

Abstract
Pfam is a widely used database of protein families and domains. This article describes a set of major updates that we have implemented in the latest release (version 24.0). The most important change is that we now use HMMER3, the latest version of the popular profile hidden Markov model package. This software is ∼100 times faster than HMMER2 and is more sensitive due to the routine use of the forward algorithm. The move to HMMER3 has necessitated numerous changes to Pfam that are described in detail. Pfam release 24.0 contains 11 912 families, of which a large number have been significantly updated during the past two years. Pfam is available via servers in the UK (http://pfam.sanger.ac.uk/), the USA (http://pfam.janelia.org/) and Sweden (http://pfam.sbc.su.se/).
INTRODUCTION
Pfam is a comprehensive database of conserved protein families. This collection of nearly 12 000 families is used extensively throughout the biological sciences, by experimental biologists researching specific proteins, computational biologists who need to organise sequences, and evolutionary biologists considering the origin and evolution of proteins. Pfam is also widely used in the structural biology community for identifying interesting new targets for structure determination.
From its inception 12 years ago, Pfam has been designed to scale with the growth in the number of new protein sequences deposited. Scalability is achieved by having a set of seed alignments, with each alignment containing a representative set of sequences that are relatively stable between releases of the database. The seed alignments are used to build profile hidden Markov models (HMMs) that can be used to search any sequence database for homologues in a sensitive and accurate fashion. Those homologues that score above the curated inclusion thresholds are aligned against the profile to make a full alignment.
Our goal is to make Pfam a comprehensive and accurate classification of all known protein sequences. The 11 912 curated families are known as Pfam-A and are found in approximately three quarters of known proteins. In order to increase our coverage further, we augment the Pfam-A family collection with a set of automatically generated families called Pfam-B. Pfam-B is derived from the ADDA domain collection (1), which is described later.
Ten years ago, a family with more than 1000 sequences was considered to be large. Today, a growing number of families contain over 100 000 sequences. Depositions from large-scale metagenomic and other sequencing projects mean that we can expect the number of known sequences to grow into the billions, from the millions that we currently have. In order to deal with this explosion in the number of known sequences, we have made fundamental changes to the Pfam infrastructure. The most important of these has been the move to a new version of the profile HMM software, HMMER (http://hmmer.janelia.org/), which we use to build and search our models. Since 1998, Pfam (version 3.0 onwards) has utilised the HMMER2 package for building profile HMMs and searching them against sequences in the underlying sequence database. The new version of HMMER (version 3) is ∼100 times faster than the previous version and shows increased sensitivity.
In this article, we will describe the major changes that underlie the new version of Pfam (release 24.0) along with the major updates we have made to the families within the collection. We also describe changes made to both the database and to the web site and its associated services.
HMMER3
An alpha version of HMMER3 was released in early 2009. The HMMER3 project has four main aims: (i) to adopt log-odds likelihood scores summed over alignment uncertainty (Forward scores) in place of optimal alignment (Viterbi) scores; (ii) to report posterior probabilities of alignment confidence; (iii) to be able to accurately and quickly calculate expectation values (E-values) for Forward scores (a previously unsolved problem); and (iv) to accelerate previous profile HMM performance by two orders of magnitude and achieve an overall speed competitive with BLAST (2). These goals have been met and the software package is now undergoing testing pending a stable 3.0 release. The outcome of aims (i) and (iii) make HMMER3 more sensitive. The increase in speed arises from a combination of approaches, including the use of vector-parallel SIMD (single instruction multiple data) instructions (SSE2 on Intel-compatible platforms; Altivec/VMX on PowerPC platforms); a new acceleration heuristic; and a ‘sparse rescaling’ method enabling the Forward and Backward HMM algorithms to be implemented in much faster calculations based on scaled probabilities rather than standard implementation in log probabilities (all to be described more fully in a future paper). The Cambridge Pfam team (especially RDF) has worked closely with the Janelia Farm HMMER team (especially SRE) on testing the alpha and beta versions of HMMER3 to ensure that it is stable enough to adopt in Pfam database-production pipelines. For the production of Pfam 24.0, HMMER3 beta 2 version was used.
A limitation of HMMER3 is that it currently computes only local alignments. HMMER2 was capable of calculating either local or glocal (complete model matching one or more times to subsequences of the target sequence) alignments. Until 2002, each family in Pfam was represented by a single profile HMM, where the curator of the family chose whether the model should be in local or glocal mode. From 2002, both local and glocal mode models were constructed from the same seed alignment. The membership of the family and those sequences that made it into the full alignment of a family were based on the union of the significant matches to the glocal and the local models. Under HMMER3, we again construct only a single model from the seed alignment, and that model is in local alignment mode. The parameterisation of HMMER3 local alignment models is sufficiently improved that a single HMMER3 local alignment model generally outperforms the union of HMMER2 local and glocal models. Still, glocal alignment is desirable, because it can sometimes increase sensitivity; when E-value statistics of glocal Forward scores are sufficiently well understood and can be calculated rapidly, a glocal alignment mode option will be restored in HMMER3.
The change to HMMER3 comes at an opportune moment, as the computational burden of making a Pfam release was increasing significantly due to a combination of searching two models per family (local and glocal), the increasing size of sequence databases and the increasing number of Pfam families.
As an example of HMMER3’s speed, it now typically takes <5 min to search a profile HMM against the latest version of pfamseq (9.4-million sequences), compared with the ∼535 min to search the corresponding model using HMMER2 (based on searches with a single Intel(R) Xeon(R) CPU, 3.00 GHz). Pfam release 23.0 took ∼60 CPU years to calculate (searching and post-processing of data), with the vast majority of this time spent in profile HMM searches with HMMER2. Pfam 24.0 took ∼10 CPU years, despite the near doubling of the sequence database since Pfam 23.0 and the adoption of a more computationally demanding procedure for calculating the trees for each family (described later).
Thresholds and compatibility
The new scoring system of HMMER3 means that all of our families based on HMMER2 needed to have their gathering thresholds re-defining. The gathering threshold is the bit score threshold that a sequence must match or exceed in order for it to be deemed significant and therefore belonging to that family. In Pfam, we manually define our thresholds such that no known false positives are permitted in any family. However, although it is possible to generate HMMER2 style profile HMMs from a HMMER3 profile HMM, the thresholds are not backward compatible. Therefore, it is important to emphasise that Pfam 24.0, based on HMMER3, is not backward compatible with HMMER2. Nevertheless, we believe that the significant increase in performance far outweighs this compatibility issue.
Envelope and alignment domain boundaries
By default, HMMER3 reports two sets of domain co-ordinates for each profile HMM match. The envelope co-ordinates delineate the region on the sequence where the match has been probabilistically determined to lie, whereas the alignment co-ordinates delineate the region over which HMMER is confident that the alignment of the sequence to the profile HMM is correct. In the Pfam-A full alignments we report only a single set of co-ordinates, the envelope co-ordinates, as these represent the sequence-segment that is aligned to the profile HMM. When an envelope exists, this is represented as insert (lower case) characters at the beginning and/or end of the alignment. On the whole, the envelopes are small extensions (<10 amino acids, mean 2.7 with SD of 7.9 at the N-terminus, and mean of 3.2 and SD of 9.1 at the C-terminus) at either end of the alignment co-ordinates. There are some cases, however, where the envelope start and end range can be significantly longer than the alignment co-ordinate range. In Pfam, we do not allow overlaps between the alignment co-ordinates of different Pfam-A domains.
FAMILY UPDATES
Numbers of new families
Since the last publication describing Pfam in 2008 (release 22.0), we have added 2871 Pfam-A families and deleted 236. Pfam 24.0 is based on UniProt version 15.6. We also provide our match-data against NCBI GenPept (version 172) and a collection of metagenomic samples.
Pfam 24.0 represents a 24% increase in the total number of families, relative to Pfam 23.0. Most of these new families have come from one of two sources: (i) a family seeded by a structure deposited in the Protein Data Bank—wwPDB (3)—that Pfam 23.0 did not cover, and (ii) Pfam-B families that we have used as a starting point for building Pfam-A, focusing particularly on Pfam-B clusters without a corresponding annotated family in InterPro (4). In addition to these, many families have been contributed via suggestions from the community.
Iteration of families in 23.0
As well as adding new families, we have also revised all 8339 families that were not part of a Pfam clan, where a clan is a collection of Pfam families which we believe to have a common evolutionarily ancestor (5). Pfam has existed since the late 1990s and many of the families added over this 12-year-period have had few, if any, updates since they were first deposited. Consequently, the seed alignments that were used to construct the profile HMMs were built from sequence databases that did not contain the diverse range of species that they do today. As the seed alignment is intended to contain representative members of a protein family, we have tried to improve our seed alignments for each family, through a process we refer to as iteration. This process starts with the full alignment for a family and attempts to make a new non-redundant seed alignment from it. During this process, any fragment matches are removed as are other sequences containing insertions and deletions that are likely to be incorrect. The alignment is typically made non-redundant so that no pair of sequences shares more than 80% sequence identity. The new profile HMM generated from this seed alignment is searched against the sequence database, and during this process, a proportion of families modified in this way should have captured new and more distant homologues. Having iterated every family not in a Pfam clan, 50% of these were found to have expanded with additional homologues, and their profile HMMs now have the more divergent sequence amino acid substitutions, insertions and deletions modelled in them. We intend to iterate families that belong to clans in the future.
Although in most cases the improvements to any one family were modest, some families gained many hundreds of new members. For example, the Rhodanese domain (Pfam accession PF00581) gained 1483 new sequences during iteration. Expansion also indicated hitherto unseen inter-family relationships, such that families could be merged together into a single entity. An example of one such merge is the uncharacterised family DUF30 (Pfam accession PF01727), which was found to be closely related to Peptidase_S7 (Pfam accession PF00949) and was therefore merged into that family.
INCREASE IN COVERAGE
One of the goals of Pfam is to be as comprehensive as we can be, so that as many sequences as possible fall into one of our families. Therefore, we closely monitor both the sequence coverage of Pfam (the proportion of sequences with at least one match to a Pfam-A) and the residue coverage (the proportion of residues that are matched by Pfam-A), particularly between one release and the next. Sequence coverage has increased by 1.41 percentage points from Pfam 23.0 to a coverage of 75.15% in Pfam 24.0. Residue coverage has increased by a similar margin, from 51.22% in Pfam 23.0 to 53.18% in Pfam 24.0, a gain of 1.96 percentage points.
Although these headline numbers regarding coverage are accurate, they do not represent a truly fair comparison between the current and previous releases. The dramatic increase in the number of sequences in the sequence database between Pfam 23.0 and 24.0 serves to mask an equally dramatic increase in coverage.
To make the comparison fairer, we can base it on the intersection between the two releases of Pfam in terms of families and sequences. Thus, if we consider only those sequences that are common to both Pfam 23.0 and Pfam 24.0, and only the families that are present in both 23.0 and 24.0, we observe an increase of 0.85 percentage points in sequence coverage and an increase of 1 percentage point in residue coverage. These increases are due to a combination of two factors: (i) improvements to our existing families achieved through the iteration process, and (ii) the increased sensitivity of HMMER3, as compared to HMMER2. Of the families that were present in both releases 23.0 and 24.0, 4782 families gained members, 2214 families did not change in size and 3104 families decreased in size. Of those 3104 families that decreased in size, only 974 lost more than 10 member sequences.
Close analysis of the families that have lost sequences reveals that the types of losses fall into three broad categories. The first is losses due simply to a difference in the distribution of sequences between families belonging to the same clan. For example, the ig (Pfam accession PF00047) and I-set (Pfam accession PF07679) families both belong to the Ig clan (Pfam clan accession CL0011); whilst the ig domain has lost 2365 sequences, the V-set has gained 1067 sequences, and with the other clan members gaining small numbers this gives comparable numbers of sequences matched amongst the clan members between the two releases.
The second category is losses largely confined to viral-specific families, such as HCV_NS1 (Pfam accession PF01560) and RVT_connect (Pfam accession PF06815), which have both lost thousands of members. This has been caused by sequence fragments, typically of <15 amino acids in length, being detected at the extreme N- and/or C-termini of families by HMMER2 but not by HMMER3. Many viral families will be affected in this way as UniProt contains many fragments of viral proteins, especially of polyproteins.
The third category results from a loss of sensitivity in a few, very specific HMMER3 models compared to their HMMER2 counterparts. These models fall into two sub-categories: (i) very short repeats, e.g. Hexapep (Pfam accession PF00132) and Ank (Pfam accession PF00023), and (ii) short, very divergent families, e.g. zf-C2H2 (Pfam accession PF00096) and HATPase_c (Pfam accession PF02518). All of the families that fall into this third category are symptomatic of the same feature, the match is not sufficiently long enough for the signal to be distinguishable from the noise. This is quite easy to understand for the very short repeats (subcategory (i) above). Those families in subcategory (ii) are a little harder to explain. All of these families were ‘tuned’ effectively to HMMER2’s glocal alignment (a complete domain with respect to the query model, local in the target sequence). Such families have typically short models containing <100 consensus residues with lots of extremely diverse sequences in the alignment (low average sequence identity). This gives rise to the situation where the average score per homologous position is low across a short model such that true homologues are just barely resolvable from the noise even when all consensus residues are aligned. In local alignments (HMMER3’s default and only mode), the probability theory assesses an extra bit score penalty of log_2 {2/[M(M + 1)]} (for the extra freedom of finding start/end positions anywhere in a model of consensus length M) which works out to ∼ −12 bits for a model of M = 100. If the true homologues were resolvable from noise by <12 bits by glocal alignment, then, in local alignments they might well become indistinguishable from the noise. As the Pfam curated gathering thresholds are set such that no known false positives are included in the family we are excluding these sequences that are in the top of the noise.
We intend to address the issuses of these poorly performing families with respect to HMMER2, by building multiple HMMs to represent the family, with each HMM being built from a seed alignment containing more closely related homologues. The new families will be grouped into a clan to represent the divergent family. Addressing this issue will be a priority of the curation process between Pfam 24.0 and 25.0.
Improved sensitivity
So, what has the improved sensitivity of HMMER3 achieved? We have so far focused on sequence coverage and have said less about residue coverage. The increase in residue coverage by nearly 2 percentage points is quite remarkable. In the last 5 years, our residue coverage between releases has fluctuated by only fractions of 1 percentage point. The current change clearly indicates that although we are not finding substantially more matches on sequences that do not already match a Pfam-A family, we are matching more residues on sequences where we do already have a Pfam-A match.
The increase in residue coverage comes from one or both of two sources, namely longer length matches and matches to additional domains on a sequence. Although the improvements to the HMMER3 local model have meant that local matches are longer, prior to release 24.0 we typically took glocal matches in preference to local ones. Our analysis of the unique domain architectures, where a domain architecture is defined as a particular combination of Pfam-A families, shows that there has been a 67% increase (from 48 634 to 72 629) in the number of distinct architectures between Pfam 23.0 and Pfam 24.0. Of the 4.9-million sequences common to the sequence databases of releases 23.0 and 24.0, 11.4% of them have changed domain architecture.
Another way in which the increased sensitivity has affected Pfam is that we have detected many more similarities between families due to sequence regions significantly matching more than one Pfam HMM. These relationships were predominantly identified after building HMMER3 models for all of the Pfam seed alignments, and searching them against the same sequence database on which Pfam 23.0 was built. These search results showed that the increased sensitivity of HMMER3 had expanded many of our families, but that ∼80 000 sequences now had overlapping matches to more than one Pfam family. Since we do not allow overlaps between alignment co-ordinates, each of these overlaps had to be resolved.
There are a number of reasons why overlaps might arise: (i) families overlap by only a few end residues, so we will trim the domain boundaries such that the two families no longer overlap, (ii) the sequence(s) that have the overlap are false positives in one or other of the families, so here we raise the threshold in that family such that the sequence is excluded, and this helps to maintain the high quality of the Pfam data, and (iii) two or more families may share full-length matches, so, depending on the degree of similarity between them, these families may be merged into one or be grouped together into a clan.
Many of the overlaps generated by HMMER3 were due to reason (iii). We always preferentially merge families if we can build a single HMM model that detects both sets of sequences. This explains why the number of deleted families at Pfam 24.0 was so high (236 families were deleted). For example, DUF223 (Pfam accession PF03027), a family of moth juvenile hormone binding proteins and Drosophila proteins of unknown function has been merged into JHBP (Pfam accession PF06585) which is a family of insect specific haemolymph juvenile hormone binding proteins. However, more often than not, the two families will be too divergent to make a single HMM, but the overlaps have allowed us to identify and/or confirm relationships between them for which we previously had insufficient evidence. Using the HMMER3 overlap data together with profile comparison data from PRC (6) and SCOOP (7), and structural data, we were able to assign many families to existing clans, and since Pfam 23.0 we have created 120 new clans. We have also been able to merge some existing clans (listed in Table 1), showing that we are bringing together groups of families previously thought to belong to different structural superfamilies.
Table 1.
Clans which have been merged between Pfam release 23.0 and Pfam release 24.0
| Clan | Description |
|---|---|
| CL0008 (DEAD-like superfamily) | All members of this clan have been moved to CL0023 (P-loop containing NTP hydrolase superfamily) |
| CL0017 (G-protein superfamily) | All members of this clan have been moved to CL0023 (P-loop containing NTP hydrolase superfamily) |
| CL0019 (Armadillo repeat superfamily) | All members of this clan have been moved to CL0020 (TPR repeat superfamily) |
| CL0024 (Reverse transcriptase superfamily) | All members of this clan have been moved to CL0027 (RNA dependent RNA polymerase superfamily) |
| CL0102 (Methyltransferase superfamily) | All members of this clan have been moved to CL0063 (FAD/NAD(P)-binding Rossmann fold superfamily) |
| CL0138 (Chemoreceptor superfamily) | All members of this clan have been moved to CL0192 (Family A G protein-coupled receptor-like superfamily) |
| CL0150 (Peptidase MX superfamily) | All members of this clan have been moved to CL0126 (Peptidase MA superfamily) |
| CL0152 (Xylose isomerase-like TIM barrel superfamily) | All members of this clan have been moved to CL0036 (Common phosphate binding-site TIM barrel superfamily) |
| CL0185 (Frizzled/OA1/CAR/Secretin receptor-like superfamily) | All members of this clan have been moved to CL0192 (Family A G protein-coupled receptor-like superfamily) |
| CL0211 (GDE-like sugar enzyme superfamily) | All members of this clan have been moved to CL0059 (Six-hairpin glycosidase superfamily) |
| CL0216 (DNA recombination protein RecA-like superfamily) | All members of this clan have been moved to CL0023 (P-loop containing NTP hydrolase superfamily) |
| CL0253 (DsbD like superfamily) | All members of this clan have been moved to CL0292 (LysE transporter superfamily) |
In Pfam release 24.0, 3131 out of 11 912 families (26.3%) belong to a clan, compared with 2009 out of 10 340 families (19.4%) in Pfam release 23.0. Notably, the families that typically belong to clans correspond to the larger families in Pfam, so that over 57.3% (up by 11.9% since Pfam 23.0) of our sequence annotations come from families falling within the clan hierarchical classification.
Proteome coverages
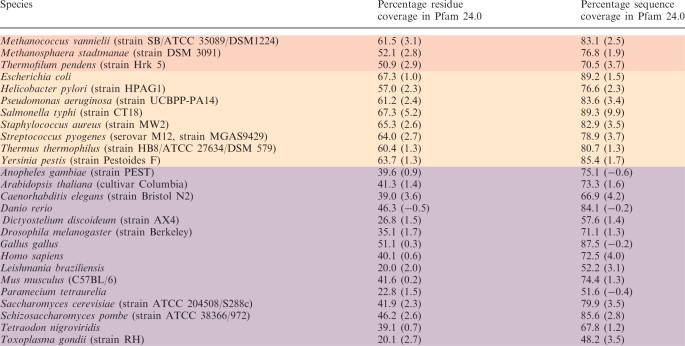
While we have focused on the highs and lows in terms of performance, there is an overall trend towards an increase in coverage. A more fine grained way of assessing the difference in coverage between Pfam 23.0 and Pfam 24.0 is to take the proteomes from completed genomes, present in both releases, and look at the differences in sequence and residue coverage of a range of these individually to see how coverage has changed (Table 2). Generally, we observe an increase that is comparable or higher to the gross coverage gains. When a similar analysis was performed in 2000 (8), the eukaryotic genomes lagged behind bacterial genomes in both sequence and residue coverage. For many of the model organims such as human and yeast, the sequence coverage is more on a par with bacterial sequence coverage. The increase in coverage is down to many factors that include better gene builds, more eukaryotic homologues in the sequence database and improved models in Pfam. Instances where the coverage has fallen between the two releases can often be attributed to new assemblies (e.g. Danio rerio) and the lag time for that new data to percolate through to the relevant databases.
Table 2.
Residue and sequence coverage of a number of complete proteomes in Pfam 24.0, with the percentage points change between Pfam releases 23.0 and 24.0 given in brackets. Archaeal species are coloured pale red, bacterial orange and eukaryotic species purple
 |
Improved Pfam-B coverage
To aid our goal of comprehensively covering the whole of sequence space, we generate a set of automated families in addition to our curated Pfam-A families. The automated families are called Pfam-B families and are built from homologous sequence clusters. Each Pfam-B is represented by a single alignment and has no associated profile HMM or accompanying annotation. In previous releases of Pfam, Pfam-B families were generated by taking clusters derived from MSP-crunch and latterly PRODOM (9) clusters, and removing regions covered by Pfam-A families to leave a set of Pfam-B clusters (10). The PRODOM database is updated infrequently which means that newly deposited sequences may not be incorporated into a PRODOM cluster for a long time. Thus if, in the meantime, Pfam updates to a more recent sequence database, the sequence coverage of Pfam contributed by Pfam-B will be much lower than it might be. The ADDA algorithm has been used from Pfam release 23.0 onwards, to generate the Pfam-B families.
ADDA is a method for automatically predicting protein sequence domains from protein sequence alignments alone. Briefly, the ADDA algorithm takes a set of non-redundant sequences (11,12) and aligns them all-versus-all using BLAST (2). Sequences are then partitioned into domains by optimising an objective function that penalises domains that (i) split alignments or (ii) overlap with alignments only partially. The resultant domains are grouped into clusters using pairwise profile–profile comparisons. The whole procedure is calibrated using SCOP (13) domains as a gold-standard.
The current ADDA core is based on a non-redundant sequence data set from a 2007 snapshot of Uniprot with 3 378 785 sequences, 6 181 472 domains and 270 191 non-singleton families. The ADDA core is projected onto the latest sequence data set using BLAT (14) with a 40% sequence identity cut-off. After mapping, ADDA assigns 8 119 847 domains to 11 626 194 sequences in 303 153 non-singleton families. On average, 83% (median: 97%) of amino acid residues in protein sequences are covered by domains. Updating the ADDA core is currently a ‘work in progress’.
For Pfam-B generation, we take clusters of multiple sequence alignments and subtract the regions covered by Pfam-A, as described previously in detail in ref. (10). We use MAFFT to create multiple sequence alignments from the sequence co-ordinates of each ADDA cluster, for clusters that contain between 2 and 1000 sequences and comprise more than 40 amino acids. This process sheds only ∼2500 ADDA clusters, most of which are already covered by Pfam-A regions. In Pfam 24.0, from the 300 422 ADDA clusters that fulfil this criterion, we have built 142 303 Pfam-B families.
In Pfam release 23.0, where ADDA was used for the first time, the sequence coverage contributed by Pfam-B increased substantially from what would have been 3.9% with PRODOM, to 11.8%. Due to the increased coverage provided by Pfam-A in release 24.0, the fact that many new families have been built starting from the largest Pfam-Bs in Pfam 23.0 and that the ADDA core is becoming more out of date, this coverage has dropped, but still provides 5.7% additional sequence coverage, and contributes 5.8% additional residue coverage. We expect future releases of Pfam to be correlated with updates to the ADDA core, and expect the coverage provided by Pfam-B to rise once more. Despite the lower than ideal coverage contribution provided from Pfam-B, the combined coverage provided by Pfam-A and Pfam-B is 80.9% for sequence coverage and 58.8% for residue coverage.
STREAMLINING OF THE PFAM PIPELINE
As part of switching to HMMER3, we have identified additional bottlenecks in the database production pipeline in terms of potential scalability and/or computational time. Our aim has been to perform the same quality control checks and post-processing on a family regardless of size (i.e. scale from 2 to 100 000 sequences), while ideally reducing the computational burden imposed by the production pipeline.
Neighbour-joining trees using FastTree
Prior to Pfam release 24.0, the ‘QuickTree’ software (15) was used to produce UPGMA-based phylogenetic trees for ordering the sequences contained in the full and seed alignments for each Pfam family. From release 23.0 onwards we started to provide the more accurate, but computationally more intensive, neighbour-joining (NJ) tree for families with <20 000 members. We were, however, unable to provide bootstrap values for the NJ trees as these were too time consuming to compute. Furthermore, for our larger families, producing the faster and computationally cheaper UPGMA tree using QuickTree could take up to 3 days, and was also proving to be memory intensive.
From release 24.0 we have moved to using the ‘FastTree’ software (16) to produce NJ trees with boostrap values. FastTree is far less memory intensive than QuickTree in addition to being considerably faster, because it uses an approximation to the maximum likelihood tree. For one of our largest families, ABC_tran (Pfam accession PF00005), the UPGMA tree generated with QuickTree for release 23.0 took over 48 h to complete, whereas the NJ tree generated for release 24.0 using FastTree took only 3 h. We are now able to provide NJ trees with bootstrap values, based on 100 replicates, for both the seed and full alignments for all our families.
Family comparisons
To detect relationships between families we perform profile–profile comparisons between all profile HMMs. In the past we used the profile comparison software ‘PRC’ (6); however, the profile–profile comparison software ‘HHsearch’ (17) is ∼10 times faster, with both pieces of software producing comparable results. Therefore, we have switched to using the faster HHsearch software. Profile–profile comparison results are shown on the family summary page, alongside results from SCOOP family comparisons. Profile–profile comparison results are also used to construct the clan relationship images (e.g. http://pfam.sanger.ac.uk/clan?acc=CL0012), where the relationship between families is depicted as a graph (families are nodes, relationships are edges between the nodes).
Data version control
All Pfam-A family and clan data files are now under version control using Subversion (SVN - http://subversion.tigris.org). We have modified our pipelines to tightly couple commits of changes to quality-control checks and population of the underlying MySQL database. We will shortly be making our SVN repository externally accessible using the HTTPS protocol. Read-access to the repository will be granted to all, thereby allowing users access to pre-released versions of our data. Furthermore, this system will open up the opportunity for trained collaborators outside the Pfam consortium to add and modify families and clans using our software. We expect this system to be fully operational by the beginning of 2010.
Changes to the underlying MySQL database
The changes caused by migration to using HMMER3 have necessitated numerous alterations to the database schema. We have also moved from using the MyISAM table-engine (http://en.wikipedia.org/wiki/MyISAM) to using InnoDB (http://www.innodb.com). Although it is not appropriate to discuss the pros and cons of different table-engines here, one of the fundamental differences between the two is that InnoDB supports foreign key relationships, i.e. there is declarative referential integrity. In addition to improving data quality-control, InnoDB also makes the schema (containing 63 tables) easier to understand and to write queries against.
NEW WEBSITE FEATURES
Interactive sequence searches
One of the primary user entry-points into the website is via a sequence search. We have already described the speed improvement achieved with HMMER3 and this improvement means that single sequence searches are now interactive. A search of a sequence of 500 amino acids takes <0.5 s against the current HMM library, using hmmscan (hmmscan is the HMMER3 program that replaces the hmmpfam program in HMMER2). Given the speed of hmmscan in HMMER3, we have decided to use an HMM-based approach for detecting matches to Pfam-Bs. In the past, Pfam-B matches were detected in query sequences by a BLAST search of the query against a fasta file containing all of the Pfam-B regions. However, this approach often gave rise to many partial Pfam-B matches to the query sequence, making these matches less useful. To improve the sensitivity and specificity of Pfam-B searches and to ensure that the search results return interactively, we have taken the 20 000 largest Pfam-Bs (Pfam-B accessions PB000001 to PB020000) and built HMMs from the Pfam-B multiple sequence aligments. The query sequence is searched against the Pfam-B HMMs with a default E-value cut-off of 0.001. When multi-threaded versions of HMMER3 become available, we anticipate expanding the Pfam-B HMM library to include all Pfam-Bs.
The sequence search results table which displays the matches to Pfam is much the same as before with the exception that we now include both the envelope and the alignment co-ordinates as well as information about whether the matched family belongs to a clan. As the user moves their mouse pointer over the ‘significant hits’ table the domain corresponding to that row is highlighted in the domain architecture graphic by a grey bar. The biggest change to the content of the results page comes when the user chooses to show the alignment between the query sequence and the HMM (see Figure 1). The query sequence is colour-coded according to the posterior-probability, given in the #PP line. The HMM is now colour-coded according to the amino acid similarity between the most probable sequence, which is shown in the #HMM line, and the query sequence, #SEQ. Identical residues are coloured cyan, while similar residues are coloured dark blue.
Figure 1.

Sequence search results page. Results page for a single sequence search, showing at the top, the graphic of the domains matched by the query sequence along its length, with any active-site or metal-binding residues marked up if present. Underneath comes, firstly, the significant matches to Pfam-A families, then the insignificant matches to Pfam-A families, followed by the significant matches to Pfam-B families. At the bottom is the expanded match results with the #HMM line coloured such that residues identical to those in the query are coloured cyan and those that are similar in dark blue, and a #PP (posterior probability) line giving the posterior-probabilities at each point such that the #SEQ, query, line is colour-coded accordingly.
Additional family page features
Displaying the domain architecture of proteins
We have adapted the way that protein domains are graphically represented on a sequence such that we are now able to depict both the envelope and the alignment co-ordinates. As before, Pfam-A families classified as type ‘repeat’ or ‘motif’ are represented by rectangles and families with type ‘family’ and ‘domain’ with a lozenge shape. Where the profile HMM match for a domain or family is only of partial length, the curved end of the lozenge/rectangle is replaced by a jagged edge, as depicted in Figure 2. Envelope regions are shown as a lighter shade of the colour used to colour the alignment region. As most envelope regions are small and we draw most of our graphics at 1 or 2 amino acids per pixel, for the majority of cases the envelope regions are barely visible. To provide more information to our users, we have expanded the information contained in the ‘tool tips’, to include both the domain and the envelope co-ordinates as well as the short descriptor of the family. In addition to indicating known and predicted active-site residues found in UniProt (18) and Pfam (19), which are represented as lollipops with a diamond-shaped head, we now also indicate the metal-binding residues defined in UniProt, represented as lollipops with a square-shaped head.
Figure 2.

New Pfam display of a protein domain architecture. Pfam-A families classified as type ‘family’ and ‘domain’ with a lozenge shape, and families with type ‘repeat’ or ‘motif’ are represented by rectangles. The alignment co-ordinates are depitcted with a solid colour, and the envelope co-ordianates in a lighter shade of this colour. Where the profile HMM match for a domain or family is only of partial length, the curved end of the lozenge/rectangle is replaced by a jagged edge. Active-site residues are marked with a lollipop with a diamond-shaped head. An example tooltip showing the domain description, co-ordinates and source is shown for the fourth domain. Note the overlapping envelopes between fourth and fifth domains.
We have re-factored the mechanisms by which the domain architectures of sequences are generated. Previously, each of the graphics would be generated as a temporary image that was served by a separate request to the web server. At busy times, we were generating thousands of temporary images per minute and serving them. To avoid the load of generating and serving the domain architecture images on the server, we now use a javascript library that we have written to render the images within the client browser.
Alignment confidence display
Another feature of HMMER3 is that as the searches are now based on the Forward algorithm, there is a probabilistic inference of alignment uncertainty. This is recorded as a posterior probability for each amino acid that is matched to a profile HMM. We have taken these posterior probabilities and converted the data into a heat map-style representation of the alignment. When the alignment of an amino acid to a match state in the profile HMM is estimated to be correct, then this amino acid is shown in upper case with its background colour shown in green. As the alignment certainty diminishes, the colour becomes closer to red. The same colouring scheme is employed for inserts, with confidently assigned inserts being coloured green. At the boundaries between inserts and matches, there tends to be less certainty about the accuracy of the alignment and hence residues here are coloured in tones closer to red. Posterior probabilities are calculated when a sequence is aligned to the profile HMM, therefore this feature is only available for our full alignments (see Figure 3) and not for our seed alignments.
Figure 3.

New alignment confidence display. The colour of the residues reflects the alignment uncertainty, and is based on the posterior probability that is calculated by HMMER3. A green residue indicates a high posterior probability which means that the alignment of the amino acid to the match/insert state in the profile HMM is very likely to be correct. Where the posterior probablity is lower, and therefore the alignment certainty decreases, the colour becomes closer to red. This allows users quickly to identify regions of the alignment where some sequences are aligned with less certainty.
HMM-logos
For each Pfam-A family we now include a graphical representation of the HMM, using the ‘logomat-m’ software (20). These HMM logos are found on a separate tab for each family and are embedded in a scrollable window.
Family protein sets
One of our most frequent user-requests of late has been the ability to download the set of UniProt sequences that belong to a particular family as a FASTA file. This has now been added as a download option, with the title ‘full-length sequences’, within the alignments section of the family web pages.
TreeFam links
TreeFam is a database of phylogenetic trees of eukaryotic genes that contains orthologue and paralogue assignments in addition to information regarding the evolutionary history of various gene families (21). We now provide reciprocal links to TreeFam from the Protein pages on the website as there is not a one-to-one relationship between Pfam-A entries and TreeFam entries. To further complicate the linking, TreeFam is based on a collection of protein sequences drawn from several different databases. To set-up the links to TreeFam, we cross reference the MD5 checksums of the protein sequences contained in Pfam with those found in TreeFam families. When a link is available, we display the TreeFam phylogenetic tree within our protein page.
Improvements to the scientific annotations
The annotation provided in Pfam for each family is a brief synopsis of the function of the family. Although we endeavour to keep these summaries up-to-date, it is becoming an ever increasingly difficult task as the numbers of families and the sequences they contain grow.
Identifying functional data from papers is time-consuming, as more often than not, protein sequence-identifiers are rarely used in any standard format. However, for protein structures, the PDB represents the single repository for such data, and consequently the PDB identifiers are a common standard format. Text-mining of full text articles for terms such as these protein-identifiers is becoming a fruitful way of identifying articles of interest. The new website will be utilising the Web services provide by BioLit (22) which allows access to metadata describing the semantic content of all open access, peer-reviewed articles (in PubMed Central) based on a PDB identifier. Whenever an article is found to contain a PDB identifier, we will use the Web services to retrieve the abstract, figures and figure legends from that article and make them available on our pages. Unfortunately, not all articles are published in open access journals, so this feature will not appear on every structure page.
The efforts from the field of structural genomics are depositing more and more structures with no associated publication. For many structural genomics targets there is additional information about the structure available in the ‘The Open Protein Structure Annotation Network’ (TOPSAN) wiki. As with BioLit, we are using Web Services to retrieve information contained within the TOPSAN wiki and display it on our protein structure pages. In addition to the retrieval of images and text, we also provide links to TOPSAN so that users can actively add to the content of the wiki. A screenshot of these two features—BioLit and TOPSAN—is shown in Figure 4.
Figure 4.

New BioLit/TOPSAN views. Left: using the webservices provided by BioLit, we display the abstract, figures and figure legends from the publication associated with a particular PDB entry (only where articles are published in open access journals). In this case, we have retrieved open access articles that reference the PDB entry 1dan. Right: using the webservices provided by TOPSAN, we display images and text from the TOPSAN wiki, and a link so that users can contribute to the TOPSAN wiki. In this example, we show the information contained in TOPSAN describing PDB entry 1kq3.
DAS ontology compliance
As described in the 2008 paper, in addition to the website and our RESTful interface, we also provide much of the data contained in Pfam via the distributed annotation system (DAS). DAS is a system for disseminating annotations and alignments of DNA or protein sequences through a simple, web-based protocol. Previously, we separated out our family annotations (both Pfam-A and Pfam-B) from other sequence annotations such as active-sites and transmembrane region predictions, such that we had two DAS features servers. Based on monitoring of our access logs and feedback from the community, we have combined the domain annotations for Pfam families and the additional sequence annotations into a single DAS features source. Furthermore, we have adapted the features response so that it conforms to the ontology standards set out for DAS feature servers (23). Making our DAS features source ontology-compliant, along with others, allows DAS clients to readily group features of similar types, thereby allowing comparisons of data without the need to make bespoke parsers for each DAS source.
LOCAL PFAM SEARCHES WITH PFAM_SCAN
To allow users to run Pfam locally we distribute a software tool called ‘pfam_scan’. The pfam_scan tool is a Perl wrapper around the HMMER package that allows protein sequences (in FASTA format) to be searched against Pfam’s library of profile HMMs, with the results post-processed in a similar fashion to that performed internally within Pfam. The original software was written almost a decade ago, and over the years has been modified as problems have been identified (and addressed) and new features implemented. The script is used widely by the Pfam user community. We have completely re-written the software to make it compatible with HMMER3. The new version of the pfam_scan script is available for download at ftp://ftp.sanger.ac.uk/pub/databases/Pfam/Tools/PfamScan.tar.gz.
To ensure consistency with Pfam, we wanted to be able to use exactly the same code for running searches on our website as our users would run when carrying out searches on their local machines. We also wanted the new code to be easier to maintain. In order to achieve these aims we have written the new code in a more modular fashion compared to the old version. This has, however, necessitated some changes in the dependencies of the software. In the past, pfam_scan was a standalone Perl script with no external dependencies other than standard Perl library modules and the HMMER programs. Rather than rewriting existing code, in the new version we have used a few modules that can be easily installed from CPAN. The modular nature of the code does mean that it can be more readily incorporated into other third party Perl scripts.
Speed
The new version of the pfam_scan script runs considerably faster than the old version for any given search. We have achieved this speed-up through both the use of HMMER3 (which gives an ∼100-fold increase in search-speeds over HMMER2) and by optimising the efficiency of the Perl wrapping code. Our benchmarks show that for a typical sequence search of 300 amino acids, against a library of ∼11 000 profile HMMs, the new pfam_scan code adds only ∼100–200 ms to the search time over and above the 1 s hmmscan run-time (benchmarks were performed on a single 2.4 GHz AMD Opteron processor).
New features and formats
The new version of pfam_scan.pl has all the functionality of the old version, including clan filtering and active-site prediction, and has some additional features which are described below:
The most important new feature is the option to search against the 20 000 largest Pfam-B families in addition to the full library of Pfam-A profile HMMs. Search times against the Pfam-A and Pfam-B profile HMM libraries are now roughly equivalent. Pfam-B accessions however, as in the past, are not stable between Pfam releases.
The default output of the script is, as in the old version, ASCII text format. The new output contains both the envelope and the alignment co-ordinates (see above for definitions) from HMMER3, and each Pfam-A match now includes its clan membership (if any), and its significance, which will be either 1 or 0 depending on whether the match scores higher than the Pfam gathering threshold, or not.
We have also added an option to write the results in JavaScript Object Notation (JSON) format. JSON is a compact, text-based data format which is most commonly used in the context of the web and javascript applications. As it is a compact and portable format, JSON can also be useful as a lightweight XML alternative.
THE XFAM BLOG
With so many changes happening to Pfam between releases 23.0 and 24.0, and because many other databases and software tools rely on Pfam annotations and tools, we needed to communicate our changes and progress to the Pfam community. An extremely effective way of doing this is through the use of a blog. In January 2009, we created the Xfam blog (http://xfam.wordpress.com/), which we share with the Rfam project (24). The blog not only allows us to explain our plans, it also allows our users to feedback comments which can open up into discussions. These discussions are recorded and are available for all to read, which is not the case when discussions happen directly with the Pfam team via e-mail.
SUMMARY
We are aware that the changes we have made to Pfam will have a significant impact on the Pfam user community, especially the lack of compatibility between Pfam 24.0 and the releases based on older versions of HMMER; however, we have tried to provide adequate communication of these changes through a variety of media. After careful evaluation of HMMER3 and the impact its adoption will have, we strongly believe that the benefits far outweigh any disadvantages. For example, sequence searches against Pfam HMM libraries can now be routinely run on modern laptops, and large-scale genome or metagenome analyses are no longer daunting in terms of their computational requirements. The increased sensitivity that our models now have is pivotal for improving the understanding of protein biology, with previously distant twilight relationships between proteins now being detectable at a level closely rivalling that found through structural comparisons.
AVAILABILITY
Pfam data can be downloaded directly from the WTSI FTP site (ftp://ftp.sanger.ac.uk/pub/databases/Pfam), either as flat files or in the form of MySQL table dumps.Pfam websites are found at WTSI (http://pfam.sanger.ac.uk/), Stockholm Bioinformatics Center (http://pfam.sbc.su.se/) and Janelia Farm (http://pfam.janelia.org/).
FUNDING
Wellcome Trust [grant numbers WT077044/Z/05/Z]; G.C. and S.R.E are supported by the Howard Hughes Medical Institute; K.F. and E.L.L.S. are funded by Stockholm University, Royal Institute of Technology and the Swedish Natural Sciences Research Council. L.H. is funded by the Academy of Finland [grant number 114498].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We are grateful for the infrastructure support provided by the Systems, Web and Database administration teams at Wellcome Trust Sanger Institute (WTSI), especially Guy Coates and Andy Bryant. Finally, we would to like to thank all of the users of Pfam who have submitted new families and/or annotation updates for existing entries.
REFERENCES
- 1.Heger A., Wilton C.A., Sivakumar A., Holm L. ADDA: a domain database with global coverage of the protein universe. Nucleic Acids Res. 2005;33:D188–D191. doi: 10.1093/nar/gki096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henrick K., Feng Z., Bluhm W.F., Dimitropoulos D., Doreleijers J.F., Dutta S., Flippen-Anderson J.L., Ionides J., Kamada C., Krissinel E., et al. Remediation of the protein data bank archive. Nucleic Acids Res. 2008;36:D426–D433. doi: 10.1093/nar/gkm937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunter S., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finn R.D., Mistry J., Schuster-Bockler B., Griffiths-Jones S., Hollich V., Lassmann T., Moxon S., Marshall M., Khanna A., Durbin R., et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madera M. Profile Comparer: a program for scoring and aligning profile hidden Markov models. Bioinformatics. 2008;24:2630–2631. doi: 10.1093/bioinformatics/btn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bateman A., Finn R.D. SCOOP: a simple method for identification of novel protein superfamily relationships. Bioinformatics. 2007;23:809–814. doi: 10.1093/bioinformatics/btm034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mistry J., Finn R. Pfam: a domain-centric method for analyzing proteins and proteomes. Methods Mol. Biol. 2007;396:43–58. doi: 10.1007/978-1-59745-515-2_4. [DOI] [PubMed] [Google Scholar]
- 9.Corpet F., Servant F., Gouzy J., Kahn D. ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000;28:267–269. doi: 10.1093/nar/28.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman A., Birney E., Durbin R., Eddy S.R., Howe K.L., Sonnhammer E.L.L. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heger A., Korpelainen E., Hupponen T., Mattila K., Ollikainen V., Holm L. PairsDB atlas of protein sequence space. Nucleic Acids Res. 2008;36:D276–D280. doi: 10.1093/nar/gkm879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park J., Holm L., Heger A., Chothia C. RSDB: representative protein sequence databases have high information content. Bioinformatics. 2000;16:458–464. doi: 10.1093/bioinformatics/16.5.458. [DOI] [PubMed] [Google Scholar]
- 13.Andreeva A., Howorth D., Chandonia J.M., Brenner S.E., Hubbard T.J., Chothia C., Murzin A.G. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howe K., Bateman A., Durbin R. QuickTree: building huge Neighbour-Joining trees of protein sequences. Bioinformatics. 2002;18:1546–1547. doi: 10.1093/bioinformatics/18.11.1546. [DOI] [PubMed] [Google Scholar]
- 16.Price M.N., Dehal P.S., Arkin A.P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 18.Wu C.H., Apweiler R., Bairoch A., Natale D.A., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mistry J., Bateman A., Finn R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics. 2007;8:298. doi: 10.1186/1471-2105-8-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuster-Bockler B., Schultz J., Rahmann S. HMM Logos for visualization of protein families. BMC Bioinformatics. 2004;5:7. doi: 10.1186/1471-2105-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruan J., Li H., Chen Z., Coghlan A., Coin L.J., Guo Y., Heriche J.K., Hu Y., Kristiansen K., Li R., et al. TreeFam: 2008 Update. Nucleic Acids Res. 2008;36:D735–D740. doi: 10.1093/nar/gkm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fink J.L., Kushch S., Williams P.R., Bourne P.E. BioLit: integrating biological literature with databases. Nucleic Acids Res. 2008;36:W385–W389. doi: 10.1093/nar/gkn317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeves G.A., Eilbeck K., Magrane M., O'D;onovan C., Montecchi-Palazzi L., Harris M.A., Orchard S., Jimenez R.C., Prlic A., Hubbard T.J., et al. The Protein Feature Ontology: a tool for the unification of protein feature annotations. Bioinformatics. 2008;24:2767–2772. doi: 10.1093/bioinformatics/btn528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S., Wilkinson A.C., Finn R.D., Griffiths-Jones S., Eddy S.R., et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]