


Abstract
fPOP (footprinting Pockets Of Proteins, http://pocket.uchicago.edu/fpop/) is a relational database of the protein functional surfaces identified by analyzing the shapes of binding sites in ∼42 700 structures, including both holo and apo forms. We previously used a purely geometric method to extract the spatial patterns of functional surfaces (split pockets) in ∼19 000 bound structures and constructed a database, SplitPocket (http://pocket.uchicago.edu/). These functional surfaces are now used as spatial templates to predict the binding surfaces of unbound structures. To conduct a shape comparison, we use the Smith–Waterman algorithm to footprint an unbound pocket fragment with those of the functional surfaces in SplitPocket. The pairwise alignment of the unbound and bound pocket fragments is used to evaluate the local structural similarity via geometric matching. The final results of our large-scale computation, including ∼90 000 identified or predicted functional surfaces, are stored in fPOP. This database provides an easily accessible resource for studying functional surfaces, assessing conformational changes between bound and unbound forms and analyzing functional divergence. Moreover, it may facilitate the exploration of the physicochemical textures of molecules and the inference of protein function. Finally, our approach provides a framework for classification of proteins into families on the basis of their functional surfaces.
INTRODUCTION
A large number of protein structures, including new structures from structural genomics projects, have already been accumulated. In most of these structures, the binding regions and key residues involved in biochemical activities are unknown. Moreover, a majority of them are in unbound (apo) forms and have no annotated functions. A starting point to understand the function of a protein is to identify its binding surface(s). Accurate assessment of binding surfaces can reveal geometric features, evolutionary history and physicochemical characteristics of proteins. Finally, well-characterized binding surfaces are useful for protein shape classification and can allow one to explore the functions of their structural homologs (1,2). However, large-scale identification, characterization, and classification of protein-binding sites are computationally challenging.
Over the past two decades, full-length sequence or fold-domain approaches such as COG (3), Pfam (4), SCOP (5) and CATH (6) have been developed to classify protein families and infer protein functions. Recent studies (7–11), however, have focused on local regions and demonstrated that the biological function of a protein is closely associated with the shape of its binding surface(s). Indeed, several structure-based methods, such as FunClust (12), 3D-SURFER (13), eF-seek (14) and SitesBase (15), have strived to identify functionally important regions in proteins. Moreover, ConSurf-DB (16), a database constructed using an evolutionary approach, provides the residue substitution rates on the protein surfaces. However, a well-characterized binding surface should include a detailed integration of geometric and evolutionary features, but most current methods do not provide such an integration, especially for unbound structures. In addition, a structural comparison between two local surfaces allows evaluating their similarities and differences to build an objective basis for inferring structural and functional relationships of proteins.
Our approach is purely geometrical and analytical. We model the shape of protein-binding surfaces instead of modeling the envelope of binding ligands. Employing the Smith–Waterman algorithm (17) and a shape matching technique, we use the spatial templates of functional pockets in our database, SplitPocket (18), to rapidly footprint the spatial pattern of an unbound surface. A major strength of this approach is that it considers the characteristics of spatial patterns, physiochemical texture and evolutionary conservation. With a fully automatic pipeline, we conduct ∼45 billion pairwise comparisons of unbound (apo) and bound (holo) forms, leading to the collection of the putative binding surfaces of ∼23 700 unbound structures in The Protein Data Bank (PDB). Although our method is targeted to predict protein-small molecule binding sites, the results indicate a potential for detecting protein–protein interactions too. Importantly, the database also includes the local structural relationships of functional homologs in protein families. These local pairwise relationships allow building structural phylogenies to understand protein functional divergence. Furthermore, a structural phylogeny allows building a computed binding profile (10) to classify protein families and to resolve some problematic issues such as enzymatic cross-reactivities, particularly in kinase families. Finally, we present site-specific measurements, highlight critical characteristics of each binding surface, and establish a bridge connecting protein structure, function and evolution.
DATA AND METHODS
Data and goal of the study
The goal of the fPOP database is to comprehensively collect PDB structures (>48 000 X-ray entries) and identify their binding surfaces. A complex structure is divided into chains. Introducing the concept of a split pocket (i.e., a pocket split by its ligand) and using a geometric approach, we have previously identified the functional pockets of selected bound forms (∼19 000 structures) and constructed the SplitPocket database (http://pocket.uchicago.edu/patch/), which contains ∼38 900 local spatial patterns (18). We now use these entries as spatial templates (Figure 1a) to footprint and identify the functional pockets in unbound forms (Figure 1c and d). We store the results in fPOP.
Figure 1.

Illustration of the fPOP shape analysis. (a) Identification of a split pocket in a bound structure as a spatial template (a collection of 38 900 spatial templates). (b) Surface segmentation of an unbound form. (c) Geometrically matching the spatial pattern of the template with those of putative pockets in the unbound form. (d) Measuring features and footprinting the binding surface of an unbound form.
Partitioning a protein according to the physicochemical texture of molecules
On the basis of the physicochemical texture of molecules, we partition the surface of a structure into putative pockets with customized probes (Figure 1b). The physicochemical texture of a surface is described in terms of atomic charge, hydrophobicity, polarity and hydrogen bond. An accurate surface-partition requires an analytical theory (19–21) and an exact algorithm (22,23) with an appropriate probe radius for each atom. Our probe radii are divided into the following four categories (11):
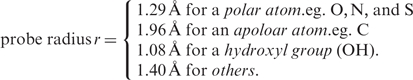 |
The assigned radius for a polar atom (O, N and S) is smaller than that for an apolar atom (C). Among all atomic types, the hydroxyl group (OH) has the smallest probe radius. With these probes, we segment a protein surface into local regions by the weighted-Delaunay triangulation (21). Having an appropriate partition, we detect all putative pockets on each individual structure by the discrete flow algorithm (20,23). For each putative pocket, we gather the set of the residues dispersed on the surface wall of the pocket. We concatenate the residues into a pocket fragment that represents a specific spatial pattern. We rank the putative pockets according to the number of amino acid residues in the pocket. Furthermore, for each pocket we obtain geometric measurements including the solvent-accessible area and the molecular volume under the specified probe radii. Basically, these identified residues on local surfaces provide the primary source for the spatial patterns. fPOP currently contains ∼1.16 million spatial patterns that are extracted from protein surfaces and can be used for further shape analysis.
f POP shape analysis
Superimposing the shapes of two spatial patterns
To evaluate the similarity between two pocket shapes, we use the Smith–Waterman algorithm to derive their local pairwise alignment. With a scheme of dynamic programming, the algorithm is carried out to deduce the optimized consensus subsequence from the alignment with the specific parameters by assigning ‘−5’ for a gap-penalty, ‘−1’ for a gap-extension and the BLOSUM62 (24) for a scoring matrix. In shape analysis, the two aligned pocket fragments are superimposed for calculating the atomic coordinate root mean square deviation (RMSD), which is minimized by optimizing the rotation matrix using the singular value decomposition (SVD). For a detailed description, see refs 7 and 11.
Footprinting the spatial patterns of unbound structures using the functional surfaces in SplitPocket
We exhaustively search for the geometric matching of a candidate pocket fragment against those of the ∼38 900 split pockets in SplitPocket. We evaluate the P-value for each candidate and declare it a binding site if the specified threshold is met (Figure 1c and d). That is, two pockets are functionally related from the geometric viewpoint if the query pattern is significantly similar to a pocket pattern (coordinate RMSD P-value ≤ 10−4 base on the receiver operating characteristic (ROC) curves of the studies of protein function inference (10,11)). In addition, we detect the split propensity of an unbound pocket at an orientation RMSD P-value ≤ 10−2. The P-values are estimated by the nonparametric statistical-based method of Binkowski et al. (7).
Characterizing the spatial pattern of a local surface
To characterize a protein functional surface, we consider the most fundamental geometric characteristics. A protein structure is a package of a large number of amino acid residues in space, but only a limited number of residues play key roles in biochemical function. Although these key residues are usually dispersed in the primary sequence (1D), they are clustered closely in a local tertiary structure (3D). Moreover, they cooperatively form a favorable micro-environment in physicochemical texture (2D) to interact with other molecules. Hence, the surface wall length, the solvent accessible area and the molecular volume are the molecular descriptors to characterize protein local structures. From on a large-scale study of ∼38 900 structures (11), we found that typically, a functional surface meets two geometric criteria. First, its wall length is >6 residues. Second, it has a molecular volume of at least 100 Å3 when its mouth is ‘open’. Hence, we use these two geometric criteria to effectively remove trivial pockets and reduce the search time.
Characterizing the evolutionary conservation of a local surface
A local protein surface can be highly conserved in evolution for function or for structure. We define the surface conservation index (SCI) for evaluating the evolutionary conservation of a protein surface patch as follows. We take advantage of the homology-derived secondary structure of proteins (HSSP; available at: http://swift.cmbi.ru.nl/gv/hssp/) constructed by Dodge et al. (25) from multiple sequence alignments with query structures. The major benefit is to obtain precomputed conservation weights of all sites in a query structure from the entropy measure of sequence variability. Denote the kth pocket fragment by 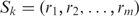 , where m is the number of residues and ri is the ith residue in the pocket fragment. We compute the position conservation (the weighted entropy score) from the HSSP. Denote the weighted entropy scores of residues normalized by the largest score of a residue on the query template in HSSP by wi, i = 1, … , m. We then normalize the sum of these normalized scores by the length (m residues) of the pocket fragment to obtain the SCI Ck for pocket k.
, where m is the number of residues and ri is the ith residue in the pocket fragment. We compute the position conservation (the weighted entropy score) from the HSSP. Denote the weighted entropy scores of residues normalized by the largest score of a residue on the query template in HSSP by wi, i = 1, … , m. We then normalize the sum of these normalized scores by the length (m residues) of the pocket fragment to obtain the SCI Ck for pocket k.
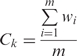 |
A surface patch (pocket) with a higher SCI usually has a higher likelihood to be a functional surface.
RESULTS
Identifying the binding sites of unbound forms is our primary task in constructing the fPOP database. We carried out the task by scanning putative pockets on each unbound structure in PDB. The goal is to determine whether a putative local surface of an unbound form has any of the split propensities sampled from similar or different folds (11) in SplitPocket. To achieve this goal, we analyze unbound forms using a large-scale computational platform.
Assessing shape similarities of functional surfaces
Footprinting the binding surface of an unbound form
We use an unbound form, the galactose-binding protein of Salmonella typhimurium (pdb1gcg), to demonstrate the general applicability of fPOP for predicting the binding surface(s) of an unbound structure. On the surface of this galactose-binding protein, we predict 13 putative pockets. We then identify the 13th pocket as the functional pocket (Figure 2a) because it has 14 similarity hits in the SplitPocket (Figure 2b). Based on the fPOP shape analysis, comparing the binding surface of the unbound form (pdb1gcg) with that of the respective bound form (pdb1gca), we find that the local RMSD between two binding surfaces is 0.7 Å, which indicates no significant conformational change. However, significant conformational changes often occur between unbound and bound forms. Examples are the triose phosphate isomerases from Saccharomyces cerevisiae. An RMSD of 4.1 Å caused by conformational changes is measured between the apo-form pdb1ypi.A (referring to chain A, Figure 2c) and the holo-form pdb2ypi (Figure 2d) using the f POP shape analysis.
Figure 2.

Predicting the binding surfaces of unbound forms. (a) The binding surface (the 13th pocket colored green with a mouth colored blue) of the galactose-binding protein (pdb1gcg) has a spatial pattern footprinted by the 16 functional surfaces of the 14 similarity hits in SplitPocket. (b) The functional surface (pdb3b6u.B) of a human motor protein is distantly related to that of the galactose-binding protein. A binding-ligand ADP (red) interacts with the split pocket (green). (c) The binding surface (the 11th pocket) of the triose phosphate isomerase (pdb1ypi.A) is correctly predicted. The fPOP shape analysis indicates that significant local conformational changes (4.1 Å RMSD) occur between the apo-form (pdb 1ypi.A) and the holo-form (pdb2ypi) in (d).
Here, we show another good example, using an unbound form from human proto-oncogene tyrosine kinases (pdb1yoj.A) to exploit the spatial homology by surface characteristics. f POP identified the 12th pocket of pdb1yoj.A as a functional surface (Figure 3a) by matching the spatial template of a remote-homologous protein (pdb3c4w.A, Figure 3b) from Bos taurus, which belongs to a specific class of G-protein-coupled receptor kinase 1 (classified by Enzyme Commission: EC 2.7.11.14). Both of the binding surfaces are responsible for adenosine triphosphate (ATP)-binding significantly involved in biological activities. However, their full-length sequence identified is <23%, whereas the similarity of the two pocket fragments is as high as 43% from an optimal alignment (Figure 3b). Moreover, the structural similarity of their functional surfaces yields a significant RMSD P-value of 4 × 10−7. Using the fPOP shape analysis, we highlight their shape similarity assessments in Figure 3.
Figure 3.

Footprinting the binding surface of a tyrosine kinase by a remote homologous protein. (a) At a significant RMSD P-value of 4 × 10−7, the binding surface (green) of pdb1yoj.A is matched with the binding pocket of pdb3c4w.A split by an ATP (red). (b) The optimal alignment of the binding surfaces between the query (pdb1yoj.A, red) and a spatial template (pdb3c4w.A, black) is used to compute their shape similarity at a RMSD of 2.3 Å. The similarity of pocket-fragments (43%) is considerably higher than that of the full-length primary sequences (22.3%). The catalytic residues (R390, A392 and N393) of pdb1yoj.A are also aligned with those (K316, E318 and N319) of pdb3c4w.A.
Functional relationships among structural homologs
The fPOP shape analysis also can reveal functional relationships among homologs. Two proteins are functionally related if the spatial patterns of their functional surfaces have the structural similarity at an RMSD P-value of <10−4, even if they are distantly related. We call such proteins ‘structural homologs’ because their homology is detected by structural comparison. With this simple criterion, we are able to obtain a structural phylogeny among homologs with branch lengths represented by the RMSD values of pairwise structural similarities (Figure 4).
Figure 4.

A structural phylogeny of binding surfaces for a subset of ATP-binding kinases.
We use the same aforementioned tyrosine kinase (pdb1yoj.A) to show that fPOP allows studying protein functional divergence among structural homologs even in the absence of sequence similarity in the superfamily. After exhaustive pairwise comparisons, we found a total of 435 homologs in PDB. Their binding surfaces are structurally related to the 12th pocket on the surface of pdb1yoj.A. Among the 435 homologs, we found that 308 PDB entries are remotely related (P-value > 10−4). To obtain an overall picture, we here select representatives (pdb1v0o.B, pdb2bfy.B, and pdb2gtn.A) from distinct species by progressive P-values of 10−4, 10−3 and 10−1, respectively. Although they are remote to the query, their binding surfaces showed subtle evolutionary conservation in spatial patterns captured by fPOP. In addition, we use pdb2src as a reference of tyrosine kinase with a catalytic domain. After extracting the binding surfaces of these five taxa, we compute a multiple pocket-sequence alignment to reconstruct a structural phylogeny (Figure 4).
Table 1 summarizes their pairwise structural, sequence, and functional relationships with the query surface (12th pocket of pdb1yoj.A). The spatial patterns of these remote homologs have experienced many substitutions, yet they have preserved a capacity to fulfill a similar biochemical function, such as ATP binding. Consequently, the geometric characteristic of spatial patterns provides valuable information for studying protein functional divergence, which may not be evident from a sequence-based comparison. Similarly, fPOP provides other biological important families such as glucose-binding, heme-binding and so forth in a systematic manner. These detailed spatial information and statistical results are accessible in fPOP.
Table 1.
Structural comparisons among remote homologs of a human tyrosine kinase (pdb1yoj.A)
| PDB | Species | Chain ID | Pocket ID | Npocket | SAA (Å2) | MV (Å3) | Full-length seq. id. (%) | Pocket- fragment seq. id. (%) | SCI | RMSD (Å) | RMSD P-value | Molecular function (EC) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1yoj | Homo sapiens | A | 12 | 20 | 263.27 | 508.48 | 100 | 100 | 0.70 | 0 | 0 | 2.7.10.2 (aka 2.7.1.121) |
| 2src | Homo sapiens | 0 | 23 | 36 | 681.01 | 986.38 | 84.6 | 64.3 | 0.71 | 1.64 | 4.5 × 10−7 | 2.7.10.2 |
| 2gtn | Mus musculus | A | 25 | 23 | 425.09 | 437.70 | 26.2 | 54.5 | 0.73 | 5.70 | 4.7 × 10−1 | 2.7.11.24 |
| 1v0o | Plasmodium falciparum | B | 25 | 20 | 381.65 | 430.93 | 25.2 | 66.7 | n/a | 3.53 | 2.2 × 10−4 | 2.7.11.22 |
| 2bfy | Xenopus laevis | B | 17 | 26 | 401.33 | 591.30 | 22.0 | 39.1 | 0.71 | 4.06 | 1.8 × 10−3 | 2.7.11.1 |
A spatial pattern is described in terms of the number of residues in the pocket (Npocket), solvent-accessible area (SAA), molecular volume (MA) and surface conservation index (SCI).
The binding surface of pdb1yoj.A is matched with those from remote homologs by structural assessments at various RMSD P-values.
Characterizing functional surfaces
In addition to the fPOP shape analysis, we further characterize protein-binding surfaces by geometric measurements and evolutionary conservation.
We use an alpha-amylase (pdb1bag) from Bacillus subtilis as a simple example to characterize its functional surface by geometric, evolutionary and physicochemical features. On the alpha-amylase surface, we predict 19 putative pockets. In Figure 5a, the 19th pocket is the functional surface split by glucose. For geometric measurements, it contains 19 residues, a solvent accessible area of 255.37 Å2 and a molecular volume of 342.27 Å3. Its mouth consists of 10 of the 19 residues that include seven hydrophobic residues (Figure 5b). Moreover, its spatial pattern carries the key residues D176, H180, Q208 and D269 (Figure 5c) with catalytic reactivities (26).
Figure 5.

Characterization of the functional surface of an alpha-amylase (pdb1bag). (a) The 19th pocket (green) is split by glucose (red). (b) The mouth of the split pocket has a hydrophobic accessible area (blue, 165.4 Å2). (c) The highest SCI (0.898) occurs in the split pocket. The spatial pattern of this functional surface consists of 19 residues with conservation weights for assessing the evolutionary characteristics. Four catalytic residues D176, H180, Q208 and D269 are highly conserved. In addition, there are 10 important residues sitting on the mouth. Among them, seven are hydrophobic residues indicated by asterisk.
Evolutionary conservation
Evolutionary conservation varies among regional surfaces, depending on their physicochemical constraints. The varied constraints result in varied substitution rates and structural divergences of the proteins (27). As a result, functionally important regions are usually conserved, although other regions may be conserved for structural stability. Here, accurate identification and characterization of spatial patterns (including functionally important residues) enable us to distinguish between different local surfaces. For example, on the alpha-amylase surface, the SCI of the functional surface (the 19th pocket) is 0.898, the highest among all putative pockets. In comparison, the SCI is 0.601 for the 18th pocket and 0.444 for the 17th pocket (Table 2). In addition, the catalytic residues of the 19th pocket such as D176 (1.00), H180 (1.00), Q208 (0.96) and D269 (1.00) are highly conserved (Figure 5c). Our findings indicate that local structures such as functional surfaces tend to be evolutionarily more conserved than other regional surfaces of the protein. Thus, SCI is a useful feature to distinguish a functional surface (binding site) from other local regions.
Table 2.
Geometric, and evolutionary characteristics of local surfaces of a bound Bacillus subtilis alpha-amylase
| Pdb1bag | Geometric features |
Evolutionary conservation | |||
|---|---|---|---|---|---|
| Pocket ID | Split | Npocket (a.a) | SAA (Å2) | MV (Å3) | SCI |
| *19th | 1 | 19 | 255.37 | 342.27 | 0.898 |
| 18th | 0 | 9 | 96.55 | 65.85 | 0.601 |
| 17th | 0 | 7 | 59.41 | 59.67 | 0.444 |
The functional surface indicated by asterisk is identified by a split pocket which has the highest SCI.
Likewise, we characterize the predicted binding surface for each unbound form with features. A typical example from the triose phosphate isomerase of S. cerevisiae is given in Table 3.
Table 3.
Characterization of putative binding surfaces of an unbound triose phosphate isomerase in yeast
| Pdb1ypi.A | Geometric features |
Evolutionary conservation | |||
|---|---|---|---|---|---|
| Pocket ID | Similarity-hits | Npocket (a.a) | SAA (Å2) | MV (Å3) | SCI |
| 12th | 0 | 18 | 300.58 | 461.62 | 0.695 |
| *11th | 46 | 13 | 167.04 | 198.02 | 0.960 |
| 10th | 0 | 10 | 80.49 | 80.71 | 0.539 |
| 9th | 0 | 7 | 30.31 | 23.00 | 0.880 |
The 11th and 12th pockets have open mouths with a molecular volume >100 Å3.
*Based on the fPOP shape analysis, the 11th pocket is the binding surface because it is matched by 46 similarity hits; it also has the highest SCI among all putative binding surfaces.
CONSTRUCTION OF THE fPOP DATABASE
Conducting a large-scale computation and collecting protein functional surfaces
The ∼38 900 functional surfaces (split pockets) in SplitPocket (18) are now used as spatial templates to footprint the putative binding surfaces in the unbound forms. To do so, we directly work on the ∼1.16 million putative pockets obtained from the 48 665 X-ray structures in PDB, including bound and unbound forms. From these putative pockets, one arduous task is to identify the binding surfaces of the unbound forms. An exhaustive way is to use the all-against-all search scheme, but it requires ∼1.2 × 1012 comparisons. Instead, we use pattern-to-pattern searches to identify the binding surfaces of each unbound form (Figure 1). We exhaustively compare the local shapes of the ∼38 900 spatial patterns in SplitPocket against each shape of the 1.16 million putative pockets (a total of 4.5 × 1010 comparisons). In total, we are able to predict ∼50 500 binding surfaces in ∼23 700 unbound structures. In ∼6000 out of the 48 655 structures in PDB, our searches do not detect any binding surfaces. These include structures that do not have similarity hits with any of the spatial templates in the current version of SplitPocket, small proteins without binding pockets and proteins with shallow depressions instead of pockets as the functional pockets (11). Thus, fPOP currently includes the predicted ∼50 500 binding surfaces of the ∼23 700 unbound forms and their structural homologs from the ∼19 000 bound forms as well as the ∼38 900 binding surfaces of the ∼19 000 selected bound forms. All geometric measurements, SCIs, spatial patterns, structural homologs and pairwise relationships with split pockets are included in the f POP system. This high-throughput computation of 45 billion pairwise comparisons was executed on a 170-processor Beowulf Linux cluster.
Prediction accuracy
In our previous study (11), we tested our method on a benchmark dataset prepared by Weisel et al. (28) and found that our method achieved a success rate of 90%. The success rate is defined as the ratio of the number of positive cases to the total number of cases studied, where a positive case is defined as the pocket-fragment identity of >40% between an unbound form and its corresponding bound form. The entries in the benchmark data set are representatives from various protein families. These results suggest that our method has a high accuracy. Of course, a certain fraction (about 10%) of our predictions is false positives. This caution should be kept in mind when using f POP.
DATA ACCESS
fPOP has a companion web interface for users to obtain spatial information. The database is freely accessible at: http://pocket.uchicago.edu/fpop/.
FUNDING
NIH grant GM30998 to (W.H.L.). Funding for open access charge: Academia Sinica, Taiwan.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Dr Jie Liang, the University of Illinois at Chicago and Dr. Andrew Binkowski, Argonne National Laboratory, for fruitful discussions.
REFERENCES
- 1.Binkowski TA, Freeman P, Liang J. pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res. 2004;32:W555–W558. doi: 10.1093/nar/gkh390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binkowski TA, Joachimiak A, Liang J. Protein surface analysis for function annotation in high-throughput structural genomics pipeline. Protein Sci. 2005;14:2972–2981. doi: 10.1110/ps.051759005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;26:320–322. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 6.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH–a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 7.Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 2003;332:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 8.Najmanovich RJ, Allali-Hassani A, Morris RJ, Dombrovsky L, Pan PW, Vedadi M, Plotnikov AN, Edwards A, Arrowsmith C, Thornton JM. Analysis of binding site similarity, small-molecule similarity and experimental binding profiles in the human cytosolic sulfotransferase family. Bioinformatics. 2007;23:e104–e109. doi: 10.1093/bioinformatics/btl292. [DOI] [PubMed] [Google Scholar]
- 9.Stark A, Sunyaev S, Russell RB. A model for statistical significance of local similarities in structure. J. Mol. Biol. 2003;326:1307–1316. doi: 10.1016/s0022-2836(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 10.Tseng YY, Dundas J, Liang J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J. Mol. Biol. 2009;387:451–464. doi: 10.1016/j.jmb.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tseng YY, Li WH. Identification of protein functional surfaces by the concept of a split pocket. Proteins. 2009;76:959–976. doi: 10.1002/prot.22402. [DOI] [PubMed] [Google Scholar]
- 12.Ausiello G, Gherardini PF, Marcatili P, Tramontano A, Via A, Helmer-Citterich M. FunClust: a web server for the identification of structural motifs in a set of non-homologous protein structures. BMC Bioinformatics. 2008;9(Suppl. 2):S2. doi: 10.1186/1471-2105-9-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sael L, Li B, La D, Fang Y, Ramani K, Rustamov R, Kihara D. Fast protein tertiary structure retrieval based on global surface shape similarity. Proteins. 2008;72:1259–1273. doi: 10.1002/prot.22030. [DOI] [PubMed] [Google Scholar]
- 14.Kinoshita K, Murakami Y, Nakamura H. eF-seek: prediction of the functional sites of proteins by searching for similar electrostatic potential and molecular surface shape. Nucleic Acids Res. 2007;35:W398–W402. doi: 10.1093/nar/gkm351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gold ND, Jackson RM. SitesBase: a database for structure-based protein-ligand binding site comparisons. Nucleic Acids Res. 2006;34:D231–D234. doi: 10.1093/nar/gkj062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldenberg O, Erez E, Nimrod G, Ben-Tal N. The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res. 2009;37:D323–D327. doi: 10.1093/nar/gkn822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 18.Tseng YY, Dupree C, Chen ZJ, Li WH. SplitPocket: identification of protein functional surfaces and characterization of their spatial patterns. Nucleic Acids Res. 2009;37:W384–W389. doi: 10.1093/nar/gkp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelsbrunner H, Facello M, Fu P, Liang J. Measuring proteins and voids in proteins. Proc. 28th Ann. Hawaii Int'l; Conf. Syst. Sci. 1995;5:256–264. [Google Scholar]
- 20.Edelsbrunner H, Facello M, Liang J. On the definition and the construction of pockets in macromolecules. Discrete Appl. Math. 1998;88:83–102. [PubMed] [Google Scholar]
- 21.Edelsbrunner H, Mucke E. Three-dimensional alpha shapes. ACM Trans. Graph. 1994;13:43–72. [Google Scholar]
- 22.Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: I. Molecular area and volume through alpha shape. Proteins. 1998;33:1–17. [PubMed] [Google Scholar]
- 23.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodge C, Schneider R, Sander C. The HSSP database of protein structure-sequence alignments and family profiles. Nucleic Acids Res. 1998;26:313–315. doi: 10.1093/nar/26.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujimoto Z, Takase K, Doui N, Momma M, Matsumoto T, Mizuno H. Crystal structure of a catalytic-site mutant alpha-amylase from Bacillus subtilis complexed with maltopentaose. J. Mol. Biol. 1998;277:393–407. doi: 10.1006/jmbi.1997.1599. [DOI] [PubMed] [Google Scholar]
- 27.Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: a Bayesian Monte Carlo approach. Mol. Biol. Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- 28.Weisel M, Proschak E, Schneider G. PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007;1:7. doi: 10.1186/1752-153X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]