


Abstract
Peptidases, their substrates and inhibitors are of great relevance to biology, medicine and biotechnology. The MEROPS database (http://merops.sanger.ac.uk) aims to fulfil the need for an integrated source of information about these. The database has a hierarchical classification in which homologous sets of peptidases and protein inhibitors are grouped into protein species, which are grouped into families, which are in turn grouped into clans. The classification framework is used for attaching information at each level. An important focus of the database has become distinguishing one peptidase from another through identifying the specificity of the peptidase in terms of where it will cleave substrates and with which inhibitors it will interact. We have collected over 39 000 known cleavage sites in proteins, peptides and synthetic substrates. These allow us to display peptidase specificity and alignments of protein substrates to give an indication of how well a cleavage site is conserved, and thus its probable physiological relevance. While the number of new peptidase families and clans has only grown slowly the number of complete genomes has greatly increased. This has allowed us to add an analysis tool to the relevant species pages to show significant gains and losses of peptidase genes relative to related species.
INTRODUCTION
The MEROPS database is a manually curated information resource for peptidases (also known as proteases, proteinases or proteolytic enzymes), their inhibitors and substrates. The database has been in existence since 1996 and can be found at http://merops.sanger.ac.uk.
The organizational principle of the database is a hierarchical classification in which homologous sets of peptidases and protein inhibitors are grouped into protein species, which are in turn grouped into families, which are grouped into clans. A family contains proteins that can be shown to be related by sequence comparison alone, whereas a clan contains proteins where the sequences are so distantly related that similarity can only be seen by comparing structures. Sequence analysis is restricted to that portion of the protein directly responsible for peptidase or inhibitor activity which is termed the ‘peptidase unit’ or the ‘inhibitor unit’, respectively. A peptidase or inhibitor unit will normally correspond to a structural domain, and some proteins will contain more than one peptidase or inhibitor domain. Examples are potato virus Y polyprotein which contains three peptidase units, each in a different family, and turkey ovomucoid, which contains three inhibitor units all in the same family. At every level in the database a well-characterized type example is chosen, to which all other members of the family or clan must be shown to be related in a statistically significant manner. The type example at the peptidase or inhibitor level is termed the ‘holotype’ (1,2).
The MEROPS database is released quarterly and users can now keep up to date with the latest MEROPS information by subscribing to the MEROPS database Blog at http://meropsdb.wordpress.com. Statistics from release 8.5 (August 2009) of MEROPS are shown in Table 1 and compared with release 7.8 from April 2007. The number of peptidase sequences has more than doubled, whereas the numbers of protein species, families and clans has increased only slightly. The number of inhibitor sequences has tripled, with the majority of increases in three families (I1, I4 and I63) due to large numbers of homologues being present in some eukaryote genomes. These increases reflect the considerable effort being put into sequencing new genomes. It also demonstrates the power of the peptidase classification to make sense of the data deluge.
Table 1.
Counts of identifiers, families and clans for peptidase and protein inhibitor homologues in the MEROPS database
| MEROPS 7.8 |
MEROPS 8.5 |
|||
|---|---|---|---|---|
| Peptidases | Inhibitors | Peptidases | Inhibitors | |
| Sequences | 66 524 | 4912 | 140 313 | 16 337 |
| Protein species | 2403 | 571 | 3215 | 678 |
| Families | 185 | 53 | 208 | 66 |
| Clans | 51 | 33 | 52 | 34 |
The numbers in the current release of MEROPS (release 8.5, August 2009) are compared with release 7.8 from April 2007.
In 2007 we published criteria for distinguishing one peptidase from another (3), and in the last two years much of our effort has been focussed on implementing these criteria in the MEROPS database. We have applied these criteria to hypothetical peptidase homologues identified by analysing completely sequenced genomes (4), allowing us to assign a MEROPS identifier where appropriate. Two of the important distinguishing criteria are the different peptidase specificities and the overall arrangement of all the domains within the proteins. The new displays discussed below make use of these criteria and enable us to identify novel peptidases.
GENOME ANALYSES
The number of completely sequenced genomes from cellular organisms now exceeds 1300. Because the genomes from several strains of the same organism have been sequenced, this represents the genomes of 780 different species. We have recently introduced a feature in the organism species pages of MEROPS for a summary analysis of the peptidase homologues. We highlight instances where the genome contains members of a peptidase family not found in 90% or more of other closely related species (an unexpected presence), or where a peptidase family is missing but present in 90% or more of other closely related species (an unexpected absence), or when the organism in question contains more or less members of a peptidase family than any other closely related species. This page is a product of a CGI program which progresses up the organism classification starting from the family level towards superkingdom, one taxon at a time, collecting the number of species with completely sequence genomes. When that number exceeds five, then the analysis is performed and the results are presented at the foot of the species page. An example analysis is shown in Figure 1.
Figure 1.

A summary analysis for the peptidase homologues from the completely sequence genome of the archaean Cenarchium symbiosum. The figure is taken from the species page in the MEROPS website. A list of peptidase homologues arranged alphabetically by MEROPS identifier is shown in the top panel and the genome analysis is shown at the bottom of the page. The peptidase portion of the proteome of C. symbiosum (12) has been compared with those of 17 other species from the class Thermoprotei. There are unexpected absences of members of peptidase families C26, C44, M38, M48, S9 and U62, and an unexpected presence of a homologue from peptidase family M3. Of the species compared, C. symbiosum had the fewest number of peptidase family M20 homologues, but the most for peptidase family S8. The large number of absent peptidase families may indicates that this endosymbiont genome is degenerate.
DOMAIN ARCHITECTURES
The images showing domain architectures have been overhauled. Because only the peptidase and inhibitor units are classified in the MEROPS database, it can be useful to compare the architectures of different proteins within the same peptidase or inhibitor family. This can now be done for all the holotypes from a family by clicking on the ‘architecture’ button on the family page. An example of a family architecture is shown in Figure 2.
Figure 2.

The domain architectures for holotypes in peptidase subfamily M12B. The figure is taken from the domain architecture page for peptidase subfamily M12B (the adamalysins) from the MEROPS website. The arrangement of regions and domains are shown for a selection of holotype proteins. The structures are arranged from the top of the page in order of MEROPS identifier. The name of the peptidase is given on the left-hand side. All the structures are drawn to the same scale. The sequence length is denoted by the pale blue line. Regions and domains as determined by MEROPS, the Pfam database and Swiss-Prot entries in the UniProt database (7), are shown as coloured rectangles on this bar. The domains that are classified within the MEROPS database are shown as slightly larger boxes, in green for a peptidase unit and grey for an inhibitor unit (not shown). The MEROPS identifier is displayed in the centre in black text. Domains derived from the Pfam database (13) are shown as smaller rectangles in crimson, with the domain name in white text. On clicking on the box, the user will be taken to the relevant Pfam entry. Regions from Swiss-Prot include signal peptides and transmembrane regions (shown as even smaller boxes in black) and propeptides (in dark grey). Active site residues (red ‘lollipops’) and metal ligands (blue ‘lollipops’) are shown along the bottom edge. Carbohydrate-binding residues (orange ‘lollipops’) and disulphide bridges (black lines connecting the cysteines) are shown along the top edge. Mouse-over text gives details of the feature displayed in all cases.
SUBSTRATES AND SPECIFICITY DISPLAYS
One of the most important distinguishing features of a peptidase is its specificity: where it will cleave a substrate protein or peptide. The MEROPS substrate cleavage collection began in 1998 with the publication of the CD version of the Handbook of Proteolytic Enzymes (5) and has now grown from 1919 cleavages in release 7.8 (April 2007) to include over 34 000 known cleavages in proteins and peptides (physiological and non-physiological) and over 2700 cleavages in synthetic substrates. Protein and peptide substrates are mapped to a UniProt identifier where possible, and the P1 residue for each cleavage [the residue on the amino side of the scissile bond (6)] mapped to a residue number within the UniProt database entry. The peptidase responsible for the cleavage is mapped to the MEROPS identifier. We have recently added cleavages to this collection that result in removal of targeting signals from proteins, including initiating methionines from cytoplasmic proteins by methionyl aminopeptidases, the signal peptides from proteins that enter the secretory pathway by signal peptidases, and removal of targeting peptides for proteins that are imported into chloroplasts, mitochondria and peroxisomes. Only those cleavages that have been experimentally verified, usually by N-terminal sequencing of the mature protein, have been included.
We have introduced ‘flags’ on the substrate pages to indicate the method used to identify the cleavage position. The flags are as follows: NT shows that the cleavage position was determined by N-Terminal sequencing, MS shows that the peptide composition was determined by mass-spectroscopy (MS) and the cleavage position computed, MU shows that the cleavage position was determined by site-directed MUtagenesis, CS indicates that the cleavage position was postulated from a consensus motif (CS) within the protein sequence. Because the substrates as used by researchers are usually mature proteins and peptides, the substrates page also includes an extra column in the table to show the residue range of the protein or peptide used in each study.
A tool has been assembled to allow the dynamic alignment of substrate protein sequences. On the assumption that a physiologically relevant cleavage will be conserved in orthologous protein sequences from closely related organisms, cleavage sites are highlighted in the alignment to show conservation or lack of it. Cleavage sites with little conservation are probably fortuitous and of no physiological significance (though in a minority of cases they may be pathological). For each substrate where cleavages are known, the corresponding UniRef50 entry (7) is found and all the UniProt protein sequences included within that entry are aligned with MUSCLE (8).
It is assumed that most cleavages in native proteins occur within surface loops and interdomain linkers. Where the tertiary structure has been solved, the secondary structural elements are indicated on the substrate alignment. An example protein substrate alignment with secondary structure indicated is shown in Figure 3.
Figure 3.

An example of a substrate protein sequence alignment. The figure is taken from the MEROPS website and shows a protein sequence alignment of human C–X–C motif chemokine 11 and its close homologues, showing conservation around the matrix metallopeptidase 8 (MMP8, M10.002) cleavage site at residue 84 (14). The sequence of the protein in which the cleavage was discovered is highlighted in green. Residues are numbered according to this sequence. The MEROPS identifiers of the peptidases known to cleave this substrate are shown below the residue numbers on the left. The arrows next to each MEROPS identifier show the residue range of the peptide fragment used in the experiment, which in most cases is the mature protein without the signal peptide (the signal peptidase cleavage at residue 22 is shown). A question mark instead of an angled bracket would indicate that the terminus has not been determined. The scissile bond symbol (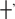 ) shows where cleavage occurs. Each symbol can be clicked, and the alignment will be highlighted to show conservation around that cleavage site. Four residues either side of each cleavage site (P4–P4′) (6) are highlighted. Completely conserved residues are highlighted in orange. Although not shown in this example, a residue highlighted in pink would not be conserved, but the amino acid would have been observed in the same position in another MMP8 substrate. Ile84 in the sequence from the European ferret (Mustela putorius fero), labelled UniProt A8DBL7, is shown with a black background because isoleucine is unknown in this position for any MMP8 substrate. The last line shows the secondary structure: an alpha helix is shown as a series of ‘a’s highlighted in red, and a strand as a series of ‘b’s highlighted in green. This example shows that MMP8 is capable of cleaving this protein substrate within an alpha helix.
) shows where cleavage occurs. Each symbol can be clicked, and the alignment will be highlighted to show conservation around that cleavage site. Four residues either side of each cleavage site (P4–P4′) (6) are highlighted. Completely conserved residues are highlighted in orange. Although not shown in this example, a residue highlighted in pink would not be conserved, but the amino acid would have been observed in the same position in another MMP8 substrate. Ile84 in the sequence from the European ferret (Mustela putorius fero), labelled UniProt A8DBL7, is shown with a black background because isoleucine is unknown in this position for any MMP8 substrate. The last line shows the secondary structure: an alpha helix is shown as a series of ‘a’s highlighted in red, and a strand as a series of ‘b’s highlighted in green. This example shows that MMP8 is capable of cleaving this protein substrate within an alpha helix.
The display showing cleavages in a selected protein depends on the user choosing the correct species from which the substrate was derived. If no cleavages are known for the user-selected protein but are known for the same protein from a different species, then an option is automatically presented to display the sequence alignment with those cleavages highlighted.
We use the MEROPS substrate cleavage collection to indicate the specificity of a peptidase. This is shown as a WebLogo (9) and a frequency matrix for the residues accepted in binding pockets P4 to P4′, provided we know of 10 or more substrates. There are over 300 peptidases for which 10 or more substrates are known. These displays are shown on the relevant peptidase summary page. However, this does not allow easy comparison of one peptidase with another. So in addition to the displays on a peptidase summary, MEROPS now includes displays to compare preferences in binding pockets S4 to S4′. These show preference in terms of all amino acids, amino acid properties and individual amino acids. The first of these shows, for each peptidase, an amino acid if it occurs in the same binding pocket in 40% or more of the substrates. So no more than two amino acids are shown for any one binding pocket. The amino acid is shown with a green background, and brighter the green, the greater the percentage of substrates with the amino acid in that binding pocket. The second display is similar but instead of showing individual amino acids, these are collected into ‘aliphatic’, ‘aromatic’, ‘acidic, ‘basic’ or ‘small’ groups. In the third option the user is prompted to select an amino acid from a pull-down menu and the display shows the percentage of substrates with the selected amino acid in each binding pocket for each peptidase. Where an amino acid has not been observed in a binding pocket, this is highlighted in black. In all three displays where no amino acid is possible (for example P4, P3 and P2 for an aminopeptidase, of P2′, P3′ or P4′ for a carboxypeptidase) the binding pocket is highlighted in grey. Figure 4 shows a portion of one of these new displays.
Figure 4.

Comparison of peptidase specificity. The figure shows a portion of a page from the MEROPS website. Peptidase preference for the amino acid proline is shown. The MEROPS identifiers and names of the peptidases are shown on the left, along with the number of substrate cleavages in the MEROPS collection. Where proline occurs in the same position in 40% or more of substrates, the cell is highlighted in green and the percentage of substrates with proline in this position is shown. Cells are only highlighted if 10 or more substrates are known for the peptidase. Where there can be no binding pocket to accommodate a substrate residue, for example in position P4, P3 and P2 for an aminopeptidase or P2′, P3′ and P4′ for a carboxypeptidase, these cells are highlighted in grey.
ALIGNMENTS AND TREES
We have been aware that as more data are collected some of our alignments are becoming very large. Not only will there be hundreds (even thousands) of sequences, but the consequences of aligning so many diverse sequences means that more gap characters are inserted and the alignments become wider. These are difficult to view on a computer screen, and on scrolling the screen, the residue numbers or sequence identifiers disappear off screen. To help to alleviate these problems, we have made our dendrograms more interactive. The nodes of the tree are now active links and on clicking on the node an alignment of all the sequences derived from that node will be displayed. This alignment also includes the family type example and the sequence numbering derived from the type example sequence. The alignment displayed is not dynamic, but is derived from the full alignment by removing any insert characters common to all the sequences. In order to make this happen, we are now including the aligned peptidase or inhibitor unit sequences and the dendrograms (in New Hampshire format) in the MySQL database.
The sequence page of the peptidase (or inhibitor) summary now includes an ALIGN VARIANTS button. Many peptidases and inhibitors are sequenced many times and variants exist, either strain-specific or the result of alternative initiation, alternative splicing of exons, allelic variation or single nucleotide polymorphisms (SNPs). Clicking on the ALIGN VARIANTS button will generate a dynamic alignment of all the variants we have collected from the primary sequence databases. Residues that differ from the sequence we have selected for inclusion in our protein sequence collection are highlighted as white text on a black background.
NEW INDEXES
Indexes are important tools to allow users to find the data they want. We have added a number of new indexes to MEROPS.
A new index of gene names has been added to the main index page (the left-hand menu). You can now search for any peptidase or protein inhibitor homologue knowing the name of its gene or its gene locus.
A new substrate menu has been added, which includes an index of substrate names to make it easier for the user to find a substrate for which we have cleavages in our collection. Substrates are arranged alphabetically by name. Names are usually taken from the UniProt description, but where the substrate is a fragment of a larger protein, the common name of the peptide is favoured over the protein name. For example, a user will find ‘substance P’ in the index as well as the source protein, ‘protachykinin-1’. The index also includes the names of synthetic substrates. The substrate menu also provides access to the pages that compare peptidase specificity.
LITERATURE
The MEROPS database includes an extensive collection of bibliographic references (over 37 000). Each of these references is tagged with the MEROPS identifier for the relevant peptidase, inhibitor, family or clan, and a list of references is given for each peptidase, inhibitor, family or clan. We have marked some of the publications that are relevant to particularly important topics by use of coloured ‘flags’. The full list of flags is shown in Table 2.
Table 2.
Flags used to mark publications that are relevant to particularly important topics and their explanation
| Explanation | |
|---|---|
| A | Assay method, |
| E | recombinant Expression, |
| I | design of small-molecule Inhibitors, |
| K | gene Knockout or other artificial genetic manipulation, |
| M | natural Mutation, allelic variant or polymorphism, |
| P | Substrate specificity, |
| R | RNA splice variation, |
| S | three-dimensional Structure, |
| T | proposed as a therapeutic Target, |
| U | suggested to have therapeutic potential itself, |
| V | Review |
DATABASE CROSS-REFERENCES
A new item has been added to the Searches menu. The MEROPS database includes many cross-references to other databases and bioinformatics resources. To make it easier for others to map their database entries to MEROPS there is a new CGI that presents the cross-references between MEROPS and any database selected from a pull-down menu. There are a considerable number of cross-references between MEROPS and primary sequence databases, so these are returned in batches of 50 000.
A distributed annotation system (DAS) server (10) has been set-up for MEROPS. This allows others to extract data directly from the MEROPS MySQL database for inclusion in their own Internet service. The user enters an accession as a parameter in the URL (usually this will be a UniProt accession, but an EMBL/GenBank ProtID will work for MEROPS) and data relating to the sequence stored in our collection will be returned. For a peptidase or protein inhibitor, this will include the MEROPS identifier, family and clan, the extent of the peptidase or inhibitor unit, active site residues (and metal ligands for metallopeptidases), the amino acid sequence and a link to a page in MEROPS for each feature. For a protein substrate, positions of known cleavages and the MEROPS identifiers of the peptidases responsible are returned. Example URL’s are:
http://das.sanger.ac.uk/das/merops/features?segment=P07858 (features for human cathepsin B)
http://das.sanger.ac.uk/das/merops/sequence?segment=P07858 (sequence for human cathepsin B)
http://das.sanger.ac.uk/das/merops/features?segment=P05067 (known cleavages for human amyloid beta A4 protein precursor)
ENHANCEMENTS TO EXISTING FEATURES
For eukaryotes with completely sequenced genomes, the chromosomal location (in megabases) of the peptidase or protein inhibitor homologue gene is now shown on the organism page. These locations are derived from the EnSEMBL database (11) by searching for entries with a cross-reference to the UniProt protein sequence database, therefore a location will not be shown for a gene from any genome where the copy number is low. However, the locations for all homologues from human and mouse are shown. For human and mouse these locations are also shown in the Genetics table of the peptidase or protein inhibitor summary. Here the locations are linked to the contig view in EnSEMBL, which shows the exon and intron structure of the gene. The name of the chromosome (or genomic scaffold) precedes the location and the strand is indicated by a plus or minus sign in parentheses after the location.
The displays of peptidase or inhibitor distribution among organisms have been enhanced. There is now mouse-over text at every node which gives the name of the taxon.
MEROPS identifiers have been added to the tables of peptidase-inhibitor interactions, and it is now possible to order the tables according to the identifier or the protein name.
COMMUNITY ANNOTATION
Facilities have been set-up for our users to contribute to annotation in MEROPS via a ‘Submissions’ button. At present there are only two submission items, both for advising us of any known protein cleavage sites that we are unaware of. The first of these is a form for the submission of a single cleavage, and the second allows the user to upload a file of known cleavage sites. The latter has been designed with proteomics experiments in mind. The information provided will allow us to map the cleavage to an entry in the UniProt database. Users are also welcome to send comments on any aspect of the MEROPS website to the following E-mail address: merops@sanger.ac.uk.
FUNDING
Wellcome Trust [grant number WT077044/Z/05/Z]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We would like to thank Pfam and Rfam colleagues for helpful discussions, and Paul Bevan, Jody Clements and Matthew Waller from the Sanger Institute web team for all their help in maintaining this resource. We would also like to thank those users who have pointed out errors and omissions, or who have suggested changes and improvements.
REFERENCES
- 1.Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J. 1993;290:205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawlings ND, Tolle DP, Barrett AJ. Evolutionary families of peptidase inhibitors. Biochem. J. 2004;378:705–716. doi: 10.1042/BJ20031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett AJ, Rawlings ND. Species of peptidases. Biol. Chem. 2007;388:1151–1157. doi: 10.1515/BC.2007.151. [DOI] [PubMed] [Google Scholar]
- 4.Rawlings ND, Morton FR. The MEROPS batch BLAST: a tool to detect peptidases and their non-peptidase homologues in a genome. Biochimie. 2008;90:243–259. doi: 10.1016/j.biochi.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. London: Academic Press; 1998. [Google Scholar]
- 6.Schechter I, Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem. Biophys. Res. Commun. 1968;32:898–902. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 7.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowell RD, Jokerst RM, Day A, Eddy SR, Stein L. The distributed annotation system. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallam SJ, Konstantinidis KT, Putnam N, Schleper C, Watanabe Y, Sugahara J, Preston C, de la Torre J, Richardson PM, DeLong EF. Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc. Natl Acad. Sci. USA. 2006;103:18296–18301. doi: 10.1073/pnas.0608549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox JH, Dean RA, Roberts CR, Overall CM. Matrix metalloproteinase processing of CXCL11/I-TAC results in loss of chemoattractant activity and altered glycosaminoglycan binding. J. Biol. Chem. 2008;283:19389–19399. doi: 10.1074/jbc.M800266200. [DOI] [PubMed] [Google Scholar]