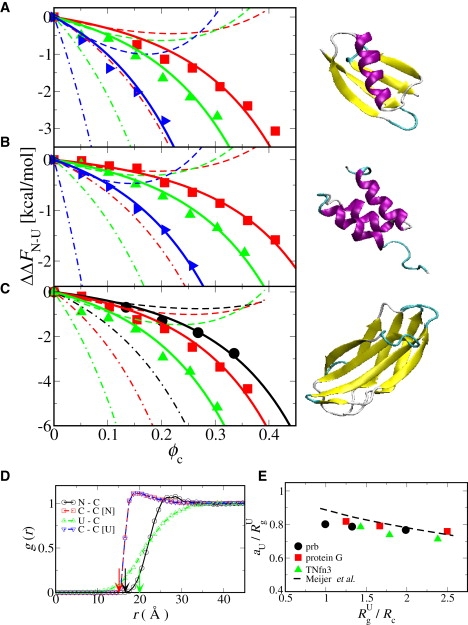
Effect of crowding on protein stability. The change in folding free energy with crowder size and packing fraction is given for (A) protein G (T = 320 K), (B) prb (T = 320 K), and (C) TNfn3 (T = 300 K); the corresponding protein structure is shown next to each plot. Symbols: simulations for Rc = 20 Å (circle), 16 Å (square), 12 Å (triangle up), and 8 Å (triangle right); (dashed lines) Zhou theory (16); (dot-dash lines) Minton theory; and (solid line) effective hard sphere radius for the unfolded state motivated by the form of the U-C g(r). Radius of gyration for the native state RgN = 9.5 (prb), 10.6 (protein G), and 13.0 (TNfn3). (D) Pair distribution functions g(r) between crowders (C) and the native (N) and unfolded (U) protein (C-C [U] and C-C [N] are the C-C g(r) in the presence of U and N protein, respectively), for prb with Rc = 8 Å, ϕc = 0.05. Arrows indicate corresponding sums of hard sphere radii. (E) Dependence of effective unfolded state radius aU on crowder size Rc; each axis is given relative to the unfolded radius of gyration RgU.
By contrast, the Minton theory predicts a stabilization much greater than found in the simulations over the full range of ϕc. This theory treats the unfolded protein (with radius of gyration ) as an equivalent hard sphere with radius . An alternative theory by Minton (13), using the so-called Gaussian cloud model for the unfolded state, works slightly better but still does not explain our simulation data over the whole range (A. P. Minton, personal communication, 2009). The use of an effective hard sphere for the unfolded chain is justified by the highly successful Asakura-Oosawa theory (38) of polymer-colloid mixtures and the statistical-mechanical description of such mixtures by Shaw and Thirumalai (39). However, as indicated by the pair distribution function g(r) between the unfolded protein and the crowders (Fig. 2), the unfolded protein is quite soft, so that a smaller choice of hard sphere radius may be more appropriate (40). We find that the ΔΔFN–U data are well described by SPT with the folded protein radius aN = , and an unfolded radius aU < . The optimal value of aU is approximately independent of ϕc as evident from the fit using a single aU for each Rc (Fig. 2, A–C). In fact aU is also only weakly dependent on Rc (Fig. 2 E) and a similar quality fit can be obtained using a single aU = 0.8 (data not shown). The slight reduction in aU with decreasing Rc results from greater penetration of the unfolded protein by the smaller crowders. This weak dependence on crowder size can be related to the analogous mapping of polymer-hard spheres mixtures onto the Asakura-Oosawa model (41), where the effective hard sphere radius of the polymer scales as (Rg/Rc)−1/3; the results of this mapping for simulations of hard spheres in a bath of ideal chains (42) are superimposed on Fig. 2 E.
We have also studied mixed macromolecular crowding with binary A:B and ternary A:B:C mixtures for the α-helical protein prb. For A:B mixtures, we calculated the change in folding free energy with the packing fraction of the type-A crowders (radius 8 Å) fixed at , and the fraction of type-B crowders (radius 12 Å) varying (Fig. 3). To test whether the effect of mixed crowding is simply additive and can be easily estimated from the pure crowder simulations, we also calculate the change in folding free energy from the following additivity Ansatz,
where index i runs over N different types of crowding particles,
is the fraction of crowder type i in the mixture, and is the volume fraction of i. We note that our expression differs from a previously proposed model in which (43)
Figure 3.
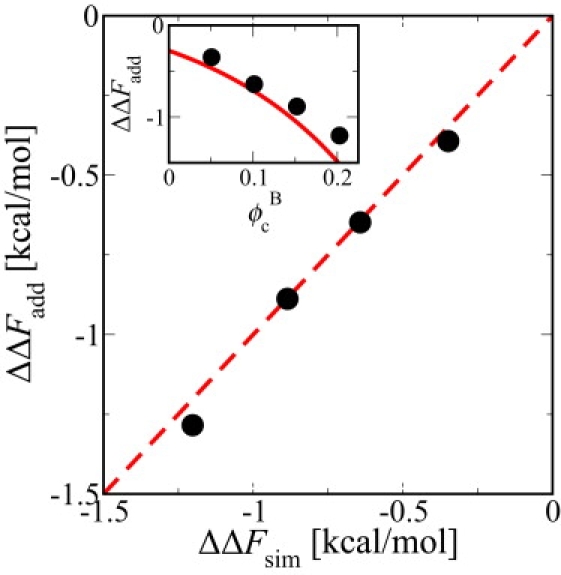
SPT prediction for binary mixtures of 8 Å and 12 Å crowders. With the packing fraction of particle type A fixed to and the fraction of particle type B, , the change in folding free energy of prb from simulations ΔΔFsim is shown as a function of change in folding free energy ΔΔFadd from the additivity Ansatz (circle), as discussed in the text. (Inset) Simulation data (circle) along with SPT model predictions (line) using the additive model.
The predictions of our additive model ΔΔFadd for A:B mixtures are in extremely good agreement with the simulation results ΔΔFsim as shown in Fig. 3. Moreover, for an A:B:C mixture of crowder radii 8, 12, and 16 Å with volume fraction = 0.05 of each component, the agreement between the simulation result (0.55 kcal/mol) and the additive model (0.54 kcal/mol) is remarkably good. Applying additivity to our SPT model predictions for single crowders also provides reasonable estimates for ΔΔF (Fig. 3, inset). Our results and the additive model for mixed crowding do not predict an optimal mixing ratio as expected from a previous theory (18). Indeed, for a mixture of crowders at given ϕc, the greatest stabilization will occur when all the crowders are of the more stabilizing (smaller) type.
From our explicit dynamical model of folding, we are able to estimate folding rates directly from mean first-passage time calculations. Starting from at least 400 different initial coordinates of prb drawn from an equilibrium unfolded ensemble (Q ≈ QU ≡ 0.2) at a given packing fraction, we calculate the average time τf = 1/kf taken to reach the folded state (Q > 0.9). We estimate the change in apparent barrier height from the folding rates as
which is justified if the position of the folding barrier and the diffusion coefficient along the reaction coordinate are unchanged (Fig. 4) (44). We also estimate directly the contribution of the barrier to the rate from the free energy surface F(Q) using
with integration bounds Q‡ ± 0.1 and QU ± 0.1 for ‡ and U, respectively. With this definition, the change of barrier height from bulk to crowded should match that calculated from the rates, if the dynamics along Q is diffusive with a constant, position-independent diffusion coefficient (44).
Figure 4.
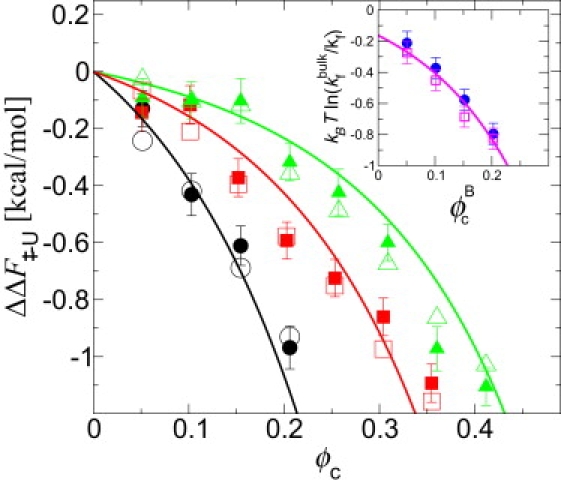
Influence of crowding on folding kinetics. The change in barrier height ΔΔF‡–U is estimated using kBT ln(kfbulk/kf) (solid symbols) and from F(Q) (open symbols) for crowders of radius 8 Å (circle), 12 Å (square), and 16 Å (triangle up); lines are the SPT predictions using transition state = 11.4 Å (prb). (Inset) Folding rates with binary crowder mixtures (A, 8 Å radius; B, 12 Å radius; ). Results from direct simulations (circle) are compared with the predictions based on simulations with a single crowder size (square) and from SPT (line).
The folding barrier height decreases monotonically with increasing ϕc in qualitative agreement with previous theoretical models (3). Indeed, it is possible to predict the change in rates using SPT with no adjustable parameters: we calculate the transition state radius a‡ from the Q umbrella simulations (Q = 0.55), and the change in barrier height from SPT, using a‡ in place of aN. The agreement between the change in barrier height estimated from the rates and from the free energy surface confirms that the barrier height rather than variations in diffusion coefficient or dynamics is the dominant contribution to kinetics. The effect of mixed macromolecular crowding on kinetics can also be obtained from single crowder simulations by assuming additivity (Fig. 4, inset).
In summary, we find that scaled particle theory provides an accurate description of the effect of macromolecular crowding on both folding stability and rates. The effect of purely repulsive crowders can be well approximated over a wide range of crowder sizes and packing fractions by treating the unfolded state as a hard sphere of fixed radius aU. In all the cases considered, we find . Our results have several important consequences. For crowders of a single size, folding rate, and stability will increase with increasing packing fraction monotonically under the relevant physiological conditions. When considering mixtures of purely repulsive crowders of different sizes the effects of crowding are additive, and the most stabilizing composition will consist completely of the smallest crowder. Therefore, any maximum in stabilization as a function of the ratio of various components at a fixed total packing fraction implies the existence of attractive interactions with at least one of the crowders (45).
Acknowledgments
We are grateful to Dr. Attila Szabo for several helpful discussions. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov), Theory Sector computing resources at Cambridge, and the National Science Foundation TeraGrid resources provided by the Texas Advanced Computing Center.
J.M. acknowledges the Human Frontier Science Program for a short-term fellowship. R.B.B. is supported by a Royal Society University Research Fellowship.
Contributor Information
Jeetain Mittal, Email: jeetain@lehigh.edu.
Robert B. Best, Email: rbb24@cam.ac.uk.
References
- 1.Ellis R.J., Minton A.P. Cell biology: join the crowd. Nature. 2003;425:27–28. doi: 10.1038/425027a. [DOI] [PubMed] [Google Scholar]
- 2.Lucent D., Vishal V., Pande V.S. Protein folding under confinement: a role for solvent. Proc. Natl. Acad. Sci. USA. 2007;104:10430–10434. doi: 10.1073/pnas.0608256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minton A.P. The effect of volume occupancy upon the thermodynamic activity of proteins: some biochemical consequences. Mol. Cell. Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- 4.Cheung M.S., Klimov D., Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung M.S., Thirumalai D. Effects of crowding and confinement on the structures of the transition state ensemble in proteins. J. Phys. Chem. B. 2007;111:8250–8257. doi: 10.1021/jp068201y. [DOI] [PubMed] [Google Scholar]
- 6.Minh D.D., Chang C.E., McCammon J.A. The influence of macromolecular crowding on HIV-1 protease internal dynamics. J. Am. Chem. Soc. 2006;128:6006–6007. doi: 10.1021/ja060483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stagg L., Zhang S.Q., Wittung-Stafshede P. Molecular crowding enhances native structure and stability of α/β protein flavodoxin. Proc. Natl. Acad. Sci. USA. 2007;104:18976–18981. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Z., Jiang J., Rajagopalan R. Effects of macromolecular crowding on biochemical reaction equilibria: a molecular thermodynamic perspective. Biophys. J. 2007;93:1464–1473. doi: 10.1529/biophysj.107.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H.-X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Ann. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudlay A., Cheung M.S., Thirumalai D. Crowding effects on the structural transitions in a flexible helical homopolymer. Phys. Rev. Lett. 2009;102:118101. doi: 10.1103/PhysRevLett.102.118101. [DOI] [PubMed] [Google Scholar]
- 11.Cheung J.K., Shen V.K., Truskett T.M. Concentration and crowding effects on protein stability from a coarse-grained model. In: Zaman M.H., editor. Statistical Mechanics of Cellular Systems and Processes. Cambridge University Press; Cambridge, UK: 2009. [Google Scholar]
- 12.Shen V.K., Cheung J.K., Errington J.R., Truskett T.M. Insights into crowding effects on protein stability from a coarse-grained model. J. Biomed. Eng. 2009;131 doi: 10.1115/1.3127259. 071002–1–071002–7. [DOI] [PubMed] [Google Scholar]
- 13.Minton A.P. Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: macromolecular crowding and protein stability revisited. Biophys. J. 2005;88:971–985. doi: 10.1529/biophysj.104.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebowitz J.L., Rowlinson J.S. Thermodynamic properties of mixtures of hard spheres. J. Chem. Phys. 1964;41:133–138. [Google Scholar]
- 15.Hansen-Goos H., Roth R., Dietrich S. Solvation of proteins: linking thermodynamics to geometry. Phys. Rev. Lett. 2007;99:128101. doi: 10.1103/PhysRevLett.99.128101. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H.-X. Protein folding in confined and crowded environments. Arch. Biochem. Biophys. 2008;469:76–82. doi: 10.1016/j.abb.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabo A., Zwanzig R., Agmon N. Diffusion-controlled reactions with mobile traps. Phys. Rev. Lett. 1988;61:2496–2499. doi: 10.1103/PhysRevLett.61.2496. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H.-X. Effect of mixed macromolecular crowding agents on protein folding. Proteins. 2008;72:1109–1113. doi: 10.1002/prot.22111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang T., Zhu Y., Gai F. Folding of a three-helix bundle at the folding speed limit. J. Phys. Chem. B. 2004;108:3694–3697. [Google Scholar]
- 20.Hamill S.J., Steward A., Clarke J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J. Mol. Biol. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- 21.McCallister E.L., Alm E., Baker D. Critical role of β-hairpin formation in protein G folding. Nat. Struct. Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 22.Nymeyer H., García A.E., Onuchic J.N. Folding funnels and frustration in off-lattice minimalist protein landscapes. Proc. Natl. Acad. Sci. USA. 1998;95:5921–5928. doi: 10.1073/pnas.95.11.5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mickler M., Dima R.I., Rief M. Revealing the bifurcation in the unfolding pathways of GFP by using single-molecule experiments and simulations. Proc. Natl. Acad. Sci. USA. 2007;104:20268–20273. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karanicolas J., Brooks C.L., 3rd The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci. 2002;11:2351–2361. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolynes P.G. Recent successes of the energy landscape theory of protein folding and function. Q. Rev. Biophys. 2005;38:405–410. doi: 10.1017/S0033583505004075. [DOI] [PubMed] [Google Scholar]
- 26.Shea J.-E., Onuchic J.N., Brooks C.L., III Energetic frustration and the nature of the transition state in protein folding. J. Chem. Phys. 2000;113:7663–7671. [Google Scholar]
- 27.Levy Y., Wolynes P.G., Onuchic J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA. 2004;101:511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chavez L.L., Onuchic J.N., Clementi C. Quantifying the roughness on the free energy landscape: entropic bottlenecks and protein folding rates. J. Am. Chem. Soc. 2004;126:8426–8432. doi: 10.1021/ja049510+. [DOI] [PubMed] [Google Scholar]
- 29.Best R.B., Hummer G. Reaction coordinates and rates from transition paths. Proc. Natl. Acad. Sci. USA. 2005;102:6732–6737. doi: 10.1073/pnas.0408098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks C.L., III, Brünger A., Karplus M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem. Phys. Lett. 1984;105:495–500. [Google Scholar]
- 31.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 32.Ferrenberg A.M., Swendsen R.H. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 1989;63:1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 33.Kumar S., Bouzida D., Rosenberg J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 34.Best R.B., Hummer G. Diffusive model of protein folding dynamics with Kramer's turnover in rate. Phys. Rev. Lett. 2006;96:228104. doi: 10.1103/PhysRevLett.96.228104. [DOI] [PubMed] [Google Scholar]
- 35.Mittal J., Best R.B. Thermodynamics and kinetics of protein folding under confinement. Proc. Natl. Acad. Sci. USA. 2008;105:20233–20238. doi: 10.1073/pnas.0807742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Coeur C., Demé B., Longeville S. Compression of random coils due to macromolecular crowding. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2009;79:031910. doi: 10.1103/PhysRevE.79.031910. [DOI] [PubMed] [Google Scholar]
- 37.Takagi F., Koga N., Takada S. How protein thermodynamics and folding mechanisms are altered by the chaperonin cage: molecular simulations. Proc. Natl. Acad. Sci. USA. 2003;100:11367–11372. doi: 10.1073/pnas.1831920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asakura S., Oosawa F. On interaction between two bodies immersed in a solution of macromolecules. J. Chem. Phys. 1954;22:1255–1256. [Google Scholar]
- 39.Shaw M.R., Thirumalai D. Free polymer in a colloidal solution. Phys. Rev. A. 1991;44:R4797–R4800. doi: 10.1103/physreva.44.r4797. [DOI] [PubMed] [Google Scholar]
- 40.Louis A.A., Bolhuis P.G., Meijer E.J. Can polymer coils be modeled as “soft colloids”? Phys. Rev. Lett. 2000;85:2522–2525. doi: 10.1103/PhysRevLett.85.2522. [DOI] [PubMed] [Google Scholar]
- 41.Louis A.A., Bolhuis P.G., Meijer E.J. Density profiles and surface tension of polymers near colloidal surfaces. J. Chem. Phys. 2002;116:10547–10555. [Google Scholar]
- 42.Meijer E.J., Frenkel D. Computer simulation of polymer-induced clustering of colloids. Phys. Rev. Lett. 1991;67:1110–1113. doi: 10.1103/PhysRevLett.67.1110. [DOI] [PubMed] [Google Scholar]
- 43.Qin S.B., Zhou H.-X. Atomistic modeling of macromolecular crowding predicts modest increases in protein folding and binding stability. Biophys. J. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kramers H.A. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 1940;7:284–303. [Google Scholar]
- 45.Du F., Zhou Z., Liang Y. Mixed macromolecular crowding accelerates the refolding of rabbit muscle creatine kinase: implications for protein folding in physiological environments. J. Mol. Biol. 2006;364:469–482. doi: 10.1016/j.jmb.2006.09.018. [DOI] [PubMed] [Google Scholar]