


Abstract
The prediction of protein function as well as the reconstruction of evolutionary genesis employing sequence comparison at large is still the most powerful tool in sequence analysis. Due to the exponential growth of the number of known protein sequences and the subsequent quadratic growth of the similarity matrix, the computation of the Similarity Matrix of Proteins (SIMAP) becomes a computational intensive task. The SIMAP database provides a comprehensive and up-to-date pre-calculation of the protein sequence similarity matrix, sequence-based features and sequence clusters. As of September 2009, SIMAP covers 48 million proteins and more than 23 million non-redundant sequences. Novel features of SIMAP include the expansion of the sequence space by including databases such as ENSEMBL as well as the integration of metagenomes based on their consistent processing and annotation. Furthermore, protein function predictions by Blast2GO are pre-calculated for all sequences in SIMAP and the data access and query functions have been improved. SIMAP assists biologists to query the up-to-date sequence space systematically and facilitates large-scale downstream projects in computational biology. Access to SIMAP is freely provided through the web portal for individuals (http://mips.gsf.de/simap/) and for programmatic access through DAS (http://webclu.bio.wzw.tum.de/das/) and Web-Service (http://mips.gsf.de/webservices/services/SimapService2.0?wsdl).
INTRODUCTION
Protein sequences are of utmost importance for studying the function and evolution of genes and genomes. Evolutionary processes of mutation and selection have shaped the protein sequence space and became manifest in the protein sequences as well as their pair-wise and group-wise similarities. Therefore, a rich collection of methods in computational biology relies on the analysis and comparison of protein sequences. Many of these intensively used methods perform sequence similarity searches [e.g. BLAST (1)] or compare protein sequences against secondary databases of protein families [e.g. InterPro (2)].
The fast increasing volume of publicly available protein sequences forges a computational dilemma for bioinformatics tasks that require repeated all-against-all calculations of sequence similarities or sequence features. Such rather straightforward but technically challenging tasks among others are the annotation of genomes or the clustering of the protein sequence space into protein families. Due to the exponential growth of the number of sequences and the quadratic complexity of the sequence similarity matrix, the computational demand of calculating an all-versus-all sequence matrix of all known proteins easily outgrows available computational resources. Due to the subsequent growth of the secondary databases, a similar problem exists for the prediction of protein domains. As a consequence, any repeated ab initio recalculation of the similarity matrix is highly ineffective due to the recalculation of the vast majority of already known sequence similarity relations. However, as the number of recently added sequences is always small compared to the bulk of know sequences, repeated recalculations—frequently performed in many sequence-based projects—waste a remarkable amount of compute sources worldwide.
The Similarity Matrix of Proteins (SIMAP) solves the computational dilemma described above by incrementally pre-calculating the sequence similarities forming the known protein sequence space (3). The comparison of new sequences versus known ones returns symmetric scores that can be updated accordingly in the existing records. Compared to other resources that pre-calculate sequence similarities [e.g. NCBI Blink (4)], the FASTA (5) and Smith–Waterman (6) based similarity calculation in SIMAP is only restricted by a static and sensitive raw score threshold without limiting the maximal number of hits per sequence. Hence the structure of the sequence similarity matrix is not influenced by the taxonomy and study biases that exist in the major protein sequence databases. The SIMAP database stores raw scores from the calculated alignments. When querying SIMAP, e-values are calculated on-the-fly according to the selected databases and taxa. To complement the pair-wise sequence similarity matrix by position specific searches against known protein families, SIMAP in addition pre-calculates sequence based features as e.g. InterPro matches (2). To maximize its coverage to provide an efficient alternative to BLAST or Interproscan calculations, the comprehensive representation of the protein sequence space is crucial for SIMAP. Recent improvements in SIMAP have addressed this requirement by further expanding the sequence space and including metagenomic sequences. Further improvements have extended the functional annotation of the protein sequence space in SIMAP by pre-calculated GO annotations and improved the data access and query tools of SIMAP.
NEW FEATURES AND IMPROVEMENTS IN SIMAP
Comprehensive coverage of the protein sequence space
SIMAP represents the known protein sequence space comprehensively and up-to-date. According to this goal, the SIMAP database is synchronized once per month with the major protein sequence databases (Table 1). The consideration of each of these databases in SIMAP is justified by providing either unique protein sequences that are not found in other databases [e.g. ENSEMBL (7)], or unique protocols for data processing [e.g. NCBI RefSeq (8)]. The continuous and rapid growth of the sequence space demands for a sophisticated high-performance computing infrastructure to pre-calculate the sequence similarities of all new sequences and their sequence-based features immediately after the import of new sequences even in case of SIMAP’s incremental implementation. The SIMAPBOINC public resource computing project (9) steadily provides compute power beyond the current need and thus enables rapid updating of SIMAP.
Table 1.
Number of protein entries and non-redundant sequences of the major protein sequence databases included in SIMAP as of September 2009
| Database | Protein entries | Non-redundant sequences |
|---|---|---|
| NCBI GenBank | 16 146 018 | 13 065 886 |
| NCBI RefSeq | 8 181 910 | 6 681 186 |
| Uniprot/TrEMBL | 8 926 016 | 7 586 794 |
| Uniprot/Swissprot | 495 880 | 416 496 |
| PDB | 139 106 | 41 445 |
| PEDANT | 5 480 442 | 5 389 911 |
| ENSEMBL | 1 094 482 | 1 062 197 |
Consistent processing and annotation of metagenomes
With the breakthrough of next generation sequencing methods and their application to environmental samples (10), metagenomic sequences have indelibly expanded the protein sequence space to non-culturable organisms and environmental communities. However, the pioneering ‘Global ocean sampling’ (GOS) project (11) so far remains the only metagenomic dataset of which protein sequences are represented in a major public sequence database [NCBI GenBank (4)]. All other metagenomes are—if at all—deposited in distributed resources as the ‘Whole Genome Shutgun’ (wgs) section of NCBI GenBank (4) or the IMG/M database (12). No standardized protocol for gene calling and the annotation of protein-coding sequences has been established so far for these data collections. As the consistent annotation of metagenomes is indispensable for any downstream comparative analysis such as comparisons of taxonomic or functional profiles between different metagenomes, an extension of SIMAP was implemented that extracts coding sequences from metagenomic sequencing reads, assembled contigs and scaffolds in a consistent way.
This part of SIMAP covering environmental sequence fragments is monthly synchronized with three major repositories of metagenomes (Table 2). Entirely redundant metagenomes are considered only once, whereas redundant representations of the same project differing in their total number of nucleotides (e.g. the whale fall samples in IMG/M and GenBank wgs) are retained. Similar to the methodology used by the GOS project (13), coding sequences are extracted from the nucleotide sequences in a multi-step procedure:
all open reading frames (ORF) exceeding a length of 90 nt are extracted from the nucleotide sequences of a metagenome,
all-against-all protein sequence similarities between all ORFs in a metagenome are calculated using the SIMAP software (3): first a FASTA (5) similarity search against the low-complexity masked sequences down to the BLOSUM50 (14) score of 80 is performed without restricting the number of hits, thereafter the alignments are re-calculated without low-complexity masking,
ORFs are weighted by the number and score of their sequence alignments; shadow ORFs are detected by their overlap with higher weighted ORFs and removed using the methodology and parameters as in the GOS project (13),
remaining ORFs having a length of at least 60 aa are imported into the main SIMAP database,
all-against-all protein sequence similarities between all ORFs of a metagenome and all other protein sequences in SIMAP are calculated as in step 2,
again, shadow ORFs are removed as in step 3.
Table 2.
Number of metagenomic samples and extracted protein-coding sequences in SIMAP as of September 2009
| Database | Metagenomic samples | Non-redundant sequences |
|---|---|---|
| Camera (JCVI) | 54 | 6 031 109 |
| NCBI Genbank/wgs section | 130 | 4 244 008 |
| IMG/M (JGI) | 65 | 2 833 359 |
Compared to the supervised gene prediction methods as used in other metagenomic resources, the procedure applied in SIMAP is not biased towards any taxonomic group (i.e. prokaryotes) and only limited by the minimal length of open reading frames in step 1 and 4. The parameters applied in this procedure ensure optimal sensitivity in detecting coding sequences both in single-exon and multi-exon genes.
The derived metagenomic ORFs have almost doubled the volume of the known protein sequence space and thus significantly added valuable information (Figure 1). However, metagenomic sequences exhibit lower accuracy compared to completely sequenced genes and genomes, show fragmentation in case of multi-exon genes and lack knowledge of their taxonomic origin. Therefore, metagenomic sequences can be excluded when retrieving data from SIMAP according to the individual requirements of the user.
Figure 1.
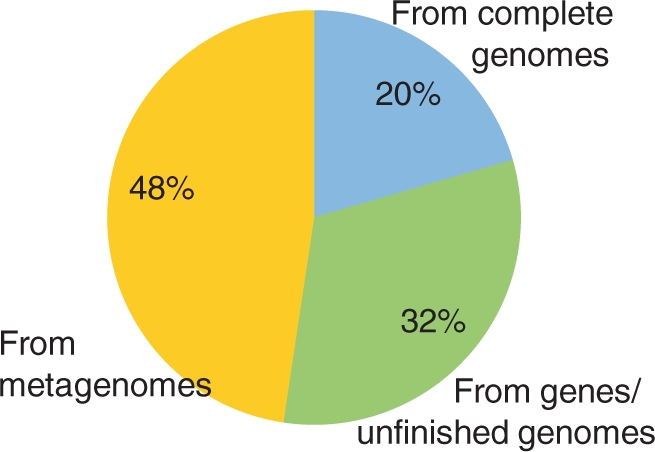
Composition of the non-redundant protein sequence space in SIMAP as of September 2009.
Functional annotation of the protein sequence space
Many computational methods to support the prediction of protein function are computationally expensive and therefore benefit from comprehensive pre-calculation and incremental updates as the basic design principles of SIMAP. SIMAP thus pre-calculates Interpro domains and features (2) for all sequences including metagenomic ORFs (15). New releases of InterPro are incorporated into SIMAP as soon as they become available; SIMAP is regularly updated to the latest InterPro version (currently 22.0).
SIMAP provides an ideal complete resource for the computation of secondary features such as the functional annotation of protein sequences based on information transfer from annotated proteins. BLAST2GO may serve as an example that provides various annotation tools for the functional classification of proteins (16,17). Blast2GO achieves the automatic functional annotation of DNA or protein sequences employing the Gene Ontology vocabulary. We have adapted the Blast2GO suite to enable the retrieval of sequence similarities from the SIMAP database instead of performing BLAST (1) searches. This step saves an enormous amount of compute-time compared to BLAST and allows annotating the complete protein sequence space of SIMAP using a few PCs within a week. We have integrated the adapted BLAST2GO program into the monthly update workflow of SIMAP in order to keep the pre-calculated BLAST2GO annotations complete and up-to-date (Table 3).
Table 3.
Pre-calculated functional annotations in SIMAP as of September 2009
| Method | Number of pre-calculated features |
|---|---|
| InterProScan | 133 829 528 |
| TargetP | 17 205 439 |
| SignalP | 11 060 831 |
| TMHMM | 15 841 454 |
| Phobius | 18 488 832 |
| Blast2GO | 190 801 556 |
High performance data access facilities
All data in SIMAP are freely available. The continuously growing size of SIMAP demands a sophisticated implementation of the database to provide versatile and rapid access to the data with respect to a broad spectrum of use cases. Based on the established database and standard middleware components of SIMAP, we have improved the performance and stability of SIMAP through clustering of two independent database and application servers. This clustering effectively uncouples production and maintenance processes. Each of the servers is ready to process more than 2 million complex queries per day.
Furthermore, we have improved the different data access facilities connecting SIMAP to its users. The versatile web portal allows searching for proteins by text or sequence queries. The matches are starting points for retrieving homologous proteins based on sequence similarity or domain architecture. Protein report pages integrate data from SIMAP including InterPro and GO annotation as well as from external resources as the PEDANT database (18). To facilitate clustering methods, all-against-all matrices of similarity scores can be downloaded for user-supplied groups of proteins.
Programmatic access to SIMAP is provided by several SOAP based Web-Services. The SimpAT (Simap Access Tools) allows easy access to the SIMAP database using Web-Service functionality. Recently, we have implemented Distributed Annotation System (DAS) services for SIMAP. These can be accessed via the URL http://webclu.bio.wzw.tum.de/das/ and provide easy and rapid access to the proteins, sequence similarities, InterPro matches and GO annotations from SIMAP. These data with the exception of the very huge similarity matrix itself can also be downloaded as flat files from the SIMAP web portal. For research projects interested in parts of the similarity matrix, we provide project specific monthly dumps upon request.
DISCUSSION
The SIMAP database is a unique fundamental resource for computational biology that consequently puts the principle of incremental pre-calculation of sequence similarities and sequence based features into practice. SIMAP as an exhaustive, up-to-date resource to inspect the sequence similarity of any known sequence enables of any type of systematic post-processing with respect to the functional or structural classification of proteins.
The recent integration of metagenomic sequences into SIMAP based on a consistent extraction of coding sequences has been beneficial to preserve the comprehensiveness of the sequence space representation in SIMAP. At the same time it makes use of the sequence similarity matrix of SIMAP to resolve overlaps and remove shadow ORFs. SIMAP represents to our knowledge the largest and most homogeneous resource for the annotation of coding sequences in metagenomes. It provides an ideal data repository and speed-up for tools as e.g. MEGAN (19) that extract taxonomic and functional information from similarities between metagenomic ORFs and known proteins in major sequence databases.
The extended functional annotation of the sequence space through the pre-calculation of GO annotations and the improved data access facilities have enhanced the potential of SIMAP in assisting biologists in answering their individual research questions as well as facilitating downstream projects in computational biology at any scale.
FUNDING
SUN Microsystems Inc. (funding a fully equipped X4500 data center server that is hosting parts of the SIMAP database, through a SUN Academic Excellence Grant), European Science Foundation (financial support for Stefan Götz through the activity entitled ‘Frontiers of Functional Genomics’). Funding for open access charge: Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the BOINCSIMAP community for donating their CPU power for the calculation of protein similarities and features. They are grateful to their colleagues at MIPS, in particular Mathias Walter, Martin Muensterkoetter and Manuel Spannagl, for many helpful discussions and suggestions.
REFERENCES
- 1.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold R, Rattei T, Tischler P, Truong MD, Stumpflen V, Mewes W. SIMAP-The similarity matrix of proteins. Bioinformatics. 2005;21:ii42–ii46. doi: 10.1093/bioinformatics/bti1107. [DOI] [PubMed] [Google Scholar]
- 4.Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2009;37:D5–D15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- 6.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Bwl. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 7.Hubbard TJP, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rattei T, Walter M, Arnold R, Anderson DP, Mewes W. Using public resource computing and systematic pre-calculation for large scale sequence analysis. Lecture Notes Comp. Sci. 2007;4360:11–18. [Google Scholar]
- 10.Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol. Biol. Rev. 2004;68:669–685. doi: 10.1128/MMBR.68.4.669-685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K. The Sorcerer II global ocean sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D, Chen I, Min A, Grechkin Y, Dubchak I. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 2008;36:D534–D538. doi: 10.1093/nar/gkm869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rattei T, Arnold R, Tischler P, Lindner D, Stumpflen V, Mewes HW. SIMAP: the similarity matrix of proteins. Nucleic Acids Res. 2006;34:D252–D256. doi: 10.1093/nar/gkj106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 17.Götz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter MC, Rattei T, Arnold R, Guldener U, Munsterkotter M, Nenova K, Kastenmuller G, Tischler P, Wolling A, Volz A. PEDANT covers all complete RefSeq genomes. Nucleic Acids Res. 2008;37:D408–D411. doi: 10.1093/nar/gkn749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]