

Abstract
Gliomas are a diverse group of brain tumors of glial origin. Most are characterized by diffuse infiltrative growth in the surrounding brain. In combination with their refractive nature to chemotherapy this makes it almost impossible to cure patients using combinations of conventional therapeutic strategies. The drastically increased knowledge about the molecular underpinnings of gliomas during the last decade has elicited high expectations for a more rational and effective therapy for these tumors. Most studies on the molecular pathways involved in glioma biology thus far had a strong focus on growth factor receptor protein tyrosine kinase (PTK) and phosphatidylinositol phosphatase signaling pathways. Except for the tumor suppressor PTEN, much less attention has been paid to the PTK counterparts, the protein tyrosine phosphatase (PTP) superfamily, in gliomas. PTPs are instrumental in the reversible phosphorylation of tyrosine residues and have emerged as important regulators of signaling pathways that are linked to various developmental and disease-related processes. Here, we provide an overview of the current knowledge on PTP involvement in gliomagenesis. So far, the data point to the potential implication of receptor-type (RPTPδ, DEP1, RPTPμ, RPTPζ) and intracellular (PTP1B, TCPTP, SHP2, PTPN13) classical PTPs, dual-specific PTPs (MKP-1, VHP, PRL-3, KAP, PTEN) and the CDC25B and CDC25C PTPs in glioma biology. Like PTKs, these PTPs may represent promising targets for the development of novel diagnostic and therapeutic strategies in the treatment of high-grade gliomas.
Keywords: Signal transduction, Tyrosine phosphorylation
Introduction
Like in many other cell types, accumulation of genetic damage in glial (precursor) cells may ultimately lead to transformation into tumor cells. The World Health Organization (WHO) classifies the resulting gliomas in many different histological subtypes and in four different malignancy grades [88]. Astrocytomas, oligodendrogliomas and mixed oligoastrocytomas are the most common glioma types. Most of these tumors are characterized by diffuse infiltrative growth in the surrounding brain parenchyma which precludes curative treatment by surgery or radiotherapy alone, and together with their relative resistance towards conventional chemotherapy this places these tumors among the most intractable human malignancies [24]. For the most frequent of these gliomas, i.e. glioblastoma multiforme (GBM), only 10% of the glioblastoma patients survive 5 years post-diagnosis when receiving combined chemo- and radio therapy as opposed to 2% for radiation treatment alone [74, 137]. Ependymomas are quite rare and are traditionally thought to originate from the ependymal cells lining the ventricular walls in the brain and the central canal in the spinal cord.
A more detailed insight in the molecular etiology of these gliomas may open up new ways to combat these tumors. Multiple studies towards that end, including cytogenetic analyses, but also genome-wide microarray expression screens, have been performed and important players have been identified. They mainly converge on aberrant growth factor signaling and deregulation of cell cycle control, processes that exploit the phosphorylation of proteins on tyrosine residues [17, 43, 106]. This reliance on protein tyrosine kinase (PTK) activity urges for a closer look on the role that is played by protein tyrosine phosphatases (PTPs), their enzymatic counterparts (Fig. 1), in glioma biology. The PTP enzyme family has emerged as an important regulator of developmental and disease-related signaling pathways [143], and multiple members are directly linked to malformation syndromes and tumorigenesis [54, 107]. So far, a systematic review on the role of PTPs in gliomagenesis is lacking. Here, following a brief discussion of the aberrant signaling pathways in glioblastomas, we will introduce the general features of the PTP family before presenting detailed information on PTP involvement in glioma biology. The PTP enzymes discussed may represent entry points for development of novel diagnostics and therapies in the treatment of high-grade gliomas.
Fig. 1.
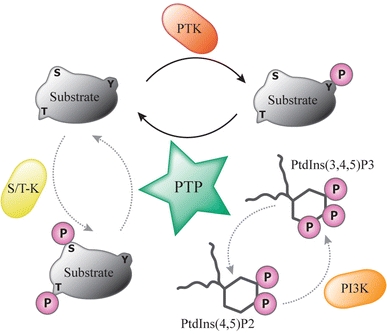
Reversibility of signaling pathways by virtue of protein tyrosine phosphatase activity. The opposing actions of PTK and protein tyrosine phosphatase (PTP) enzymes provide the cell with a functional diad that regulates the activity of the mutual substrates through phosphorylation of tyrosine (Y) residues in a reversible manner. Note that phosphorylation itself may well regulate the activity of PTKs and PTPs themselves. Therefore, PTPs do not simply repress or undo PTK activity; they may synergize or cooperate in situations where tyrosine phosphorylation boosts PTP activity or dephosphorylation activates the PTK. Some PTPs have a so-called dual specificity; in addition to their activity towards phosphotyrosine they are also able to dephosphorylate serine (S) and/or threonine (T) residues in protein substrates that have been phosphorylated by serine/threonine kinases (S/T-K). A subset of these dual-specific PTPs, most notably the tumor suppressor protein PTEN, even demonstrate the potential to dephosphorylate phospholipid substrates like phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3) that is produced by phosphatidylinositol-3-kinase (PI3K)-mediated phosphorylation of PtdIns(4,5)P2 (phosphatidylinositol-4,5-bisphosphate)
Affected processes in gliomas
The sequential changes that transform glial (precursor) cells into tumor cells fit well with the cancer cell hallmarks encountered in many other tumor systems [52]. First, cancer cells have acquired the ability to be self-sufficient in providing growth signals while reducing their sensitivity to growth-inhibitory signals. The molecules and pathways involved in gliomas include alterations in Ras and PI3K (phosphatidylinositol-3-kinase) signaling pathway components, which regulate proliferation, survival and differentiation. Especially, genes encoding the receptor PTKs for epidermal growth factor (EGFR) and platelet-derived growth factor (PDGFR) are frequently mutated, and also alterations in the fibroblast growth factor (FGFR) and hepatocyte growth factor/scatter factor (cMET) receptor PTK genes have been documented [17]. Proteins involved in the subsequent signaling downstream of these PTK receptors have been linked to gliomagenesis as well (Fig. 2). Mutations in two tumor suppressor genes, NF1 and PTEN (Phosphatase and tensin homolog on chromosome 10), are found in a considerable portion of glioblastomas and also genetic changes in Akt and Ras have been documented [17]. The involvement of PTEN and other PTP family members will be described in more detail later on.
Fig. 2.
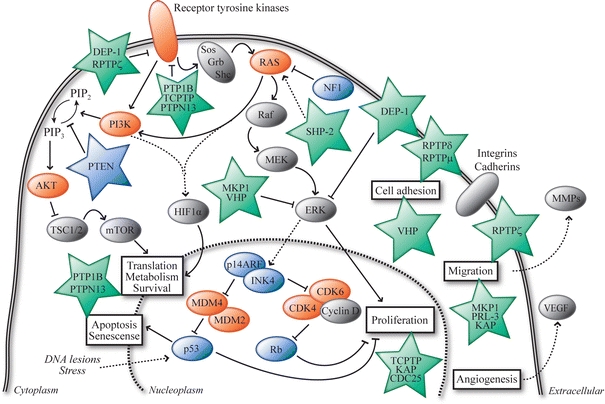
Interplay of PTP signaling and the major pathways affected in glioblastomas. The Ras/PI3K, p53 and Rb signaling pathways, which are often altered in glioblastomas, are shown in a cellular context. For simplicity, only major factors that are described in the text are depicted. Proteins shown in vermillion are frequently hyperactive in gliomas (due to amplification/mutation) while proteins in blue are frequently hypoactive or even inactive (as a result of mutation/deletion). The (mostly green) asterisks represent the receptor-type and non-transmembrane PTPs (protein names are used) that are discussed in the text and in Table 1, and the ways how they feed into the Ras/PI3K, p53 and Rb pathways and affect cellular processes (boxed text) is depicted. Arrows indicate interactions that result in stimulation, lines ending with a perpendicular bar indicate inhibitory interactions. Dashed lines reflect indirect interaction pathways. PIP2 phosphatidylinositol-4,5-bisphosphate, PIP3 phosphatidylinositol-3,4,5-triphosphate
Secondly, glioma cells tend to evade apoptosis and senescence. The TP53 gene, encoding a key regulator of cell cycle progression, DNA repair, cellular senescence, apoptosis and angiogenesis [152], is frequently inactivated at an early stage in gliomagenesis. Alternatively, other important players that affect p53 functioning, e.g. the p53 activator p14ARF or the negative p53 regulators MDM2 and MDM4 (Fig. 2) are deleted or amplified in gliomas, respectively. In addition, glioma cells may exploit several other mechanisms to evade apoptosis, including abrogated PI3K signaling (Fig. 2), genetic alterations in death receptor and mitochondria-dependent pathways via Bcl2-like-12 [135, 136], and inactivation of the retinoblastoma (Rb) tumor suppressor pathway (Fig. 2). The Rb pathway controls the cell cycle entry step into the DNA replication phase [46]. Many of the involved proteins are genetically altered in GBMs: the CDK4 and CDK6 genes are often amplified, while Rb is deleted or mutated in ~11% of the glioblastomas [17]. Normally, continued proliferation of somatic cells leads to senescence due to successive shortening of chromosome ends, the telomeres [14]. High-grade gliomas bypass telomere shortening, hence senescence, either by restoring embryonal telomerase activity [128] or by exploiting a telomere maintenance mechanism that involves recombination-based interchromosomal exchanges of DNA segments [55].
Thirdly, gliomas elicit angiogenesis and invade surrounding tissues. GBMs are among the most highly vascularized solid tumor types [66] and this may be due to mutations in the key glioma genes PTEN and EGFR that feed into the HIF1α pathway [68]. HIF1α is a transcription factor that normally accumulates under hypoxic conditions and then activates factors involved in angiogenesis and cell survival [127], including the vascular endothelial growth factor (VEGF) and VEGF receptor families (Fig. 2). In glioma specimens, this activation may be independent of hypoxia [110] and often gives rise to abnormal microvasculature that results in thrombosis and microhemorrhages, paradoxically causing hypoxia and eventually tumor necrosis. ‘Dispersion’ of glioma cells in the surrounding brain tissue is distinct from the invasion and metastasis as displayed by other tumors [24], but still many parallels exist. Like in epithelial tumors, several integrins, including αvβ3, are upregulated in gliomas [82]. In addition, the expression of the N-cadherin cell adhesion molecule and its associated protein β-catenin is increased at the borders of GBMs [149]. In addition, the levels of matrix metalloproteases (MMPs) and non-MMP proteases, instrumental in degrading surrounding extracellular matrix, have been reported to be elevated in gliomas [146]. Low-grade gliomas usually have normal protease levels [153, 157], but still display an invasive phenotype, suggesting that increased protease activity is not required for glioma dispersal.
Protein tyrosine phosphatases
Reversible tyrosine phosphorylation of proteins (Fig. 1) plays an important role in the regulation, proliferation and differentiation of cells and the development and function of tissues and organisms [143]. The exploitation of this signaling mechanism to drive gliomagenesis is reflected in the altered activities of PTK growth factor receptors and their downstream effectors that have been observed in tumor specimens (as depicted in Fig. 2) and warrants a closer look on the role of the catalytic opponents of PTKs, the PTPs.
There are 107 genes in the human genome that belong to the PTP superfamily of enzymes and, based on the sequence homology of their catalytic domains and these have been categorized into four different classes [6]. Class I comprises 38 so-called ‘classical’ PTPs, i.e. enzymes that exclusively dephosphorylate phosphotyrosine residues, as well as 61 dual-specificity PTPs (DSPs). As suggested by the name, DSPs can also dephosphorylate phosphoserine and phosphothreonine residues, and some even display a preference for phosphatidylinositol phosphates and mRNAs as substrates [115]. The 38 classical PTPs can be further subdivided into transmembrane, receptor-like (RPTPs) and non-receptor-type PTPs [54]. In the human genome, there is only a single Class II gene (ACP1). It encodes the low-molecular weight PTP, termed LMPTP, that is specific for phosphotyrosine residues [117]. Class III comprises three CDC25 homologs that dephosphorylate tyrosine and threonine residues within cyclin-dependent kinases (CDKs), which participate in cell cycle regulation [120]. Class IV consists of the eyes absent (Eya) proteins, which recognize phosphorylated tyrosine, or dual serine and tyrosine residues and function as transcriptional regulators [65]. Recently, Eya was shown to dephosphorylate tyrosine phosphorylated histone H2AX, thereby regulating the recruitment of pro-apoptotic and/or DNA repair factors to sites of DNA damage [27, 78].
Each PTP class is believed to originate from a distinct ancestral gene and as such the clear similarity in the dephosphorylation mechanism they exploit provides an impressive example of convergent evolution. A common feature of the PTP classes I, II and III is the PTP signature motif (H/V)C(X)R(S/T) in their catalytic domain [138]. The cysteine residue is essential for catalytic activity; the target phosphate group is transferred from the substrate onto this catalytic site cysteine, producing a thiol intermediate, and is subsequently released via hydrolysis [8]. The Eya Class IV phosphatases use a slightly different mechanism, in which aspartate instead of cysteine plays a crucial role in a metal-dependent reaction. Outside the catalytic domain, PTPs are very diverse in their structure. Multiple additional protein domains, such as protein–protein interaction or phospholipid-binding motifs, help to diversify their functions (Fig. 3).
Fig. 3.
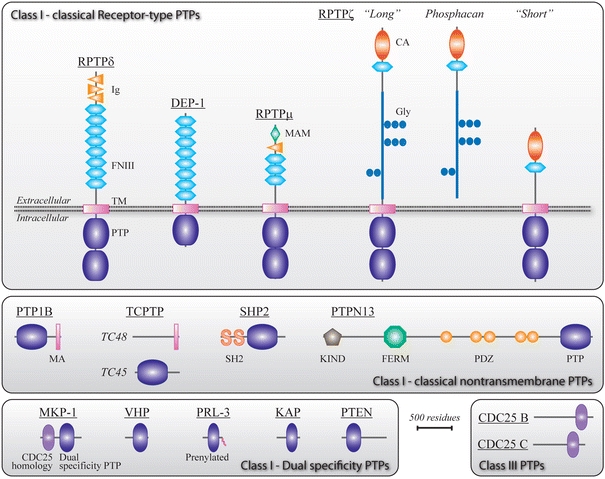
Domain structure of the PTP superfamily members implicated in glioma biology. Schematic representations of class I transmembrane receptor type and intracellular ‘classical’ PTPs are given in the upper and middle part of the figure, respectively. The lower part shows the class I dual-specific phosphatases and the class III CDC25 family members. Thus far, class II and class IV type PTPs have not been implicated in gliomagenesis. Alternative splicing of genes PTPRZ and PTPN2 leads to expression of three variants of RPTPζ [87] and two of TCPTP [60], respectively. PTP protein names are used here (Table 1), and isoform names are in italics. Protein domain names and acronyms are indicated and the curly tail in PRL-3 represents its C-terminal prenylation. CA carbonic anhydrase-like domain, FERM band 4.1/ezrin/radixin/moesin homology domain, FNIII fibronectin type III repeat, Glyc glycosylation sites encoded within PTPRZ exon 12, Ig immunoglobulin-like repeat, KIND kinase non-catalytic C-lobe domain, MA membrane-associated domain, MAM meprin/A5-protein/PTPμ homology domain, PDZ PSD-95/discs-large/ZO-1 homology domain, PTP catalytic protein tyrosine phosphatase domain, SH2 Src homology 2 domain, TM transmembrane domain. Drawings are to scale (bar corresponds to 500 amino acid residues)
PTPs in tumorigenesis
Because multiple PTKs have been identified as proto-oncogenes, it was initially believed that many PTPs might be rapidly uncovered as tumor suppressors. However, it turned out that there is a considerable redundancy with regard to PTP functioning. Furthermore, PTPs may not simply oppose PTK actions but, as some kinases are (auto)inhibited by phosphorylation, could also act in synergy. Thus, next to some tumor suppressors, the PTP superfamily also harbors proto-oncogenes [107]. Before turning to the involvement of PTPs in gliomagenesis, the evidence implicating PTPs in other tumors will be summarized.
In fact, PTEN represents the only PTP that can unambiguously be termed a tumor suppressor, i.e. the gene is frequently deleted or mutated in tumor specimens [70]. Ironically, it is not PTEN’s protein dephosphorylation capacity, but its ability to dephosphorylate D3-phosphoinositosides—and thereby dampen PI3K-Akt signaling—that is crucial for maintaining tissue homeostasis [90, 102]. Several other PTP genes are deleted or mutated in cancer tissues as well, but the frequency is usually quite low and causal relationships often have not been assessed. A screen encompassing 87 out of the 107 human PTP genes revealed mutations for six of them, all encoding classical PTPs (PTPRF, PTPRG, PTPRT, PTPN3, PTPN13 and PTPN14) in 26% of the colon cancer samples analyzed [154]. Alterations in these genes were also identified in lung, breast and gastric cancer samples, but at an even lower frequency. For PTPRT, the most frequently mutated PTP out of these six, growth suppressive activity was indeed demonstrated in cell transfection experiments, underscoring the relevance of this PTP in cell growth regulation [154]. The majority of cancer-related PTPRT mutations affects the extracellular domain that mediates homophilic cell–cell interactions, suggesting that mutation of this phosphatase supports tumor migration [159]. The signaling pathways that are regulated by PTPRT remain to be disclosed, but recently the signal transducer and activator of transcription 3 (STAT3), that plays an important role in a variety of hematopoietic malignancies and solid tumors [158], was identified as a PTPRT substrate [163]. Extensive sequence analysis screens have also revealed genetic alterations in some other classical PTP genes (PTPRJ, PTPN6, PTPRO, PTPN1), implicating these as potential tumor suppressors [107].
As stated above, some PTPs might even function as oncogenes. Currently, the only PTP gene for which this has been demonstrated is PTPN11 that encodes the classical PTP SHP2 [20]. SHP2 (Fig. 3) is normally in an inactive conformation due to an intramolecular interaction of its N-terminal SH2 domains with the catalytic PTP domain at its C-terminus. SHP2 mutations as observed in hematological malignancies and some solid tumors disrupt this interaction, thereby constitutively activating the phosphatase [85, 141]. Then, this synergizes with growth factor and cytokine stimuli that run via the Ras-ERK, AKT and STAT5 signaling routes [99], all pathways that have important roles in growth, proliferation and survival. How exactly SHP2 is stimulating these ligand-gated signaling pathways, i.e. which specific targets it dephosphorylates, remains controversial, but the dephosphorylation and consequent activation of Src family kinases is a likely scenario [20]. Indications that other PTPs also have oncogenic potential are more indirect. Several classical PTP genes are overexpressed in human cancers, including PTPRA, PTPRH, PTPRF, PTPN1, PTPN6 and PTPN7, but these may reflect homeostatic adaptation to enhanced kinase activities [107]. Further studies are needed to address the contribution of these PTPs in tumor development in a more direct way.
Linking PTPs to glioma biology
It has also been investigated whether PTPs are relevant players in glioma development. Indeed, 15 out of the 107 PTP genes are implicated in some way. In Table 1, the observational data on these PTPs and gliomagenesis are summarized and on the following pages, their involvement is discussed in more detail (in the subheadings first the official gene name and between brackets the common protein name will be given). Undoubtedly, the number of studies and the fraction of PTPs that are involved in gliomagenesis will increase in the coming years. However, it is to be expected that genome-wide microarray datasets will only make a limited contribution to this, due to the very low expression levels of most PTP genes; hence, more PTP-focused approaches are required.
Table 1.
Overview of protein tyrosine phosphatases associated with glioma biology
| Genea | Protein (synonyms) | Chromosomal location | Alteration | Glioma type | WHO grade | Model system | References |
|---|---|---|---|---|---|---|---|
| Classical class I PTPs (8 out of 38 genes) | |||||||
| PTPRD | RPTPδ | 9p23–p24.3 | Del/Mut | OD, AC | II, III and IV | CM | [61, 130, 150] |
| PTPRJ | DEP1 (CD148, RPTPη) | 11p11.2 | 11p ampb | AG | I | CM | [114] |
| PTPRM | RPTPμ | 18p11.2 | LOE | GBM | IV | CM | [15] |
| PTPRZ | RPTPζ | 7q31.3 | OE | GBM | IV | CM | [147] |
| PTPN1 | PTP1B | 20q13.1–13.2 | Act | GBM | IV | PCL + TG | [4] |
| PTPN2 | TCPTP (MPTP, PTP-S) | 18p11.3–11.2 | Dephosph EGFRvIII | GBM | IV | CL | [73] |
| PTPN11 | SHP2 (SH-PTP2, Syp, PTP1D, PTP2C, SH-PTP3) | 12q24.1 | Mutb | OD | II | CM | [91] |
| PTPN13 | PTPN13 (PTP-BAS, FAP-1, PTP1E, RIP, PTPL1, PTP-BL) | 4q21.3 | OE | GBM | IV | CM | [39] |
| Dual-specific class I PTPs (DSPs, 5 out of 61 genes) | |||||||
| DUSP1 | MKP-1 (3CH134, PTPN10, erp, CL100/HVH1) | 5q34 |
DR OE |
GBM | IV |
CL + Etop CL + DM |
[86] [84] |
| DUSP26 | VHP (‘similar to RIKEN cDNA 0710001B24’) | 2q37.3 | DR | GBM | IV | CM | [140] |
| PTP4A3 | PRL-3 | 8q24.3 | OE | Gliomas | II, III and IV | CM | [75] |
| CDKN3 | KAP | 14q22 | Ab splicing | Prim GBM | IV | CM | [161] |
| PTEN | PTEN (MMAC1, TEP1) | 10q23.3 | Mut / Del / Epigenetic | Prim and sec GBM | IV | CM |
and references therein |
| Class III PTPs (2 out of 3 genes) | |||||||
| CDC25B | CDC25B | 20p13 | OE | AC, GBM | III, IV | CM | [104] |
| CDC25C | CDC25C | 5q31 | DR | GBM | IV | CL + AM | [44] |
Genes are listed in the order used in Alonso et al. [6]
Note that thus far no PTPs from classes II (LmPTP) and IV (Eya) have been implicated in glioma biology
Ab aberrant, Act activation, AG angiocentric glioma, amp amplification, AC astrocytoma, AM ansamycin, CL cell line, CM clinical material, Del homozygous deletion, Dephosph dephosphorylates, DM dexamethasone, DR down-regulated, Etop etoposide, GBM glioblastoma, LOE loss of expression, Mut mutation, OD oligodendroglioma, PCL primary cell line, prim primary, Sec secondary, TG troglitazone
aSome PTP genes give rise to multiple protein isoforms
bFound in one patient only
PTPRD (RPTPδ)
RPTPδ, a cell adhesion molecule-like RPTP with fibronectin type III (FNIII) and immunoglobulin-like (Ig) repeats in its extracellular domain (Fig. 3), is mainly expressed in brain. Mouse model studies revealed its importance in hippocampal learning and memory [145]. PTPRD is located on chromosome 9p23–24.1, a genomic region that is often lost during the progression from low- to high-grade gliomas of the astrocytoma and oligodendroglioma types [61]. Two recent studies on GBMs report focal homozygous deletions at the PTPRD locus that do not impart surrounding genes [130, 150]. Also, missense or nonsense mutations in the PTPRD gene or hypermethylation of the PTPRD promoter were frequently encountered [130, 150], and the loss of RPTPδ expression predicts for poor prognosis in these patients [150]. PTPRD mutations and deletions are also common among other tumors [29, 31, 125, 133]. In melanomas and lung carcinomas, these mutations are distributed all over the PTPRD gene. The alterations that were detected in GBM samples more locate to the RPTPδ extracellular part than to the intracellular PTP domains [130, 150]. The re-introduction of functional RPTPδ in GBM cell lines harboring PTPRD mutations or deletions leads to reduced proliferation and an increase in apoptotic cells [130]. Transfection experiments pointed to STAT3 as an RPTPδ substrate [150] and, interestingly, aberrant activation of STAT3 is commonly observed in GBM specimens [116]. Thus, RPTPδ has a tumor suppressor function in a variety of tissues. In glial cells, this activity may require its putative cell adhesion potential as well as phosphatase activity (Fig. 2).
PTPRJ (DEP-1)
PTPRJ encodes the transmembrane protein DEP-1 that consists of a single intracellular PTP domain and eight FNIII repeats in its long extracellular part (Fig. 3). DEP-1 is able to counteract the signaling of several RTKs, including PDGFR, VEGFR2 and MET [77, 108, 139], pointing to a role as tumor suppressor (Fig. 2). Indeed, it was shown to negatively regulate cellular growth [69]. In glioma cells, DEP-1 is instrumental in the growth-inhibitory effect of somatostatin by dephosphorylating and inactivating the MAP kinase ERK [93, 94]. DEP-1 expression is detectable in only a subset of gliomas and expression levels correlate well with somatostatin’s anti-proliferative effects [93, 94]. Not much is known about possible alterations in the PTPRJ gene in glioma specimen. Thus far, an amplification of the chromosome 11p11.2 region, which contains the PTPRJ gene, has been detected in an angiocentric glioma [114]. Angiocentric gliomas are grade I tumors that contain features of both astrocytic and ependymal differentiation [88]. PTPRJ amplification would not correspond with a suggested tumor suppressor function for DEP-1 but, since only one case has been described, clearly more angiocentric glioma samples need to be studied before conclusions can be drawn.
PTPRM (RPTPμ)
The cell surface receptor RPTPμ is a homophilic cell–cell adhesion molecule expressed in neuronal, glial and endothelial cells (Fig. 3). RPTPμ not only forms an adhesive contact itself, but it also regulates cell adhesion by dephosphorylating components of the cadherin–catenin complexes. In addition, the rigidity of the extracellular part of the molecule is thought to dictate the location of this phosphatase in cell–cell spacings [7] in line with a role for this PTP in cell contact signaling processes. When compared with normal brain tissue and low-grade astrocytomas, full-length RPTPμ protein expression is lost specifically in GBM [15]. To investigate whether this influenced cell adhesive and migratory characteristics, RPTPμ knockdown experiments were performed in a GBM cell line. Reduced RPTPμ levels resulted in morphological changes and an increased migration in vitro, and in a mouse xenograft model of intracranially injected GBM cells RPTPμ knockdown caused morphological heterogeneity in the grafts [15]. These data put forward RPTPμ as a ‘migration suppressor’ with regard to the diffuse infiltrative growth pattern observed in human gliomas (Fig. 2). A more recent study revealed that the RPTPμ downregulation in GBM results from proteolytic breakdown which releases an active PTP fragment in the cytosol [16]. Interestingly, both overexpression of RPTPμ and shRNA-mediated reduction of the RPTPμ intracellular fragments suppressed migration and growth factor-independent survival of glioblastoma cells. Apparently, decisions in glioma cells on proliferation versus cell death or adhesion versus migration are dependent on the relative levels of full-length RPTPμ and RPTPμ intracellular domain. Importantly, application of a peptide inhibitor of RPTPμ phosphatase activity also resulted in a reduction of glioma cell migration [16], warranting further studies towards RPTPμ targeted therapeutics.
PTPRZ (RPTPζ)
The classical PTP gene that currently has the strongest links with gliomagenesis is PTPRZ, which encodes RPTPζ [80]. Originally, the cDNA was cloned by two different groups, who termed the encoded protein PTPζ [79] and RPTPβ [83], respectively. This is causing quite some confusion in the literature, also because the name RPTPβ is nowadays linked to the protein encoded by the gene PTPRB [6]. As a result of alternative splicing, PTPRZ encodes three variants that may have different roles in glial differentiation (Fig. 3). Long and short transmembrane RPTPζ isoforms are present in glial precursor cells. In more mature glia, a secreted version called phosphacan is expressed, which lacks the PTP part and consists of the RPTPζ extracellular domain [18]. Pleiotrophin (PTN) has been identified as a ligand for RPTPζ [89]. It induces cell migration, at least in part by binding and clustering RPTPζ, and thereby inhibiting its phosphatase activity [42, 96]. Fibroblasts that overexpress PTN acquire a transformed phenotype suggesting that PTN may function as an oncogene [21]. In line with this, several carcinoma patients showed increased PTN levels in both serum and tumor material [131]. PTN is overexpressed in GBM tissues as well [147] and may exert an oncogenic effect through the inactivation of RPTPζ.
The role of RPTPζ in glioma development is as yet unclear. PTPRZ overexpression in tumor material is correlated with increased malignancy [101, 147]. Some other cancer variants also display increased PTPRZ expression [38]. Although in normal adult brain it is mainly phosphacan that is expressed, all three PTPRZ isoforms can be found in gliomas. Knockdown of RPTPζ leads to a decrease in GBM growth, both in vitro and in vivo [148], but the mechanism that may explain this growth advantage resulting from RPTPζ overexpression remains unclear. After all, excess of RPTPζ would be expected to function as a potential PTN scavenger, thus neutralizing PTN’s oncogenic abilities. The tumor-promoting effect of RPTPζ may be explained by mechanisms independent of PTN, for instance through the effects of other RPTPζ ligands that include growth factors, extracellular matrix proteins and neuronal cell adhesion molecules [111]. Neuronal adhesion molecule binding by RPTPζ is thought to influence neuron–glial cell interactions and cell migration, e.g. by promoting transcriptional activity of NFκB [87] which is implicated in the integration of signals involved in adhesion and migration. In addition, a recent study revealed that the endothelial integrin αvβ3 not only interacts with both PTN and RPTPζ, but in doing so mediates the stimulatory effect of PTN on cell migration and angiogenesis [97]. Thus, the overexpression of RPTPζ may provide an advantage for GBM cells by promoting their migration, even on endothelial cell tracks [142]. Alternatively, because proteolytic cleavage involving MMPs and presenilin/γ-secretase was shown to produce an RPTPζ intracellular fragment that displays nuclear localization [23], even more direct effects at the transcriptional level should be considered. Finally, altered RPTPζ levels have a bearing on neurotrophic signaling pathways in view of the recent identification of tropomyosin-related kinase A, TrkA, a nerve growth factor receptor that is highly expressed in the developing nervous system, as an in vivo RPTPζ substrate [129]. Also because high RPTPζ levels were reported in oligodendrogliomas as opposed to (oligo)astrocytomas within the grade II gliomas [49], a further evaluation of RPTPζ’s contribution to gliomagenesis is warranted.
PTPN1 (PTP1B)
The cytoplasmic, ER membrane-associated phosphatase PTP1B (Fig. 3) is overexpressed in several tumors, including mammary and ovary carcinomas, but it is unclear as yet whether it should be viewed as a potential oncogene or a tumor suppressor [107]. PTP1B can act as a positive regulator of Ras signaling, partly by modulating p120RasGAP levels [33] and it is also able to activate the oncogene protein Src [11]. Importantly, the elimination of PTP1B activity in MMTV-neu induced mammary tumors in mice significantly delayed tumor growth and prevented lung metastasis, whereas PTP1B overexpression in mammary tissue led to spontaneous breast cancer development [9, 67]. In contrast, PTP1B effectively inhibits signaling of several oncogenic tyrosine kinases [50], while inactivation of the gene in TP53 knockout mice accelerates the spontaneous development of lymphomas [32].
In gliomas, no amplification of the PTPN1 gene has been found [118]. However, PTP1B may well be of relevance for glioma therapy. The treatment of tumor cells derived from a primary GBM patient with troglitazone led to PTP1B activation and subsequent dephosphorylation of STAT3, a negative regulator of Fas-mediated apoptosis that controls anti-apoptotic FLIP and Bcl-2 protein levels [4]. Thus, PTP1B activity may facilitate at least one way of tumor cell death. It should be noted, however, that most recently it was found that STAT3 not only functions in transcription regulation in the nucleus in response to cytokine and tyrosine kinase oncoprotein signaling, but that in addition it is required in mitochondria for the alterations in energy metabolism that support Ras-dependent malignant transformation of cells [47, 155]. Reasoning along these lines, PTP1B activity could potentially counteract STAT3s moonlighting activity in mitochondria and in this way hamper the glioma cell’s adaptation to a more glycolytic, hypoxic growth state (Fig. 2). Taken together, depending on the cell type, PTP1B has distinct functions, but current data are more in favor of a tumor suppressive rather than an oncogenic role in GBM. Investigations on PTP1B as a drug target [162] principally relate to diabetes [34], and several inhibitors have been tested clinically.
PTPN2 (TC-PTP)
TC-PTP is an intracellular PTP that is ubiquitously expressed and that is highly similar in amino acid sequence to PTP1B (Fig. 3). As a consequence, these PTPs have different as well as overlapping substrates and thus may fulfill complementary as well as redundant roles in health and disease [34]. Alternative splicing of PTPN2 transcripts leads to two protein isoforms; a 48 kDa variant localized at the endoplasmic reticulum and a 45 kDa variant, TC45, that lacks the hydrophobic C-terminus [19, 28, 100]. TC45 is usually found in the nucleus but upon proper stimuli it can also reside in the cytoplasm. Interestingly, TC45 causes a reduced proliferation of U87MG glioblastoma cells that are engineered to express the glioma-associated truncated protein EGFRvIII when compared with normal U87MG cells [73]. This might well be caused by inhibition of ERK and PI3K signaling due to direct dephosphorylation of EGFRvIII (Fig. 2). Injection of TC45- and EGFRvIII-overexpressing U87MG cells in the brains of nude mice revealed that also in vivo TC45 expression leads to reduced growth of EGFRvIII-expressing tumors [73]. Although no altered TC45 activity levels in gliomas have been reported to date, the potential of TC-PTP to counteract EGFRvIII activity in vivo may be of therapeutic value.
PTPN11 (SHP-2)
So far, PTPN11 is the only PTP that was proven to function as an oncogene in certain tumors [20, 107]. It contains two Src homology type 2 (SH2) domains that function as phosphotyrosine-binding domains (Fig. 3). Activating germline PTPN11 mutations are found in patients with Noonan syndrome, a developmental disorder characterized by an increased risk of malignancies. Somatic mutations that activate PTPN11 occur in several types of hematologic malignancies, most notably juvenile myelomonocytic leukemia. The role of SHP-2 in RAS/ERK signaling (Fig. 2) is thus well established in tumor development. Germline mutations that impair phosphatase activity and turn PTPN11 into a dominant-negative mutant are causative of LEOPARD (lentigines, electrocardiogram abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and deafness) syndrome, which also predisposes to the development of cancers such as myelodysplastic syndrome, acute myelogenous leukemia or neuroblastoma. It remains an intriguing conundrum why Noonan and Leopard syndrome mutations result in partly similar pathologies despite their quite opposite effects on SHP2 catalytic function [45].
The expression of SHP-2 in the U87MG GBM cell line resulted in increased AKT phosphorylation upon EGF stimulation [156], underscoring that SHP-2 also facilitates growth factor signaling in glial cells. PTPN11 mutations in gliomas are rather uncommon [64, 91] but other components in the RAS/PI3K pathway, like EGFR, NF1 and RAS, are genetically altered in the majority (88%) of GBMs [17]. Only a single PTPN11 mutation, in a grade II oligodendroglioma patient, has been described thus far [91]. This missense mutation in the SH2 domain of SHP-2 promotes its phosphatase activity and thus resembles the Noonan syndrome type of activating mutations. Although PTPN11 can display oncogenic behavior in other tumor types, and plays an important positive role in Ras signaling in gliomas as well, it does not represent a specific target in these tumors.
PTPN13 (PTPN13)
The large cytosolic PTP PTPN13 (Fig. 3) appears to be endowed with tumor suppressive as well as oncogenic potential [2]. Support for a tumor suppressive role comes from mutation screens in colon, breast, lung and ovarian cancer specimens [119, 154] and our finding that high-risk human papillomavirus (HPV) protein E6-induced degradation of PTPN13 in squamous epithelial cells contributes to oncogenic transformation [132], at least in part by augmenting the Ras/Erk signaling pathway. In line with this, PTPN13 inactivating mutations and enhanced Erk activity was detected in HPV-negative head and neck squamous cell carcinomas [56]. Contrasting with the above, several studies have pointed to a tumor-promoting activity of PTPN13. The PTPN13 gene is a target for the transcription regulator fusion protein EWS-FLI1 and the resulting overexpression of PTPN13 in Ewing’s sarcoma cells boosts cell growth and motility [1]. Furthermore, PTPN13 expression in a variety of tumors provides the cancer cells with a survival mechanism, by inhibiting Fas-induced apoptosis [51]; and, finally, PTPN13 may be instrumental in tumor cell survival through its interactions with p75NTR, TRPM2 and IκBα, all proteins that modulate cell stress signaling [2].
PTPN13’s link to FAS-induced apoptosis appears relevance for glioma biology (Fig. 2). FAS is a death receptor that, upon activation by its ligand, induces caspase cascades leading to cleavage of proteins and apoptosis. PTPN13 attenuates FAS receptor cell surface levels by inhibiting the export of the FAS receptor from intracellular stores to the cell membrane [62]. PTPN13 expression is specifically upregulated in GBM tissues [39] and knockdown of PTPN13 in GBM cell lines indeed results in increased FAS-mediated apoptosis. In addition, PTPN13 directly interacts with and dephosphorylates FAS in a FAS ligand-dependent manner, thereby reducing the ability of glioma cells to undergo FAS-mediated apoptosis [39]. Hence, PTPN13 might play a role in the aforementioned apoptosis resistance that is displayed by gliomas and that complicates their treatment with chemo- and radiotherapy.
DUSP1 (MKP-1)
Dual-specificity MAP kinase phosphatases (MKPs) are capable of dephosphorylating phosphotyrosine as well as phosphothreonine residues of MAP kinases and thus influence important cellular processes such as proliferation, differentiation, apoptosis and survival [115]. MKP-1 (Fig. 3) is induced by growth factors and dephosphorylates the MAP kinases JNK (Jun N-terminal Kinase), p38 and ERK1/2. The apoptosis-inducing effect of the chemotherapeutic drug etoposide in a glioma cell line was shown to be PKCδ-dependent, involving the ubiquitin-mediated degradation of MKP-1 and resulting in increased ERK1/2 phosphorylation [86]. Likewise, MKP-1 levels are decreased when glioma cells are treated with cadmium, another apoptosis-inducing compound that also leads to increased MAP kinase phosphorylation [72]. On the other hand, the inhibitory effect of dexamethasone and rosiglitazone on glioma cell invasiveness rather depends on an increase in MKP-1 levels in glioma cells [63, 84]. Thus, both anti-apoptotic and anti-migratory aspects are to be considered for MKP-1 as a potential target in glioma therapy (Fig. 2).
DUSP26 (VHP)
The gene DUSP26 is expressed in brain and retina. It encodes VHP (Fig. 3), a dual-specificity phosphatase with unclear function. VHP has been implicated in MAP kinase dephosphorylation and in upregulation of cell–cell adhesion [140]. The latter is the result of VHP’s ability to dephosphorylate Kap3, a subunit of the KIF3 motor complex that is involved in the transport of cadherin/catenin components from intracellular vesicles to the cell membrane at sites of cell–cell contact. If VHP promotes cell–cell adhesion, a process that is lost in diffusively infiltrating gliomas, one may expect impaired VHP activity in GBM material. Indeed, in eight out of nine cases, a quantitative PCR analysis of mRNA levels showed that DUSP26 transcript levels were decreased in GBM patient samples when compared with normal brain tissue [140]. This may be taken as evidence that VHP is an important regulator of cellular adhesion in glial cells (Fig. 2) and urges for studies involving additional samples and functional read-outs. In anaplastic thyroid cancer samples, however, DUSP26 expression levels were found to be upregulated. The DUSP26 overexpression stimulated the growth of these cells, most likely through the dephosphorylation of p38 and thus inhibition of p38-mediated apoptosis, whereas it had little effect on Erk1/2 MAP kinases [160]. This would rather point to DUSP26 as being an oncogene.
PTP4A3 (PRL-3)
Phosphatase of regenerating liver (PRL) enzymes is unique in the PTP family because of their C-terminal CAAX prenylation motif (Fig. 3). Not much is known about the nature of their substrates [115]. In dividing cells, PRLs are located at the mitotic spindle, but in interphase cells they are membrane associated. It is conceivable that this cell cycle-dependent localization confines PRL access to substrates. Several studies tie PRL members to oncogenic events such as angiogenesis, cell invasion, motility and metastasis [122], and proposed modes of action include stimulation of Src, Rho or PI3K signaling pathways. The PRL-3 protein was found to display a conspicuous expression pattern during gliomagenesis. In normal brain tissue and grade I gliomas, no PRL-3 is detectable, whereas grade II gliomas display low PRL-3 levels. In high-grade glioma tissues strong PRL-3 expression is observed [75]. Interestingly, the PRL-3 levels correlate with that of several matrix metalloproteinases that are instrumental in the proteolytic degradation of the extracellular matrix, suggesting that PRL-3 is associated with glioma invasion (Fig. 2).
CDKN3 (KAP)
KAP was identified as a cell cycle regulating protein because of its ability to dephosphorylate Cdk2, thereby inhibiting G1-S phase progression [48, 53]. KAP also binds two other cell cycle regulators, Cdk3 and cdc2, but it remains to be determined whether these also serve as KAP substrates. It therefore came as a surprise that astrocytomas display increased CDKN3 messenger levels that correlate well with increasing malignancy grade and decreased patient survival [161]. A closer examination revealed that in primary GBM tissues CDKN3 transcripts are spliced differently, leading to the expression of a dominant-negative KAP variant. Thus, the aberrant splice variant interferes with normal KAP functioning and, as a consequence, increases the Cdk2-dependent proliferation of GBM cells (Fig. 2). The reduction in KAP activity also has a bearing on the migration of glioma cells, but this reflects KAP phosphatase-dependent regulation of cdc2 protein levels and hence cdc2-dependent cell motility [161].
PTEN (PTEN)
The paradigm of PTP involvement in glioma development is PTEN. PTEN is a dual-specificity phosphatase (Fig. 3) and is thus able to dephosphorylate phosphorylated serine, threonine and tyrosine residues in vitro, with a preference for extremely acidic substrates [103]. Germline mutations in PTEN give rise to a whole series of seemingly unrelated syndromes including Cowden disease, an autosomal dominant disorder characterized by multiple hamartomas and predisposition to breast, thyroid and endometrial carcinomas [36]. Intriguingly, it is PTEN’s capability to dephosphorylate phosphaditylinositol-3,4,5-triphosphate (PIP3) [90] (Figs. 1, 2) that appears most relevant in vivo and has turned PTEN into the second-most inactivated tumor suppressor protein in human cancers [70]. Key in this notion was a missense mutation in a Cowden patient, which disrupted the phosholipid phosphatase activity of PTEN but did not affect its protein phosphatase activity [102]. Thus, PTEN counteracts PI3K by preventing the PIP3-mediated recruitment of the serine/threonine kinase Akt to the cell membrane. As a consequence, Akt will not be activated by a submembranous kinase and will not be able to phosphorylate its many target proteins that stimulate cellular growth, proliferation and survival (Fig. 2).
Ample reports, from its discovery onwards [134], have documented PTEN inactivation, either by mutation (homozygous) deletion or through epigenetic mechanisms, in high-grade gliomas [43]. Mice lacking PTEN expression in astrocytes show an increased proliferation of these cells [41]. In addition, PTEN loss contributes to the angiogenic process featured in high-grade gliomas via upregulation of VEGF transcript levels [113]. Several members of the JNK family show increased levels in PTEN-deficient glioma cells as well, resulting in the simultaneous upregulation of the JNK and PI3K pathway that feed into cellular processes such as proliferation, survival, DNA repair and apoptosis [151]. Intriguingly, in a recent study, the microRNA miR-26a was disclosed as a direct regulator of PTEN expression: it appeared to be frequently amplified in human glioma specimens with monoallelic PTEN loss, and miR-26a-mediated PTEN repression-enhanced tumor formation in a murine glioma model [58]. This epigenetic mechanism for PTEN downregulation in glioma corroborates the Akt signaling pathway as a major Achilles’ heel in the development of these tumors (Fig. 2). For further details on PTEN involvement in gliomagenesis, we refer to recent dedicated reviews [22, 35, 76].
CDC25B and CDC25C
The human class III PTP subfamily comprises three CDC25 variants that are all involved in cell cycle regulation [120]. They positively regulate different stages of mitosis, and are often found upregulated in human cancers, where they cause aberrant cell cycle regulation and genetic instability [13]. CDC25B (Fig. 3) has been linked to glioma progression based on its high expression in WHO grades III and IV astrocytomas when compared with the levels in lower grade tumors, and may serve as a prognostic marker in astrocytoma patients [104]. Also, CDC25C may have bearing for glioma biology as revealed by experiments involving the potential anticancer antibiotic ansamycin. Ansamycin inhibits the activation of Hsp90 and thus results in degradation of Hsp90 target proteins and ultimately to cell cycle arrest and apoptosis [57, 71, 95]. Ansamycin treatment of glioma cell lines resulted in the downregulation of CDC25C and Cdc2 levels, both proteins that are involved in G2–M transition [44]. In lung cancer cell lines, a similar effect was observed [126]. CDC25C downregulation, therefore, could explain the ansamycin-induced cell cycle arrest and apoptosis observed in glioma cell lines. Future experiments will have to reveal whether a similar effect on CDC25C can be exploited for glioma treatment in vivo. All three CDC25 proteins have been targets for pharmaceutical drug discovery, but no inhibitors have so far been tested clinically.
PTP signaling significance in glioma biology
The above illustrates that several PTPs are involved in gliomagenesis, whereas the contribution of other PTPs is as yet hypothetical (Table 1; Fig. 2). Especially, the results obtained with permanent glioma cell lines should be interpreted with care, since these model systems only partially represent the molecular and pathological characteristics of glial tumors.
A number of PTPs appear frequently mutated in glioma tumor specimens, providing strong evidence for a causal contribution to gliomagenesis. PTEN is inactivated by mutation, deletion or gene silencing in a large portion of these tumors, and several recent reviews are dedicated to its involvement in gliomagenesis [3, 22, 35, 76]. Also PTPRD is located in a region that is often deleted in astrocytomas (including GBMs) and oligodendrogliomas, suggestive of an important role for PTPRD in glial cell functioning. PTPRD-deficient mice, however, while presenting neuropathological symptoms [145], do not display increased glial tumor susceptibility. Genomic alterations in glioma samples have also been detected for PTPRJ and PTPN11, but since this concerns single cases it remains uncertain whether these mutations play a role in gliomagenesis or are merely the consequence of genetic instability in these tumors. The precedent of PTPN11 activating mutations in leukemias speaks in favor of SHP-2 as an oncogenic protein in sporadic gliomas. The status of PTPRJ as a cancer susceptibility gene [121] could as yet not be backed by knockout studies; DEP-1-deficient mice did not show an increased tumor incidence, not even in colon [144].
Several PTPs have been linked to gliomagenesis based on the stage-specific changes in their messenger and/or protein expression levels. So far, it remains unclear by which mechanism such PTP level changes are brought about and whether the observed changes are functionally relevant in an in vivo tumor setting. The changes in the availability of crucial transcription factors or in the accessibility of PTP gene regulatory sequences through epigenetic mechanisms may play a role at the transcriptional level, but also factors that impinge on mRNA and/or protein stability may post-transcriptionally modulate PTP levels and favor tumor growth. In line with this, small compounds, such as etoposide, ansamycins and cadmium induce the specific degradation of certain PTPs and result in reduced proliferation and increased apoptosis in glioma cell models. A clinically relevant hallmark of gliomas is their very infiltrative growth pattern. Therefore, PTPs that are known or suspected to be involved in adhesion and migration of cells deserve special attention. Three of the PTP genes that display altered expression levels in the highly malignant GBM subtype have been linked to cellular adhesion: PTPRD, PTPRM and DUSP26 (Table 1). Also, PTPRZ, DUSP1, PTP4A3 and CDKN3, all implicated in the control of cell migration, demonstrate aberrant expression levels in GBM specimens (Table 1). It is tempting to speculate that these alterations contribute to the adhesive and migratory changes of glioma cells (Fig. 2).
Confounding issues in glioma research
So far, the acquired insight into the molecular mechanisms underlying gliomagenesis has not yet resulted in markedly improved treatment modalities for GBM patients. Several reasons may account for this, including the often extensive and diffuse infiltrative growth in the brain parenchyma, the difficult ‘druggability’ of this tumor type due to the blood–brain barrier (BBB), and the lack of easily accessible and clinically relevant glioma model systems for research purposes. For instance, most glioma cell models do not recapitulate the diffuse infiltrative growth pattern upon introduction in the mouse brain [25, 37, 112]. Furthermore, neither the amplification of EGFR, which is found in approximately 50% of the GBM patients, nor the expression of EGFRvIII is present in most GBM cell lines [10, 105, 109]. The use of short term, primary cultures does not solve the problem because significant genetic alterations have been found after only a small number of passages in glioma cultures [30, 81]. To create research models that better recapitulate GBM features, including diffuse infiltrative growth, xenograft models have been developed through orthotopic (the tissue from which the tumor originated) implantation of (primary) glioma cells into immune-suppressed or immune-deficient mice [5, 25, 40]. A drawback of these xenograft models is that grafts usually lack accurate angiogenesis and that contributions by the immune system cannot be considered [37]. The use of genetically modified animal models that spontaneously develop gliomas, therefore, seems a powerful approach [5, 59] and quite recently the generation of a mouse GBM model in a region- and cell type-specific manner using intracerebral lentiviral transduction was reported [92].
However, even in such powerful models, the BBB, which regulates the composition of the central nervous system interstitial fluid by allowing low-molecular weight and lipid compounds to pass while at the same time restricting access of water-soluble compounds and macromolecules, may well hamper the diffusion of chemotherapeutic agents to glioma cells [98]. Consequently, progress in drug discovery for glioblastoma treatment has been slow. DNA-alkylating agents lomustine and carmustine were first marketed over 30 years ago, while temozolomide is a more recent addition in the same drug class [124, 137]. New insights into glioma’s molecular drivers have led to the approval by the FDA, in an accelerated review early 2009, of the use of bevacizumab (avastin) against refractory GBM. In addition, a number of clinical trials are ongoing or planned. Table 2 lists currently approved and experimental drugs for glioblastoma. Their mode of action is rather diverse and given the number of compounds that target PTKs or their downstream effectors one may expect PTP targeting drugs to enter the pipeline in the near future.
Table 2.
Approved and experimental drugs for glioblastoma
| Drug name | Mode of action | Status | Notes |
|---|---|---|---|
| Lomustine | Alkylating agent | Marketed | |
| Carmustine | Alkylating agent | Marketed | Also sold as slow-release polymeric wafer system under Gliadel® |
| Temozolomide | Alkylating agent | Marketed | Prodrug, indicated for newly diagnosed glioblastoma multiforme |
| Bevacizumab (Avastin) | Anti-VEGF | Approved in 2009 | |
| Nimotuzumab | Anti-EGFR | Phase 3 | |
| TM601 | Tumor cell binder, antiangiogenic | Phase 3 planned | Synthetic form of the scorpion venom peptide chlorotoxin; also as radiolabelled formulations |
| MPC-6827 (Azixa) | Microtubule-destabilizing agent | Phase 2 | |
| CDX-110 | EGFRvIII vaccine | Phase 2 | |
| Cotara | 131I-radiolabelled mAb conjugate | Phase 2 | |
| XL184 | RTK inhibitor | Phase 2 | Targets Met, VEGFR2, Kit, Flt-3 and Tie-2 |
| TLN-4601 | Peripheral benzodiazepine receptor (PBR) ligand | Phase 2 | Inhibitor of the RAS-mitogen-activated protein kinase (MAPK) pathway |
| CYT997 | Vascular targeting agent and tubulin inhibitor | Phase 2 | |
| BSI-201 | PARP-1 inhibitor | Phase 2 | |
| Banoxantrone (AQ4 N) | Topoisomerase inhibitor and DNA intercalator | Phase 2 | Prodrug, activated by hypoxia |
| ICT-121 | CD133 peptide vaccine | Phase 1 | Trial planned 2010 |
| ICT-107 | Dendritic cell-based vaccine | Phase 1 | Uses patient-derived, specifically primed dendritic cells |
| MP-470 | Multiple TRKi and Rad51 DNA repair inhibitor | Phase 1 |
BioWorld® Today (AHC Media LLC), Thomson Pharma® IDdb, Pharmaprojects, Company and NIH websites
Experimental treatments are usually tested in combination with approved therapies
This table shows that more recently developed compounds are targeting specific signaling pathways rather than the general DNA damaging effect of the earlier drugs
The potential therapeutic and diagnostic use of PTP signaling pathways in gliomas
Glioma patients are still facing a very poor prognosis and, in view of the limited durability of surgical and local irradiation treatments and small number of applicable drugs (Table 2), research efforts aim at the identification of novel targets. PTPs that are linked to glioma development and progression thus represent potential starting points in glioma diagnostic and therapeutic strategies. A prime candidate is of course the PTEN tumor suppressor that is so frequently inactivated in GBM. To overcome this loss of function, the PI3K pathway needs to be suppressed and several inhibitors of PI3K signaling are being evaluated for clinical use. Rapamycin is a widely used drug that inhibits mTOR, a downstream target of AKT [12]. Unfortunately, a recent phase I clinical trial revealed that rapamycin treatment leads to increased AKT phosphorylation in PTEN-deficient GBM patients, probably due to the relief from a negative feedback loop created by mTOR [26]. This indicates that PI3K signaling should be targeted more upstream to become useful in anti-glioma strategies.
Apoptosis resistance of gliomas provides yet another hurdle on the road towards effective therapies against these tumors. Several PTPs have been associated with apoptosis signaling pathways in gliomas (Fig. 2), which potentially makes them therapeutic targets. For instance, the finding that in glioma cell lines the anti-diabetic and anti-inflammatory drug troglitazone activates PTP1B and, as a consequence, downregulates the anti-apoptotic FLIP and Bcl2 proteins [4] urges for a follow-up in in vivo glioma models. Likewise, compounds that specifically inhibit PTPN13, the negative regulator of FAS-mediated apoptosis in glioma cell lines [39], may enhance the sensitivity of glial tumor cells to undergo apoptosis, e.g. following radio- or chemotherapy.
Targeting of gliomas via the blood stream implies successful transfer over the BBB, an obstacle that prevents adequate effect of many agents in diseases of the central nervous system. In the design of small compound inhibitors for glioma-relevant PTPs, one therefore has to take the BBB into account. Likewise, the exploitation of (monoclonal) antibodies, nowadays common agents to fight cancer growth, to specifically target glioma cells in the brain is limited by the BBB [123]. To circumvent this problem, antibodies that for example inhibit EGFR and VEGFR signaling might be injected directly intracerebrally. A promising start for the application of PTP-directed antibodies in glioma therapies was obtained using a monoclonal antibody against the extracellular domain of the short RPTPζ transmembrane isoform [38]. When coupled to the cytostatin saporin, the antibody killed U87MG glioma cells in vitro and it significantly delayed in vivo tumor growth of a U87MG xenograft. As mentioned before, such cell-based models do not recapitulate all pathobiological features of gliomas and the therapeutic potential of this approach needs further investigation. Irrespective, these results urge for further studies on the use of monoclonal antibodies against receptor-type PTPs as tumor-directed agents in glioma therapies. In addition, in view of the often altered expression levels for several of the PTPs discussed here in GBM specimens when compared with normal glial tissue, the assessment of PTP expression levels in tumor tissue may well serve to refine GBM staging, aid in the design of combinatorial treatment protocols and/or support the monitoring of treatment responses.
Concluding remarks
Ample evidence indicates an important role for PTPs in development and progression of tumors, including gliomas. Alterations in multiple PTP genes and their products have been noted in gliomas. However, careful assessment of their exact functional relevance in glioma biology is needed. Functional studies on PTP signaling in other tumor systems will of course be of value but, in view of the unique growth and dispersion characteristics of gliomas, studies using tumor models that faithfully mimic glioma biology are mandatory. This will ultimately allow a better validation of glioma treatment modalities and enable improvement in the poor prognosis glioma patients still face today.
Acknowledgments
We apologize to all colleagues whose original work could not be referred to due to space constraints. This work was supported in part by a European Research Community Fund (MRTN-CT-2006-035830).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- BBB
Blood–brain barrier
- CDK
Cyclin-dependent kinase
- DSPs
Dual-specificity PTPs
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- EGFRvIII
EGFR variant III
- Eya
Eyes absent
- FNIII
Fibronectin type III
- GBM
Glioblastoma multiforme
- HPV
Human papillomavirus
- JNK
Jun N-terminal kinase
- LEOPARD
Lentigines, electrocardiogram abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and deafness
- MKPs
MAP kinase phosphatases
- MMPs
Matrix metalloproteases
- PDGFR
Platelet-derived growth factor receptor
- PI3K
Phosphatidylinositol-3-kinase
- PIP3
Phosphaditylinositol-3, 4, 5-triphosphate
- PRL
Phosphatase of regenerating liver
- PTEN
Phosphatase and tensin homolog on chromosome 10
- PTK
Protein tyrosine kinase
- PTN
Pleiotrophin
- PTP
Protein tyrosine phosphatase
- Rb
Retinoblastoma
- VEGF
Vascular endothelial growth factor
References
- 1.Abaan OD, Levenson A, Khan O, Furth PA, Uren A, Toretsky JA. PTPL1 is a direct transcriptional target of EWS-FLI1 and modulates Ewing’s Sarcoma tumorigenesis. Oncogene. 2005;24:2715–2722. doi: 10.1038/sj.onc.1208247. [DOI] [PubMed] [Google Scholar]
- 2.Abaan OD, Toretsky JA. PTPL1: a large phosphatase with a split personality. Cancer Metastasis Rev. 2008;27:205–214. doi: 10.1007/s10555-008-9114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abounader R. Interactions between PTEN and receptor tyrosine kinase pathways and their implications for glioma therapy. Expert Rev Anticancer Ther. 2009;9:235–245. doi: 10.1586/14737140.9.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akasaki Y, Liu G, Matundan HH, Ng H, Yuan X, Zeng Z, Black KL, Yu JS. A peroxisome proliferator-activated receptor-gamma agonist, troglitazone, facilitates caspase-8 and -9 activities by increasing the enzymatic activity of protein-tyrosine phosphatase-1B on human glioma cells. J Biol Chem. 2006;281:6165–6174. doi: 10.1074/jbc.M505266200. [DOI] [PubMed] [Google Scholar]
- 5.Aldape K, Colman H, James CD. Models of malignant glioma. Drug Discov Today Dis Models. 2006;3:191–196. [Google Scholar]
- 6.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 7.Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, van der Merwe PA, Jones EY. Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science. 2007;317:1217–1220. doi: 10.1126/science.1144646. [DOI] [PubMed] [Google Scholar]
- 8.Barford D, Jia Z, Tonks NK. Protein tyrosine phosphatases take off. Nat Struct Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- 9.Bentires-Alj M, Neel BG. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007;67:2420–2424. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- 10.Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, Bigner DD. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–8022. [PubMed] [Google Scholar]
- 11.Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439–41446. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- 12.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 13.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 14.Bryan TM, Cech TR. Telomerase and the maintenance of chromosome ends. Curr Opin Cell Biol. 1999;11:318–324. doi: 10.1016/S0955-0674(99)80043-X. [DOI] [PubMed] [Google Scholar]
- 15.Burgoyne AM, Palomo JM, Phillips-Mason PJ, Burden-Gulley SM, Major DL, Zaremba A, Robinson S, Sloan AE, Vogelbaum MA, Miller RH, Brady-Kalnay SM (2009) PTPmu suppresses glioma cell migration and dispersal. Neuro Oncol. doi:10.1215/15228517-15222009-15228019 [DOI] [PMC free article] [PubMed]
- 16.Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, Robinson S, Sloan AE, Miller RH, Brady-Kalnay SM. Proteolytic cleavage of protein tyrosine phosphatase μ regulates glioblastoma cell migration. Cancer Res. 2009;69:6960–6968. doi: 10.1158/0008-5472.CAN-09-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Network Cancer Genome Atlas Research. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canoll PD, Petanceska S, Schlessinger J, Musacchio JM. Three forms of RPTP-beta are differentially expressed during gliogenesis in the developing rat brain and during glial cell differentiation in culture. J Neurosci Res. 1996;44:199–215. doi: 10.1002/(SICI)1097-4547(19960501)44:3<199::AID-JNR1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 19.Champion-Arnaud P, Gesnel MC, Foulkes N, Ronsin C, Sassone-Corsi P, Breathnach R. Activation of transcription via AP-1 or CREB regulatory sites is blocked by protein tyrosine phosphatases. Oncogene. 1991;6:1203–1209. [PubMed] [Google Scholar]
- 20.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan AK, Li YS, Deuel TF. Pleiotrophin transforms NIH 3T3 cells and induces tumors in nude mice. Proc Natl Acad Sci USA. 1993;90:679–682. doi: 10.1073/pnas.90.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng CK, Fan Q-W, Weiss WA. PI3K signaling in glioma: animal models and therapeutic challenges. Brain Pathol. 2009;19:112–120. doi: 10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow JP, Fujikawa A, Shimizu H, Suzuki R, Noda M. Metalloproteinase- and gamma-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J Biol Chem. 2008;283:30879–30889. doi: 10.1074/jbc.M802976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007;114:443–458. doi: 10.1007/s00401-007-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claes A, Schuuring J, Boots-Sprenger S, Hendriks-Cornelissen S, Dekkers M, van der Kogel AJ, Leenders WP, Wesseling P, Jeuken JW. Phenotypic and genotypic characterization of orthotopic human glioma models and its relevance for the study of anti-glioma therapy. Brain Pathol. 2008;18:423–433. doi: 10.1111/j.1750-3639.2008.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cool DE, Tonks NK, Charbonneau H, Walsh KA, Fischer EH, Krebs EG. cDNA isolated from a human T-cell library encodes a member of the protein-tyrosine-phosphatase family. Proc Natl Acad Sci USA. 1989;86:5257–5261. doi: 10.1073/pnas.86.14.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox C, Bignell G, Greenman C, Stabenau A, Warren W, Stephens P, Davies H, Watt S, Teague J, Edkins S, Birney E, Easton DF, Wooster R, Futreal PA, Stratton MR. A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci USA. 2005;102:4542–4547. doi: 10.1073/pnas.0408593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Witt Hamer PC, Van Tilborg AA, Eijk PP, Sminia P, Troost D, Van Noorden CJ, Ylstra B, Leenstra S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene. 2008;27:2091–2096. doi: 10.1038/sj.onc.1210850. [DOI] [PubMed] [Google Scholar]
- 31.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dube N, Bourdeau A, Heinonen KM, Cheng A, Loy AL, Tremblay ML. Genetic ablation of protein tyrosine phosphatase 1B accelerates lymphomagenesis of p53-null mice through the regulation of B-cell development. Cancer Res. 2005;65:10088–10095. doi: 10.1158/0008-5472.CAN-05-1353. [DOI] [PubMed] [Google Scholar]
- 33.Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci USA. 2004;101:1834–1839. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dube N, Tremblay ML. Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: from diabetes, obesity to cell cycle, and cancer. Biochim Biophys Acta. 2005;1754:108–117. doi: 10.1016/j.bbapap.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 35.Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–5430. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- 36.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 37.Finkelstein SD, Black P, Nowak TP, Hand CM, Christensen S, Finch PW. Histological characteristics and expression of acidic and basic fibroblast growth factor genes in intracerebral xenogeneic transplants of human glioma cells. Neurosurgery. 1994;34:136–143. [PubMed] [Google Scholar]
- 38.Foehr ED, Lorente G, Kuo J, Ram R, Nikolich K, Urfer R. Targeting of the receptor protein tyrosine phosphatase beta with a monoclonal antibody delays tumor growth in a glioblastoma model. Cancer Res. 2006;66:2271–2278. doi: 10.1158/0008-5472.CAN-05-1221. [DOI] [PubMed] [Google Scholar]
- 39.Foehr ED, Lorente G, Vincent V, Nikolich K, Urfer R. FAS associated phosphatase (FAP-1) blocks apoptosis of astrocytomas through dephosphorylation of FAS. J Neurooncol. 2005;74:241–248. doi: 10.1007/s11060-004-7202-x. [DOI] [PubMed] [Google Scholar]
- 40.Fomchenko EI, Holland EC. Mouse models of brain tumors and their applications in preclinical trials. Clin Cancer Res. 2006;12:5288–5297. doi: 10.1158/1078-0432.CCR-06-0438. [DOI] [PubMed] [Google Scholar]
- 41.Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- 42.Fukada M, Fujikawa A, Chow JP, Ikematsu S, Sakuma S, Noda M. Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett. 2006;580:4051–4056. doi: 10.1016/j.febslet.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 43.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Morales P, Carrasco-Garcia E, Ruiz-Rico P, Martinez-Mira R, Menendez-Gutierrez MP, Ferragut JA, Saceda M, Martinez-Lacaci I. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene. 2007;26:7185–7193. doi: 10.1038/sj.onc.1210534. [DOI] [PubMed] [Google Scholar]
- 45.Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet. 2006;15(Spec. No. 2):R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- 46.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 47.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 49.Hagerstrand D, Smits A, Eriksson A, Sigurdardottir S, Olofsson T, Hartman M, Nister M, Kalimo H, Ostman A. Gene expression analyses of grade II gliomas and identification of rPTPbeta/zeta as a candidate oligodendroglioma marker. Neuro Oncol. 2008;10:2–9. doi: 10.1215/15228517-2007-041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J Biol Chem. 2003;278:739–744. doi: 10.1074/jbc.M210194200. [DOI] [PubMed] [Google Scholar]
- 51.Halle M, Tremblay ML, Meng TC. Protein tyrosine phosphatases: emerging regulators of apoptosis. Cell Cycle. 2007;6:2773–2781. doi: 10.4161/cc.6.22.4926. [DOI] [PubMed] [Google Scholar]
- 52.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 53.Hannon GJ, Casso D, Beach D. KAP: a dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc Natl Acad Sci USA. 1994;91:1731–1735. doi: 10.1073/pnas.91.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendriks WJ, Elson A, Harroch S, Stoker AW. Protein tyrosine phosphatases: functional inferences from mouse models and human diseases. Febs J. 2008;275:816–830. doi: 10.1111/j.1742-4658.2008.06249.x. [DOI] [PubMed] [Google Scholar]
- 55.Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, Wharton SB, Jellinek DA, Arbuckle SM, Yoo J, Robinson BG, Learoyd DL, Stalley PD, Bonar SF, Yu D, Pollock RE, Reddel RR. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res. 2005;11:217–225. [PubMed] [Google Scholar]
- 56.Hoover AC, Strand GL, Nowicki PN, Anderson ME, Vermeer PD, Klingelhutz AJ, Bossler AD, Pottala JV, Hendriks W, Lee JH. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene. 2009;28:3960–3970. doi: 10.1038/onc.2009.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–4009. [PubMed] [Google Scholar]
- 58.Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T, Holland EC. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 2009;19:132–143. doi: 10.1111/j.1750-3639.2008.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ibarra-Sanchez MJ, Simoncic PD, Nestel FR, Duplay P, Lapp WS, Tremblay ML. The T-cell protein tyrosine phosphatase. Semin Immunol. 2000;12:379–386. doi: 10.1006/smim.2000.0220. [DOI] [PubMed] [Google Scholar]
- 61.Idbaih A, Carvalho Silva R, Criniere E, Marie Y, Carpentier C, Boisselier B, Taillibert S, Rousseau A, Mokhtari K, Ducray F, Thillet J, Sanson M, Hoang-Xuan K, Delattre JY. Genomic changes in progression of low-grade gliomas. J Neurooncol. 2008;90:133–140. doi: 10.1007/s11060-008-9644-z. [DOI] [PubMed] [Google Scholar]
- 62.Ivanov VN, Lopez Bergami P, Maulit G, Sato TA, Sassoon D, Ronai Z. FAP-1 association with Fas (Apo-1) inhibits Fas expression on the cell surface. Mol Cell Biol. 2003;23:3623–3635. doi: 10.1128/MCB.23.10.3623-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jan H-J, Lee C-C, Lin Y-M, Lai J-H, Wei H-W, Lee H-M. Rosiglitazone reduces cell invasiveness by inducing MKP-1 in human U87MG glioma cells. Cancer Lett. 2009;277:141–148. doi: 10.1016/j.canlet.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 64.Janzarik WG, Kratz CP, Loges NT, Olbrich H, Klein C, Schafer T, Scheurlen W, Roggendorf W, Weiller C, Niemeyer C, Korinthenberg R, Pfister S, Omran H. Further evidence for a somatic KRAS mutation in a pilocytic astrocytoma. Neuropediatrics. 2007;38:61–63. doi: 10.1055/s-2007-984451. [DOI] [PubMed] [Google Scholar]
- 65.Jemc J, Rebay I. The eyes absent family of phosphotyrosine phosphatases: properties and roles in developmental regulation of transcription. Annu Rev Biochem. 2007;76:513–538. doi: 10.1146/annurev.biochem.76.052705.164916. [DOI] [PubMed] [Google Scholar]
- 66.Jouanneau E. Angiogenesis and gliomas: current issues and development of surrogate markers. Neurosurgery. 2008;62:31–50. doi: 10.1227/01.NEU.0000311060.65002.4E. [DOI] [PubMed] [Google Scholar]
- 67.Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, Kennedy BP, Muller WJ, Tremblay ML. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39:338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 68.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keane MM, Lowrey GA, Ettenberg SA, Dayton MA, Lipkowitz S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996;56:4236–4243. [PubMed] [Google Scholar]
- 70.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 71.Kim HR, Lee CH, Choi YH, Kang HS, Kim HD. Geldanamycin induces cell cycle arrest in K562 erythroleukemic cells. IUBMB Life. 1999;48:425–428. doi: 10.1080/713803539. [DOI] [PubMed] [Google Scholar]
- 72.Kim S-M, Park J-G, Baek W-K, Suh M-H, Lee H, Yoo SK, Jung K-H, Suh S-I, Jang B-C. Cadmium specifically induces MKP-1 expression via the glutathione depletion-mediated p38 MAPK activation in C6 glioma cells. Neurosci Lett. 2008;440:289–293. doi: 10.1016/j.neulet.2008.05.064. [DOI] [PubMed] [Google Scholar]
- 73.Klingler-Hoffmann M, Fodero-Tavoletti MT, Mishima K, Narita Y, Cavenee WK, Furnari FB, Huang HJ, Tiganis T. The protein tyrosine phosphatase TCPTP suppresses the tumorigenicity of glioblastoma cells expressing a mutant epidermal growth factor receptor. J Biol Chem. 2001;276:46313–46318. doi: 10.1074/jbc.M106571200. [DOI] [PubMed] [Google Scholar]
- 74.Knisely JP, Baehring JM. A silver lining on the horizon for glioblastoma. Lancet Oncol. 2009;10:434–435. doi: 10.1016/S1470-2045(09)70124-X. [DOI] [PubMed] [Google Scholar]
- 75.Kong L, Li Q, Wang L, Liu Z, Sun T. The value and correlation between PRL-3 expression and matrix metalloproteinase activity and expression in human gliomas. Neuropathology. 2007;27:516–521. doi: 10.1111/j.1440-1789.2007.00818.x. [DOI] [PubMed] [Google Scholar]
- 76.Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther. 2008;7:1321–1325. doi: 10.4161/cbt.7.9.6954. [DOI] [PubMed] [Google Scholar]
- 77.Kovalenko M, Denner K, Sandstrom J, Persson C, Gross S, Jandt E, Vilella R, Bohmer F, Ostman A. Site-selective dephosphorylation of the platelet-derived growth factor beta-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J Biol Chem. 2000;275:16219–16226. doi: 10.1074/jbc.275.21.16219. [DOI] [PubMed] [Google Scholar]
- 78.Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, Kim SJ, Tonks NK. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009;284:16066–16070. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krueger NX, Saito H. A human transmembrane protein-tyrosine-phosphatase, PTP zeta, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc Natl Acad Sci USA. 1992;89:7417–7421. doi: 10.1073/pnas.89.16.7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamprianou S, Harroch S. Receptor protein tyrosine phosphatase from stem cells to mature glial cells of the central nervous system. J Mol Neurosci. 2006;29:241–255. doi: 10.1385/JMN:29:3:241. [DOI] [PubMed] [Google Scholar]
- 81.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 82.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- 83.Levy JB, Canoll PD, Silvennoinen O, Barnea G, Morse B, Honegger AM, Huang JT, Cannizzaro LA, Park SH, Druck T, et al. The cloning of a receptor-type protein tyrosine phosphatase expressed in the central nervous system. J Biol Chem. 1993;268:10573–10581. [PubMed] [Google Scholar]
- 84.Lin Y-M, Jan H-J, Lee C-C, Tao H-Y, Shih Y-L, Wei H-W, Lee H-M. Dexamethasone reduced invasiveness of human malignant glioblastoma cells through a MAPK phosphatase-1 (MKP-1) dependent mechanism. Eur J Pharmacol. 2008;593:1–9. doi: 10.1016/j.ejphar.2008.06.111. [DOI] [PubMed] [Google Scholar]
- 85.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 86.Lomonaco SL, Kahana S, Blass M, Brody Y, Okhrimenko H, Xiang C, Finniss S, Blumberg PM, Lee HK, Brodie C. Phosphorylation of protein kinase Cdelta on distinct tyrosine residues induces sustained activation of Erk1/2 via down-regulation of MKP-1: role in the apoptotic effect of etoposide. J Biol Chem. 2008;283:17731–17739. doi: 10.1074/jbc.M801727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lorente G, Nelson A, Mueller S, Kuo J, Urfer R, Nikolich K, Foehr ED. Functional comparison of long and short splice forms of RPTPbeta: implications for glioblastoma treatment. Neuro Oncol. 2005;7:154–163. doi: 10.1215/S1152851704000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase zeta/RPTPbeta, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) J Biol Chem. 1996;271:21446–21452. doi: 10.1074/jbc.271.35.21446. [DOI] [PubMed] [Google Scholar]
- 90.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 91.Martinelli S, Carta C, Flex E, Binni F, Cordisco EL, Moretti S, Puxeddu E, Tonacchera M, Pinchera A, McDowell HP, Dominici C, Rosolen A, Di Rocco C, Riccardi R, Celli P, Picardo M, Genuardi M, Grammatico P, Sorcini M, Tartaglia M. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet Cytogenet. 2006;166:124–129. doi: 10.1016/j.cancergencyto.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 92.Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15:110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Massa A, Barbieri F, Aiello C, Arena S, Pattarozzi A, Pirani P, Corsaro A, Iuliano R, Fusco A, Zona G, Spaziante R, Florio T, Schettini G. The expression of the phosphotyrosine phosphatase DEP-1/PTPeta dictates the responsivity of glioma cells to somatostatin inhibition of cell proliferation. J Biol Chem. 2004;279:29004–29012. doi: 10.1074/jbc.M403573200. [DOI] [PubMed] [Google Scholar]
- 94.Massa A, Barbieri F, Aiello C, Iuliano R, Arena S, Pattarozzi A, Corsaro A, Villa V, Fusco A, Zona G, Spaziante R, Schettini G, Florio T. The phosphotyrosine phosphatase eta mediates somatostatin inhibition of glioma proliferation via the dephosphorylation of ERK1/2. Ann N Y Acad Sci. 2004;1030:264–274. doi: 10.1196/annals.1329.033. [DOI] [PubMed] [Google Scholar]
- 95.McIlwrath AJ, Brunton VG, Brown R. Cell-cycle arrest and p53 accumulation induced by geldanamycin in human ovarian tumour cells. Cancer Chemother Pharmacol. 1996;37:423–428. doi: 10.1007/s002800050407. [DOI] [PubMed] [Google Scholar]
- 96.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci USA. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mikelis C, Sfaelou E, Koutsioumpa M, Kieffer N, Papadimitriou E. Integrin alpha(v)beta(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. Faseb J. 2009;23:1459–1469. doi: 10.1096/fj.08-117564. [DOI] [PubMed] [Google Scholar]
- 98.Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–273. [PubMed] [Google Scholar]
- 99.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H, Akashi K, Gilliland DG, Neel BG. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 100.Mosinger B, Jr, Tillmann U, Westphal H, Tremblay ML. Cloning and characterization of a mouse cDNA encoding a cytoplasmic protein-tyrosine-phosphatase. Proc Natl Acad Sci USA. 1992;89:499–503. doi: 10.1073/pnas.89.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Muller S, Kunkel P, Lamszus K, Ulbricht U, Lorente GA, Nelson AM, von Schack D, Chin DJ, Lohr SC, Westphal M, Melcher T. A role for receptor tyrosine phosphatase zeta in glioma cell migration. Oncogene. 2003;22:6661–6668. doi: 10.1038/sj.onc.1206763. [DOI] [PubMed] [Google Scholar]
- 102.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nakabayashi H, Hara M, Shimizu K. Prognostic significance of CDC25B expression in gliomas. J Clin Pathol. 2006;59:725–728. doi: 10.1136/jcp.2005.029546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 108.Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278:5728–5735. doi: 10.1074/jbc.M210656200. [DOI] [PubMed] [Google Scholar]
- 109.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39:29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 110.Parliament MB, Allalunis-Turner MJ, Franko AJ, Olive PL, Mandyam R, Santos C, Wolokoff B. Vascular endothelial growth factor expression is independent of hypoxia in human malignant glioma spheroids and tumours. Br J Cancer. 2000;82:635–641. doi: 10.1054/bjoc.1999.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peles E, Schlessinger J, Grumet M. Multi-ligand interactions with receptor-like protein tyrosine phosphatase beta: implications for intercellular signaling. Trends Biochem Sci. 1998;23:121–124. doi: 10.1016/s0968-0004(98)01195-5. [DOI] [PubMed] [Google Scholar]
- 112.Pilkington GJ, Bjerkvig R, De Ridder L, Kaaijk P. In vitro and in vivo models for the study of brain tumour invasion. Anticancer Res. 1997;17:4107–4109. [PubMed] [Google Scholar]
- 113.Pore N, Liu S, Haas-Kogan DA, O’Rourke DM, Maity A. PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Cancer Res. 2003;63:236–241. [PubMed] [Google Scholar]
- 114.Preusser M, Hoischen A, Novak K, Czech T, Prayer D, Hainfellner JA, Baumgartner C, Woermann FG, Tuxhorn IE, Pannek HW, Bergmann M, Radlwimmer B, Villagran R, Weber RG, Hans VH. Angiocentric glioma: report of clinico-pathologic and genetic findings in 8 cases. Am J Surg Pathol. 2007;31:1709–1718. doi: 10.1097/PAS.0b013e31804a7ebb. [DOI] [PubMed] [Google Scholar]
- 115.Pulido R, Hooft van Huijsduijnen R. Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. Febs J. 2008;275:848–866. doi: 10.1111/j.1742-4658.2008.06250.x. [DOI] [PubMed] [Google Scholar]
- 116.Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 117.Ramponi G, Stefani M. Structural, catalytic, and functional properties of low M(r), phosphotyrosine protein phosphatases. Evidence of a long evolutionary history. Int J Biochem Cell Biol. 1997;29:279–292. doi: 10.1016/s1357-2725(96)00109-4. [DOI] [PubMed] [Google Scholar]
- 118.Reichardt W, Jung V, Brunner C, Klein A, Wemmert S, Romeike BF, Zang KD, Urbschat S. The putative serine/threonine kinase gene STK15 on chromosome 20q13.2 is amplified in human gliomas. Oncol Rep. 2003;10:1275–1279. [PubMed] [Google Scholar]
- 119.Revillion F, Puech C, Rabenoelina F, Chalbos D, Peyrat JP, Freiss G. Expression of the putative tumor suppressor gene PTPN13/PTPL1 is an independent prognostic marker for overall survival in breast cancer. Int J Cancer. 2009;124:638–643. doi: 10.1002/ijc.23989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rudolph J. Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry. 2007;46:3595–3604. doi: 10.1021/bi700026j. [DOI] [PubMed] [Google Scholar]
- 121.Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikos T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, Groot PC, Lindeman J, Mooi WJ, Meijjer GA, Scholten G, Dauwerse H, Paces V, van Zandwijk N, van Ommen GJ, Demant P. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- 122.Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C, Kinzler KW, Vogelstein B. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–1346. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 123.Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- 124.Sathornsumetee S, Rich JN. Designer therapies for glioblastoma multiforme. Ann N Y Acad Sci. 2008;1142:108–132. doi: 10.1196/annals.1444.009. [DOI] [PubMed] [Google Scholar]
- 125.Sato M, Takahashi K, Nagayama K, Arai Y, Ito N, Okada M, Minna JD, Yokota J, Kohno T. Identification of chromosome arm 9p as the most frequent target of homozygous deletions in lung cancer. Genes Chromosomes Cancer. 2005;44:405–414. doi: 10.1002/gcc.20253. [DOI] [PubMed] [Google Scholar]
- 126.Senju M, Sueoka N, Sato A, Iwanaga K, Sakao Y, Tomimitsu S, Tominaga M, Irie K, Hayashi S, Sueoka E. Hsp90 inhibitors cause G2/M arrest associated with the reduction of Cdc25C and Cdc2 in lung cancer cell lines. J Cancer Res Clin Oncol. 2006;132:150–158. doi: 10.1007/s00432-005-0047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- 128.Shervington A, Cruickshanks N, Wright H, Atkinson-Dell R, Lea R, Roberts G, Shervington L. Glioma: what is the role of c-Myc, hsp90 and telomerase? Mol Cell Biochem. 2006;283:1–9. doi: 10.1007/s11010-006-2495-z. [DOI] [PubMed] [Google Scholar]
- 129.Shintani T, Noda M. Protein tyrosine phosphatase receptor type Z dephosphorylates TrkA receptors and attenuates NGF-dependent neurite outgrowth of PC12 cells. J Biochem. 2008;144:259–266. doi: 10.1093/jb/mvn064. [DOI] [PubMed] [Google Scholar]
- 130.Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, Ressom H, Jean W, Bigner D, Yan H, Samuels Y, Waldman T. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68:10300–10306. doi: 10.1158/0008-5472.CAN-08-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Souttou B, Juhl H, Hackenbruck J, Rockseisen M, Klomp HJ, Raulais D, Vigny M, Wellstein A. Relationship between serum concentrations of the growth factor pleiotrophin and pleiotrophin-positive tumors. J Natl Cancer Inst. 1998;90:1468–1473. doi: 10.1093/jnci/90.19.1468. [DOI] [PubMed] [Google Scholar]
- 132.Spanos WC, Hoover A, Harris GF, Wu S, Strand GL, Anderson ME, Klingelhutz AJ, Hendriks W, Bossler AD, Lee JH. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J Virol. 2008;82:2493–2500. doi: 10.1128/JVI.02188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O’Meara A, Breatnach F. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006;66:3673–3680. doi: 10.1158/0008-5472.CAN-05-4154. [DOI] [PubMed] [Google Scholar]
- 134.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 135.Stegh AH, Kesari S, Mahoney JE, Jenq HT, Forloney KL, Protopopov A, Louis DN, Chin L, DePinho RA. Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma. Proc Natl Acad Sci USA. 2008;105:10703–10708. doi: 10.1073/pnas.0712034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stegh AH, Kim H, Bachoo RM, Forloney KL, Zhang J, Schulze H, Park K, Hannon GJ, Yuan J, Louis DN, DePinho RA, Chin L. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007;21:98–111. doi: 10.1101/gad.1480007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 138.Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. Protein tyrosine phosphatases: structure-function relationships. Febs J. 2008;275:867–882. doi: 10.1111/j.1742-4658.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- 139.Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, Abrahamson DR, Drake CJ, Shirasawa T, Daniel TO. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol. 2003;23:1817–1831. doi: 10.1128/MCB.23.5.1817-1831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tanuma N, Nomura M, Ikeda M, Kasugai I, Tsubaki Y, Takagaki K, Kawamura T, Yamashita Y, Sato I, Sato M, Katakura R, Kikuchi K, Shima H. Protein phosphatase Dusp26 associates with KIF3 motor and promotes N-cadherin-mediated cell–cell adhesion. Oncogene. 2009;28:752–761. doi: 10.1038/onc.2008.431. [DOI] [PubMed] [Google Scholar]
- 141.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 142.Teodorczyk M, Martin-Villalba A. Sensing invasion: Cell surface receptors driving spreading of glioblastoma. J Cell Physiol. 2010;222:1–10. doi: 10.1002/jcp.21901. [DOI] [PubMed] [Google Scholar]
- 143.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 144.Trapasso F, Drusco A, Costinean S, Alder H, Aqeilan RI, Iuliano R, Gaudio E, Raso C, Zanesi N, Croce CM, Fusco A. Genetic ablation of Ptprj, a mouse cancer susceptibility gene, results in normal growth and development and does not predispose to spontaneous tumorigenesis. DNA Cell Biol. 2006;25:376–382. doi: 10.1089/dna.2006.25.376. [DOI] [PubMed] [Google Scholar]
- 145.Uetani N, Kato K, Ogura H, Mizuno K, Kawano K, Mikoshiba K, Yakura H, Asano M, Iwakura Y. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19:2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Uhm JH, Dooley NP, Villemure JG, Yong VW. Mechanisms of glioma invasion: role of matrix-metalloproteinases. Can J Neurol Sci. 1997;24:3–15. doi: 10.1017/s0317167100021028. [DOI] [PubMed] [Google Scholar]
- 147.Ulbricht U, Brockmann MA, Aigner A, Eckerich C, Muller S, Fillbrandt R, Westphal M, Lamszus K. Expression and function of the receptor protein tyrosine phosphatase zeta and its ligand pleiotrophin in human astrocytomas. J Neuropathol Exp Neurol. 2003;62:1265–1275. doi: 10.1093/jnen/62.12.1265. [DOI] [PubMed] [Google Scholar]
- 148.Ulbricht U, Eckerich C, Fillbrandt R, Westphal M, Lamszus K. RNA interference targeting protein tyrosine phosphatase zeta/receptor-type protein tyrosine phosphatase beta suppresses glioblastoma growth in vitro and in vivo. J Neurochem. 2006;98:1497–1506. doi: 10.1111/j.1471-4159.2006.04022.x. [DOI] [PubMed] [Google Scholar]
- 149.Utsuki S, Sato Y, Oka H, Tsuchiya B, Suzuki S, Fujii K. Relationship between the expression of E-, N-cadherins and beta-catenin and tumor grade in astrocytomas. J Neurooncol. 2002;57:187–192. doi: 10.1023/a:1015720220602. [DOI] [PubMed] [Google Scholar]
- 150.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, Liau L, Cloughesy TC, Mischel PS, Sander C, Taylor B, Schultz N, Major J, Heguy A, Fang F, Mellinghoff IK, Chan TA. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci USA. 2009;106:9435–9440. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, Golub T, Mellinghoff IK, Davis RJ, Wu H, Sawyers CL. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007;11:555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 152.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 153.Wang M, Wang T, Liu S, Yoshida D, Teramoto A. The expression of matrix metalloproteinase-2 and -9 in human gliomas of different pathological grades. Brain Tumor Pathol. 2003;20:65–72. doi: 10.1007/BF02483449. [DOI] [PubMed] [Google Scholar]
- 154.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 155.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu CJ, O’Rourke DM, Feng GS, Johnson GR, Wang Q, Greene MI. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. 2001;20:6018–6025. doi: 10.1038/sj.onc.1204699. [DOI] [PubMed] [Google Scholar]
- 157.Yamamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K, Rao VH, Tomonaga M, Nicolson GL, Rao JS. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res. 1994;54:3656–3661. [PubMed] [Google Scholar]
- 158.Yu H, Jove R. The STATs of cancer: new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 159.Yu J, Becka S, Zhang P, Zhang X, Brady-Kalnay SM, Wang Z. Tumor-derived extracellular mutations of PTPRT/PTPrho are defective in cell adhesion. Mol Cancer Res. 2008;6:1106–1113. doi: 10.1158/1541-7786.MCR-07-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene. 2007;26:1178–1187. doi: 10.1038/sj.onc.1209899. [DOI] [PubMed] [Google Scholar]
- 161.Yu Y, Jiang X, Schoch BS, Carroll RS, Black PM, Johnson MD. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007;67:130–138. doi: 10.1158/0008-5472.CAN-06-2478. [DOI] [PubMed] [Google Scholar]
- 162.Zhang S, Zhang ZY. PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov Today. 2007;12:373–381. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 163.Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, Wang ZJ. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci USA. 2007;104:4060–4064. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]