

Abstract
O-GlcNAcylation augments vascular contractile responses and O-GlcNAc-proteins are increased in the vasculature of DOCA-salt rats. Since Endothelin-1 (ET-1) plays a major role in vascular dysfunction associated with salt-sensitive forms of hypertension, we hypothesized that ET-1-induced changes in vascular contractile responses are mediated by O-GlcNAc modification of proteins. Incubation of rat aortas with ET-1 (0.1 μM) produced a time-dependent increase in O-GlcNAc levels and decreased expression of O-GlcNAc transferase (OGT) and β-N-acetylglucosaminidase (OGA), key enzymes in the O-GlcNAcylation process. Overnight treatment of aortas with ET-1 increased phenylephrine (PE) vasoconstriction [Emax (mN)= 19±5 vs. 11±2 vehicle]. ET-1 effects were not observed when vessels were previously instilled with anti-OGT antibody or after incubation with an OGT inhibitor (ST045849, 100μM). Aortas from DOCA-salt rats, which exhibit increased prepro-ET-1, displayed increased contractions to PE and augmented levels of O-GlcNAc proteins. Treatment of DOCA-salt rats with an ETA antagonist abrogated augmented vascular levels of O-GlcNAc and prevented increased PE vasoconstriction. Aortas from rats chronically infused with low doses of ET-1 (2pmol/Kg/min) exhibited increased O-GlcNAc-proteins and enhanced PE responses [Emax (mN) = 18±2 vs. 10±3 control]. These changes are similar to those induced by PugNAc, an inhibitor of OGA. Systolic blood pressure (mmHg) was similar between control and ET-1-infused rats (117±3 vs. 123±4; respectively). We conclude that ET-1 indeed augments O-GlcNAc levels and that this modification contributes to the vascular changes induced by this peptide. Increased vascular O-GlcNAcylation by ET-1 may represent a mechanism for hypertension-associated vascular dysfunction or other pathological conditions associated with increased levels of ET-1.
Keywords: β-N-acetylglucosamine (O-GlcNAc), endothelin-1, vascular reactivity
INTRODUCTION
O-Linked attachment of β-N-acetyl-glucosamine (O-GlcNAc) on serine and threonine residues of nuclear and cytoplasmic proteins is a highly dynamic and ubiquitous post-translational modification that plays a key role in altering the function, activity, subcellular localization and stability of target proteins [1–3]. The attachment of a single β-N-acetyl-glucosamine is catalyzed by the enzyme O-GlcNAc transferase (OGT or uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase; UDP-NAc transferase) and the hydrolytic cleavage of O-GlcNAc is catalysed by β-N-acetylglucosaminidase (OGA) [3].
Although it is clear that OGlcNAcylation plays a critical role in the regulation of cell function, there is a paucity of information on the vascular effects of O-GlcNAcylation. Preliminary evidence from our laboratory suggests that augmented vascular O-GlcNAc proteins increases reactivity to constrictor stimuli. Accordingly, incubation of arterial segments with PugNAc [O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino- N-phenylcarbamate], which blocks OGA activity by mimicking the enzyme-stabilized transition state [4, 5], leads to increased reactivity to phenylephrine and serotonin [6].
In addition, the vascular content of O-GlcNAc-proteins is augmented in arteries from DOCA-salt (deoxycorticosterone acetate and salt) rats, a mineralocorticoid model of hypertension. Interestingly, increased levels of O-GlcNAc (induced by PugNAc treatment) leads to changes in mechanisms that are commonly observed in hypertensive animals, such as decreased production of nitric oxide as a consequence of decreased activity of endothelial nitric oxide synthase [7], suggesting that augmented O-GlcNAc may contribute to abnormal vascular reactivity in mineralocorticoid hypertension [8].
Endothelin-1 (ET-1) is the most abundant and important peptide from the endothelin family. Its production is increased in the vasculature of salt-sensitive forms of hypertension, including DOCA-salt hypertensive rats [9–11]. ET-1 not only induces vasoconstriction, but it also activates transcriptional factors responsible for the coordinated increase in many cytokines and enzymes, thus enhancing inflammation, oxidative stress and tissue damage [9, 10], which are all important in hypertension-associated vascular dysfunction.
In the present study, we hypothesized that ET-1 augments vascular contractile responses via increased O-GlcNAc modification of proteins. To address our hypothesis, we determined whether ET-1-induced augmented vascular reactivity can be modified by inhibition of the O-GlcNAc pathway. The effects of ET-1 on vascular function and O-GlcNAc levels were determined in vitro and in vivo, and the contribution of O-GlcNAc to ET-1 effects was assessed by pharmacologic and molecular inhibition of the O-GlcNAc pathway.
METHODS
Animals
Male Wistar rats (8–10 weeks-old, 230–250g; Harlan Laboratories, Indianapolis, IN) were used in this study. All procedures were performed in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Medical College of Georgia Committee on the Use of Animals in Research and Education. The animals were housed on a 12-h light/dark cycle and fed a standard chow diet with water ad libitum.
ET-1 incubation procedures
After euthanasia, thoracic aortas were removed and cleaned from fat tissue in an ice-cold physiological salt solution. Arterial segments were incubated in Eagle’s Minimum Essential Medium (EMEM) containing L-glutamine (1%), fetal bovine serum (FBS, 10%), penicillin (0.5%) and streptomycin (0.5%). Rings were incubated with either vehicle (H2O), or ET-1 (0.1 μM) for 1, 3, 6, 12 or 24h, to verify the optimal time-point of induction of O-GlcNAc proteins, as well as changes in the expression of OGT and OGA. In other experiments, incubations were performed for 24 hours with: vehicle (H2O), PugNAc [O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino- N-phenylcarbamate; 100μM] or ET-1 (0.1 μM). In some experiments, arteries were pre-treated (3 hours) with an OGT inhibitor [ST045849; 3-(2-adamantanylethyl)-2-[(4-chlorophenyl)azamethylene]-4-oxo-1,3-thiazaperhyd roine-6-carboxylic acid; 100μM], an ETA antagonist (atrasentan, 1 μM) or neutralizing antibody against OGT (as described below). After the 3 hours, aortas were simultaneously incubated with ET-1 plus the OGT inhibitor, the ETA antagonist, or the neutralizing antibody against OGT, for 24 hours. After incubation, aortas were used to evaluate vascular function or protein expression.
Antibody delivery by the Chariot technique
Antibodies against OGT (Santa Cruz Biotechnology, CA) were intracellularly delivered by the Chariot technique (Chariot Protein Delivery Reagent, Active Motif). This transfection reagent is able to deliver antibodies into cells while preserving their ability to localize to the proper cellular compartment and to recognize antigens within the cell [12–14] and in our experiments was used to directly inhibit OGT protein. Chariot/antibody complexes were prepared and used according to the manufacturer’s instructions. Briefly, aortic rings were incubated in Eagle’s Minimum Essential Medium (EMEM) containing L-glutamine (1.0%), fetal bovine serum (FBS, 10%), and penicillin and streptomycin (0.5%) for 30 minutes at 37°C. For each transfection, 12 μL of Chariot in 100 μL of 40% dimethyl sulfoxide were mixed with 6 μg of antibody in 100 μL of phosphate buffered saline (PBS) and incubated at room temperature for 30 minutes to allow the complex to form. The aortas were transferred to a sterile 24-well cell culture plate, overlaid with 200 μL of Chariot/antibody complex, and mixed gently. EMEM (400μL) was added, and the tissues were incubated for 1 hour at 37°C. Then, additional EMEM (750μL) was added, and tissues were further incubated for 2 hours at 37°C. After this period, rings were mounted in the myograph, and functional studies were performed.
Infusion of low doses of ET-1
Rats were anesthetized with 5% isoflurane in 100% O2 and maintained during surgery with 1.9–2.1% isoflurane in 100% O2 through a nose cone. The depth of anesthesia was verified by noting the absence of physical responses to firm paw pinch and corneal probing. Osmotic minipumps (ALZET Osmotic Pumps, CA) were implanted subcutaneously to deliver ET-1 (2 pmol/kg per minute; Phoenix Pharmaceuticals, CA) or sodium chloride (0.9%) through a catheter in the jugular vein for 14 days. Aortas from ET-1 infused or control rats were freshly removed to evaluate vascular function or protein expression.
DOCA-salt hypertension
DOCA-salt hypertension was induced as previously described [15]. Briefly, rats were unilaterally nephrectomyzed (Uni) and deoxycorticosterone-acetate (DOCA; 200 mg/Kg) silastic pellets were implanted subcutaneously in the scapular region. DOCA rats received water containing 1% NaCl and 0.2% KCl for 5 weeks. Control rats were also uninephrectomized, received silastic pellets without DOCA and tap water. Animals simultaneously received either the ETA antagonist atrasentan (5 mg/day/kg of body weight, p.o. per gavage) or vehicle for 5 weeks. Aortas from DOCA-salt, treated or not with ETA antagonist, and UNI rats were freshly removed to evaluate vascular function or protein expression.
Systolic blood pressure measurements
Systolic blood pressure (SBP) was measured in non anaesthetized animals by tail cuff using a RTBP1001 rat-tail blood pressure system (Kent Scientific Corporation, CT).
Euglycemic-hyperinsulinemic clamp
Rats were fasted overnight before being anesthetized and evaluated for insulin sensitivity using the euglycemic-hyperinsulinemic clamp method [16]. Catheters were placed in the femoral artery for blood sampling and femoral and jugular veins for infusion of insulin and glucose. Blood samples were obtained at 5-minute intervals (Novolin; 30 mU/kg/min; Novo Nordisk Pharmaceuticals, Princeton, NJ). Glucose infusion (100 mg/ml glucose in saline) was adjusted to maintain a plasma glucose at 125 mg/dl within 60 minutes, and maintained for an additional 30 minutes. The final 7 readings were averaged and reported as the glucose infusion rate (mg/kg/min).
Vascular functional studies
In one set of experiments, aortas were functionally evaluated after incubation with ET-1 for 24, with or without treatments, as previously described. In other set of experiments, vascular function was accessed in aortas freshly harvested from ET-1 infused rats (14 days) or DOCA-salt rats. In this case, after euthanasia, thoracic aortas were removed and cleaned from fat tissue in an ice-cold physiological salt solution. Aortic rings (4 mm in length) were mounted between 2 stainless-steel wires in standard organ chambers (model 610M, Danish MyoTech) for isometric tension recording, as described previously [17]. After stabilization, arterial integrity was assessed first by stimulation of vessels with 120 mM of potassium chloride (KCl) and, after washing and a new stabilization period, the presence of the endothelium was verified by contracting the segments with phenylephrine (PE; 1μM) followed by stimulation with acetylcholine (ACh; 10μM). Concentration-response curves to PE (1nM to 100μM) and serotonin [5-hydroxytriptamine (5-HT), 1nM to 100μM] were performed and responses are represented as percentage of KCl-induced contraction.
Western blot analysis
Proteins (60 μg) extracted from endothelium-intact aortas were separated by electrophoresis, and Western blots performed as previously described [10]. Antibodies used were: anti-O-GlcNAc antibody, CTD 110.6 (1:2000; Pierce Biotechnology, USA), O-GlcNAc transferase (OGT) [1:400, Santa Cruz antibodies] and OGA antibody which was kindly provided by Dr. Sidney Whiteheart (1:1000, University of Kentucky). OGA is a 106kDa heterodimer complex containing a 54-kDa α-subunit and a 51-kDa β-subunit [18].
Aortas from three different experimental conditions (incubated with ET-1, from ET-infused rats and from DOCA-salt rats) were used to perform western blots experiments. Analyses of the bands were performed with software (Un-ScanIT Gel 6.1) that evaluates the density profile extraction and band analysis for entire lanes. The result of the sum of the bands was normalized by beta-actin and expressed as arbitrary units.
Real time RT-PCR for prepro-ET-1
Gene expression of prepro-ET-1 was determined as previously described [11]. Briefly, total RNA was extracted using the RNeasy kit (Qiagen Sciences, MD, USA) and one microgram of total RNA was reverse transcribed in a final volume of 50μL using the high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA, USA). Primers for preproendothelin-1 (preproET-1) (N° Rn00561129_m1) mRNA were obtained from Applied Biosystems. Real-time reverse transcriptase polymerase chain reaction (qPCR) reactions were performed using the 7,500 fast Real-Time PCR system (Applied Biosystems) in a total volume of 20 mL reaction mixture following the manufacturer’s protocol, using the TaqMan® fast universal PCR master mix (Applied Biosystems), and 0.1 mM of each primer. Relative gene expression for preproET-1 mRNA was normalized to samples from Uni rats (calibrator) and results were calculated with the ΔΔCt method and expressed as n-fold differences in preproET-1 gene expression relative to 18S rRNA and to the calibrator.
Data analysis
The results are shown as mean ± SEM and n represents the number of animals used in the experiments. Contractions were recorded as changes in the displacement (mN) from baseline. Relaxation is expressed as percent change from the PE-contracted levels. Concentration response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 4.0; GraphPad Software Inc., San Diego, CA, U.S.A.) and two pharmacological parameters were obtained: the maximal effect generated by the agonist (or Emax) and the negative logarithm of the concentration of agonist that produces 50% of the maximum response [−log EC50 (or pD2)]. Statistical analyses of Emax and pD2 values were performed using one-way ANOVA or Student’s t-test. Post hoc comparisons were performed using Newman-Keuls’s test. Western blot data were analyzed by one-sample t test and the P value was computed from the t ratio and the numbers of degrees of freedom. Values of P<0.05 were considered statistically significant.
RESULTS
The incubation of aortas with ET-1 (0.1 μM) produced a time-dependent increase in vascular O-GlcNAc levels (Fig. 1A). No differences on the O-GlcNAc levels were observed between aortas freshly harvested or after 24 hours incubation with vehicle. An inverse temporal correlation was observed for OGT (Fig. 1B) and OGA (Fig. 1C) expression.
Figure 1. ET-1-incubation at different time points produces a time-dependent increase in vascular O-GlcNAc levels, and decreases OGT and OGA expression in rat aorta.

Aortas were incubated with ET-1 (0.1 μM), for 1, 3, 6, 12 or 24 hours and western blots were performed to evaluate O-GlcNAC-proteins, OGT and OGA expression. On the top, representative Western blot images of (A) O-GlcNAc-proteins, (B) OGT and (C) OGA; on the bottom, corresponding bar graphs showing the relative expression of O-GlcNAc, OGT and OGA after normalization to β-actin expression. Results are presented as mean ± SEM for n=4 in each experimental group. *, p<0.05 vs. vehicle (H2O).
After 24 hours of treatment with ET-1, arteries from Wistar rats displayed increased vascular reactivity to the alpha1-adrenergic agonist PE and developed force levels were similar to those exhibited by aortas incubated with PugNAc, an inhibitor of OGA (Fig. 2A and Table 1). No differences in KCl-induced contraction were observed among the groups [Emax (mN) 22.6±1.9 vehicle, 22.8±1.8 ET-1 and 21.67±1.6 PugNAc]. Additionally, differences in PE-induced contraction, after ET-1 incubation for 24 hours, persisted in arteries without endothelium [Emax (% KCl) 117.1±4.8 vehicle vs. 174.6±11 ET-1].
Figure 2. ET-1 augments PE-induced vasoconstriction.

ET-1 or PugNAc incubation for 24h increases vascular contraction to (A) PE and (B) 5-HT in rat thoracic rats aorta vs. vehicle (methanol; n=5). ET-1-infusion for 14 days increases (C) PE-induced in arteries from Wistar rats (n=5). Experimental values of contraction were calculated relative to the contractile response produced by KCl 120mM, which was taken as 100%. Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (H2O) or control.
Table 1.
Emax and pD2 values for phenylephrine in arteries from rats incubated with ET-1 or from rats infused with ET-1.
| Phenylephrine | ||
|---|---|---|
| Incubation (24h) | Emax | pD2 |
| Vehicle | 69±6 | 6.5±0.1 |
| PugNAc | 115±5* | 6.7±0.06 |
| ET-1 | 107±5* | 7.1±0.1* |
| Infusion (14 days) | ||
| Control | 77±5 | 7.4±0.06 |
| ET-1-infusion | 103±3* | 6.9±0.06* |
Values are means ± SEM for N=6 experiments in each group. Contractile response to PE are represented as percentage of KCl-induced contraction.
p<0.05 vs. vehicle or control.
Serotonin-induced contraction was increased in aortas after ET-1 incubation (Fig. 2B), when compared with control arteries [pD2 = 6.3±0.1 vehicle vs. 6.9±0.1 ET-1; Emax (% KCl) = 79.0±7 vehicle vs. 101±1 ET-1; n=5], showing that O-GlcNAcylation does not interfere specifically with alpha1-adrenergic-mediated responses.
In vivo ET-1 infusion in Wistar rats for 14 days enhanced aortic contractile responses to PE in comparison to their respective controls (Fig. 2C and Table 1). No differences in KCl-induced contraction were observed among the groups [Emax (mN) 22.7±2 control vs. 25±1 ET-1]. In addition, ET-1 infusion in Wistar rats augmented O-GlcNAc levels of vascular proteins (Fig. 3A). ET-1 infusion for 2 weeks did not change vascular OGT expression (Fig. 3B), but decreased OGA expression (Fig. 3C). SBP (mmHg) was similar between control and ET-1-infused rats (117±3 vs. 123±4; n=5, respectively; Table 2). ET-1 had no effect on fasting glucose levels (99.7±2 vs. 102±7.4, mg/dL in control and ET-1 infusion, respectively) or body weight (Table 2). In addition, the euglycemic-hyperinsulinemic clamp test demonstrated that ET-1 had no effect on insulin sensitivity; glucose infusion rate was 6.0 ± 0.6 and 6.8 ± 0.6 mg/kg/min in control (n=4) and ET-1-infused (n=3) rats, respectively.
Figure 3. ET-1 infusion in vivo for 14 days augments O-GlcNAc levels in aortas, and decreases vascular expression of OGA.
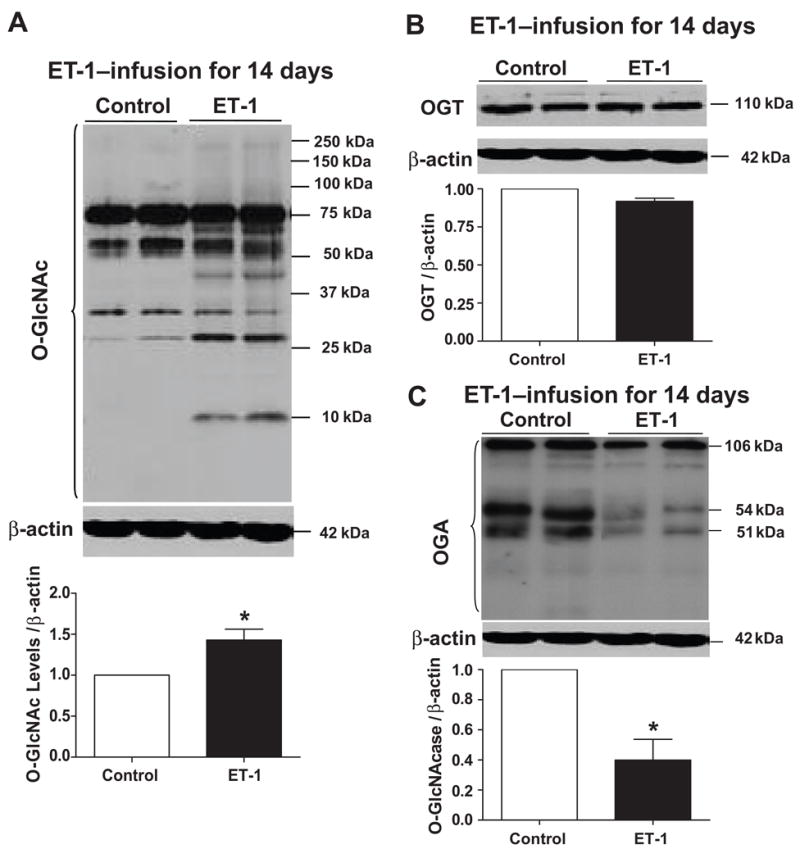
On the top, representative Western blot images of (A) O-GlcNAc-proteins, (B) OGT and (C) OGA; on the bottom, corresponding bar graphs showing the relative expression of O-GlcNAc, OGT and OGA after normalization to β-actin expression. Results are presented as mean ± SEM for n=4 in each experimental group. *, p<0.05 vs. control [(rats infused with sodium chloride (0.9%)].
Table 2.
Systolic blood pressure and body weight in rats infused with ET-1 or submitted to DOCA-salt treatment of or DOCA-salt hypertension, treated or not with atrasentan.
| Parameter | Control | ET-1-infused | Uni | DOCA | DOCA+atrasentan |
|---|---|---|---|---|---|
| SBP (mm Hg) | 117.5±3 | 123.4±4 | 124.9± 4 | 163.6 ± 6* | 137.5±6* |
| Body weight (g) | 385±10 | 380±9 | 391±13* | 320±7 | 309±12* |
SBP - Systolic blood pressure.
P < 0.05 vs. respective control, Values are means ± SEM for N = 6 in each group.
The selective inhibition of OGT, with ST045849 [3-(2-adamantanylethyl)-2-[(4-chlorophenyl)azamethylene]-4-oxo-1,3-thiazaperhydroine-6-carboxylic acid] (TimTecLLC) [19] resulted in decreased vascular O-GlcNAc levels (Fig. 4A) and also attenuated the effects of ET-1 on vascular reactivity (Fig. 4B).
Figure 4. ET-1 effects on O-GlcNAc-protein levels and vascular reactivity are not observed when vessels are previously transfected with antibodies against OGT or incubated with OGT inhibitor.

Treatment with (A,B) the OGT inhibitor as well as (C,D) neutralizing antibodies anti-OGT [Chariot (OGT)] decrease vascular O-GlcNAc levels. OGT inhibition (A,C) reduced vascular contraction and (B,D) decreased O-GlcNAc-proteins levels, upon ET-1 incubation for 24 hours. (B,D) On the top, Western blot image of O-GlcNAc-proteins; on the bottom, corresponding bar graphs showing the relative O-GlcNAc-proteins after normalization to β-actin expression. Experimental values of contraction were calculated relative to the contractile response produced by KCl 120mM, which was taken as 100%. Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (DMSO).
As shown in figure 4, the effects of ET-1 on O-GlcNAc-protein levels and vascular reactivity were not observed when vessels were previously instilled with antibodies against OGT (Fig. 4C and 4D, respectively), intracellularly delivered by a transfection system (ActiveMotif USA). Incubation with an IgG anti-rabbit antibody was used as an additional control and did not modify ET-1-induced effects (data not shown).
We sought to determine whether ET-1 activation is a key element for increased vascular O-GlcNAc-protein levels and, consequently, increased vascular reactivity in mineralocorticoid hypertension. To address this question, we used a pharmacological approach: treatment of DOCA-salt rats with an ETA receptor antagonist (atrasentan; 5mg.Kg−1.day−1). At 5 weeks of treatment, SBP (mmHg) was higher in DOCA-salt in comparison to Uni rats (Uni: 124.9 ± 3.6 mmHg vs. DOCA: 163.6 ± 6.4 mmHg, n=6; Table 2). DOCA-salt rats exhibited decreased body weight in comparison to Uni (Table 2). Prepro-ET-1 gene expression was augmented in aortas from DOCA-salt rats (fold of change: 2.1±0.4 vs. 1 control) and ETA blockade with atrasentan did not prevent increased preproET-1 mRNA expression (fold of change: 1.8±0.1), as determined by qPCR. Treatment with atrasentan attenuated, but did not normalize, blood pressure in DOCA-salt rats (137.5 ± 5.74 mmHg, n=6; Table 2) and did not change body weight in DOCA-salt animals (Table 2). On the other hand, the ETA antagonist abrogated augmented vascular levels of O-GlcNAc in DOCA-salt rats (Fig. 5A) and also prevented increased contractile responses to PE in aorta from these animals (Fig. 5B). These results suggest that ETA receptor activation plays a role on ET-1-induced vascular effects. They are further reinforced by in vitro experiments, where atrasentan (1μM) attenuated the effects of ET-1-incubation on O-GlcNAc-protein levels and vascular reactivity (Fig. 5C and 5D, respectively).
Figure 5. ETA antagonist prevents augmented vascular levels of O-GlcNAc in vivo and in vitro and also abrogates increased contractile responses to PE.

(A) Treatment of DOCA-salt rats with ETA antagonist prevents augmented vascular O-GlcNAc levels and (B) increased contractile responses to PE. ETA antagonist attenuated the effects of ET-1 incubation for 24 hours on vascular (C) O-GlcNAc levels and (D) increased contractile responses to PE. (A,C), on the top, Western blot image of O-GlcNAc-proteins; on the bottom, corresponding bar graphs showing the relative O-GlcNAc-proteins after normalization to β-actin expression. (B,D), experimental values of contraction were calculated relative to the contractile response produced by KCl 120mM, which was taken as 100%. Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (H2O); †, p<0.05 vs. Uni.
DISCUSSION
O-GlcNAc has important implications for the regulation of protein structure and function and the interplay with other post-translational modifications, such as phosphorylation [20–22]. However, with respect to vascular function, O-GlcNAc is a relatively unexplored area. Our preliminary studies showed that O-GlcNAc is increased in the vasculature from DOCA-salt hypertensive rats [8]. Considering that ET-1, which is increased in this model of salt-dependent hypertension [9, 10], contributes to vascular dysfunction in arteries from DOCA-salt rats, we sought to investigate whether O-GlcNAc underlies effects of ET-1 on vascular function.
Our data show for the first time that ET-1 augments vascular O-GlcNAcylation and that this pos-translational modification contributes to the vascular changes produced by this peptide. O-GlcNAcylation of nuclear and cytoplasmic proteins is regulated by OGT and OGA. We demonstrated that ET-1 produces a time-dependent and transient decrease in OGT vascular expression. Interestingly, PugNAc incubation for 24h, which increases vascular content of O-GlcNAc-proteins in arteries from control animals [6, 23], also decreases OGT expression [6]. Decreased OGT may represent a compensatory mechanism for the augmented vascular levels of O-GlcNAc proteins. On the other hand, aortas from rats chronically infused with low doses of ET-1, which exhibit increased O-GlcNAc-proteins, did not exhibit changes in vascular OGT expression at 14 days. This may be related to the transient changes in OGT expression produced by ET-1. A possible explanation is that OGT expression was measured at a time point (after 14 days) where expression has already returned to basal levels (as is the case for OGT expression after 24h incubation with ET-1 in vitro).
OGA expression was also decreased after treatment with ET-1, similarly to what has been demonstrated in aortas incubated with PugNAc [6]. In addition, ET-1 infusion for 14 days decreased OGA expression in thoracic aortas. Increased vascular O-GlcNAcylation upon in vivo and in vitro treatment with ET-1 may be mediated by decreased OGA expression/activity.
In order to clarify whether ET-1-induced augmented vascular O-GlcNAc protein content plays a role in the vascular functional changes induced by this peptide, we determined the vascular contraction to the alpha1-adrenergic agonist PE in various conditions. Treatment with ET-1 for 24h enhanced contractile responses of rat aortas to PE. Developed force levels were similar to those exhibited by aortas incubated with PugNAc. Furthermore, ET-1 infusion in Wistar rats enhanced aortic contractile responses to PE in comparison to their respective controls as has been shown in other studies [9, 24]. Of importance, chronic ET-1 infusion did not produce consistent increases in blood pressure, in agreement with a recent report by Wang and Wang [25], glucose levels or insulin sensitivity.
Recent work by Clarke and coworkers [14] has demonstrated that antibodies can be targeted to selective signaling cascades in smooth muscle cells of isolated ring segments of arteries. We have performed experiments using arteries incubated for 24 hours using this innovative approach to target signaling cascades, as an additional method to complement our pharmacological studies. In our experiments, Chariot protein transfection reagent was used to deliver anti-OGT antibodies to intact aortic rings. The effects of ET-1 are not observed when vessels are previously transfected with antibodies against OGT. Empty Chariot did not significantly change actions of ET-1. This result is further supported by experiments where arteries were incubated with a selective OGT inhibitor: ST045849. This pharmacological inhibitor attenuated both the effects of ET-1 in O-GlcNAc-proteins levels and vascular reactivity. Based on these observations, we conclude that elevated levels of O-GlcNAc by ET-1 represent a common mechanism underlying the adverse effects of the peptide on vascular function.
We have previously demonstrated that increased levels of O-GlcNAc augments responses to contractile stimuli [6] and that O-GlcNAcylation is associated with the development of increased vascular contractility in DOCA-salt hypertension [8]. Interestingly, ET-1, which expression is increased in DOCA-salt hypertension [9, 10] is associated with vascular dysfunction. Here, we showed that ET-1 exposition in vitro and in vivo not only augments vasoconstriction to PE, but also increases O-GlcNAc-protein levels and modulates OGT and OGA expression. Accordingly, we demonstrated that treatment with an ETA receptor antagonist prevents changes in O-GlcNAc levels acutely and chronically induced by ET-1. In addition, treatment with atrasentan was able to normalized O-GlcNAc levels as well as prevented increased contractile response to PE-stimulation in arteries from DOCA-salt rats.
One may suggest that atrasentan reduced protein modification via a decrease in blood pressure. However, we found that atrasentan attenuated, but did not normalize, blood pressure in DOCA-salt rats. Additionally, ET-1-infusion for 14 days did not result in changes in blood pressure. It seems that in this case, O-GlcNAc protein modification does not correlate directly with blood pressure but does correlate with PE reactivity. The use of other anti-hypertensive agent that does not affect the endothelin-1 system may help to further elucidate this suggestion.
In this sense is well established that increased contractile responsiveness of the vasculature, as a result of increase contraction or decreased relaxation, is a hallmark of hypertension [26]. We speculate that ET-1 modulates signaling proteins, via O-GlcNAc modification, that are important for vascular tone control, such as NOS, protein kinase C (PKC), members of the MAPKs (mitogen-activated protein kinases) family and small G proteins are target for O-GlcNAcylation. Therefore, O-GlcNAC modification of proteins induced by ET-1 ET-1 may contribute to augmented vascular response to constrictor stimuli.
Additionally, increasing evidence suggests that O-GlcNAcylation can modulate protein function by interfering with protein phosphorylation. This suggestion takes into consideration that both modifications occur on serine and threonine residues, are dynamically added and removed from proteins in response to cellular signals, and alters the function and association of the modified protein [20–22]. The relationship between O-GlcNAc and phosphorylation is obviously complex, but may be a key to understanding the function of O-GlcNAc addition. The reciprocal interplay between O-GlcNAc and phosphorylation may represent an additional link between ET-1-activated signal transduction mechanisms and the effects of O-GlcNAc on the vasculature. Conversely, phosphorylation has been shown to directly affect OGT expression and activity [27].
In conclusion, our data provide evidence that ET-1 augments O-GlcNAc levels and this modification contributes to the vascular effects of ET-1. We propose that modulation of increased vascular O-GlcNAcylation by ET-1 may represent a novel mechanism contributing to the vasoactive properties of this potent peptide. Definition of O-GlcNAc-modified vascular proteins will contribute to a better understanding of how this post-translational modification affects vascular reactivity in physiological and pathophysiological conditions.
Acknowledgments
SOURCE OF FUNDINGS
This study was supported by grants from the National Institutes of Health (HL-74167), CAPES - Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior and Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP), Brazil.
Footnotes
DISCLOSURES
The authors declared no disclosures.
PERSPECTIVES
Arterial hypertension often co-exists with diabetes, and more than 80% of patients with type 2 diabetes mellitus develop hypertension. Most of the deleterious effects associated with abnormal O-GlcNAcylation have been described in diabetic or hyperglycemic conditions, but we have recently shown that O-GlcNAcylation, which augments vascular contractile responses, is increased in the vasculature of hypertensive animals. The present study showing that O-GlcNAcylation plays a role on ET-1-induced vascular responses further suggest that this post translational modification is a key regulator of vascular function. Important insight into the pathological processes leading to vascular dysfunction can be gained from elucidating the mechanisms by which O-GlcNAcylation disrupts vascular function.
References
- 1.Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 2.Fulop N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res. 2007;73:288–297. doi: 10.1016/j.cardiores.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, Chen YF, Darley-Usmar V, Oparil S, Chatham JC. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. 2009;296:H13–28. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musicki B, Kramer MF, Becker RE, Burnett AL. nactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. PNAS. 2005;102:11870–11875. doi: 10.1073/pnas.0502488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitworth CE, Macauley MS, Stubbs KA, Dennis RJ, Taylor EJ, Vocadlo DJ, et al. Analysis of PugNag and NAG-thiazoline as transition state analogues for human O-GlcNAcase: Mechanistic and structural insights into inhibitor selectivity and transition state poise. J Am Chem Soc. 2007;129:635–644. doi: 10.1021/ja065697o. [DOI] [PubMed] [Google Scholar]
- 6.Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Fortes ZB, Carvalho MHC, Webb RC, Tostes RC. Increased vascular O-GlcNAcylation augments reactivity to constrictor stimuli. JASH. 2008;2:410–417. doi: 10.1016/j.jash.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106:466–472. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 8.Lima VV, Giachini FR, Choi H, Carneiro FS, Carneiro ZN, Fortes ZB, Carvalho MH, Webb RC, Tostes RC. Impaired vasodilator activity in deoxycorticosterone acetate-salt hypertension is associated with increased protein O-GlcNAcylation. Hypertension. 2009;53:166–174. doi: 10.1161/HYPERTENSIONAHA.108.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol. 2005;43:19–29. doi: 10.1016/j.vph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Tostes RC, Muscara MN. Endothelin receptor antagonists: another potential alternative for cardiovascular diseases. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:287–301. doi: 10.2174/1568006054553390. [DOI] [PubMed] [Google Scholar]
- 11.Carneiro FS, Nunes KP, Giachini FR, Lima VV, Carneiro ZN, Nogueira EF, Leite R, Ergul A, Rainey WE, Clinton Webb R, Tostes RC. Activation of the ET-1/ETA pathway contributes to erectile dysfunction associated with mineralocorticoid hypertension. J Sex Med. 2008;5:2793–2807. doi: 10.1111/j.1743-6109.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- 12.Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 13.Keller M, Lidington D, Vogel L, Peter BF, Sohn HY, Pagano PJ, Pitson S, Spiegel S, Pohl U, Bolz SS. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J. 2006;20:702–704. doi: 10.1096/fj.05-4075fje. [DOI] [PubMed] [Google Scholar]
- 14.Clarke CJ, Forman S, Pritchett J, Ohanian V, Ohanian J. Phospholipase C-delta1 modulates sustained contraction of rat mesenteric small arteries in response to noradrenaline, but not endothelin-1. Am J Physiol Heart Circ Physiol. 2008;295:H826–834. doi: 10.1152/ajpheart.01396.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chitaley K, Webb RC, Dorrance AM, Mills TM. Decreased penile erection in DOCA-salt and stroke prone-spontaneously hypertensive rats. Int J Impot Res. 2001;13 (Suppl 5):S16–20. doi: 10.1038/sj.ijir.3900773. [DOI] [PubMed] [Google Scholar]
- 16.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 17.Giachini FRC, Carneiro FS, Lima VV, Carneiro ZN, Carvalho MHC, Fortes ZB, Webb RC, Tostes RC. Pyk2 mediates increased adrenergic contractile responses in arteries from DOCA-salt mice. JASH. 2008;2:431–438. doi: 10.1016/j.jash.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- 19.Dehennaut V, Lefebvre T, Sellier C, Leroy Y, Gross B, Walker S, Cacan R, Michalski JC, Vilain JP, Bodart JF. O-linked N-acetylglucosaminyltransferase inhibition prevents G2/M transition in Xenopus laevis oocytes. J Biol Chem. 2007;282:12527–12536. doi: 10.1074/jbc.M700444200. [DOI] [PubMed] [Google Scholar]
- 20.Musicki B, Kramer MF, Becker RE, Burnett AL. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc Natl Acad Sci U S A. 2005;102:11870–11875. doi: 10.1073/pnas.0502488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta. 2006;1761:599–617. doi: 10.1016/j.bbalip.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. 2001;40:7845–7852. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 23.Xing D, Feng W, Not LG, Miller AP, Zhang Y, Chen YF, Majid-Hassan E, Chatham JC, Oparil S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. Am J Physiol Heart Circ Physiol. 2008;295:H335–342. doi: 10.1152/ajpheart.01259.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giachini FR, Callera GE, Carneiro FS, Tostes RC, Webb RC. Therapeutic targets in hypertension: is there a place for antagonists of the most potent vasoconstrictors? Expert Opin Ther Targets. 2008;12:327–339. doi: 10.1517/14728222.12.3.327. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wang DH. Prevention of endothelin-1-induced increases in blood pressure: role of endogenous CGRP. Am J Physiol Heart Circ Physiol. 2004;287:H1868–1874. doi: 10.1152/ajpheart.00241.2004. [DOI] [PubMed] [Google Scholar]
- 26.Bohr DF, Dominiczak AF, Webb RC. Pathophysiology of the vasculature in hypertension. Hypertension. 1991;18:III69–75. doi: 10.1161/01.hyp.18.5_suppl.iii69. [DOI] [PubMed] [Google Scholar]
- 27.Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem. 2008;283:33935–33941. doi: 10.1074/jbc.M806199200. [DOI] [PMC free article] [PubMed] [Google Scholar]