

Abstract
Objective:
A consistently identified risk factor for Alzheimer disease (AD) is family history of dementia, with maternal transmission significantly more frequent than paternal transmission. A history of maternal AD may be related to AD-like glucose consumption in cognitively healthy subjects. In this cross-sectional study, we tested whether cognitively healthy people with a family history of AD have less gray matter volume (GMV), an endophenotype for late-onset AD, than individuals with no family history, and whether decreases in GMV are different in subjects with a maternal family history.
Methods:
As part of the Kansas University Brain Aging Project, 67 cognitively intact individuals with a maternal history of late-onset AD (FHm, n = 16), a paternal history of AD (FHp, n = 8), or no parental history of AD (FH−, n = 43), similar in age, gender, education, and Mini-Mental State Examination score, were scanned at 3 T. We used voxel-based morphometry to examine GMV differences between groups, controlling for age, gender, and apoE4.
Results:
Cognitively healthy individuals with a family history of late-onset AD had significantly decreased GMV in the precuneus, middle frontal, inferior frontal, and superior frontal gyri compared with FH− individuals. FHm subjects had significantly smaller inferior frontal, middle frontal, precuneus, and lingual gyri compared with FH− and FHp subjects.
Conclusions:
Overall, maternal family history of Alzheimer disease (AD) in cognitively normal individuals is associated with lower gray matter volume in AD-vulnerable brain regions. These data complement and extend reports of cerebral metabolic differences in subjects with a maternal family history.
GLOSSARY
- AD
= Alzheimer disease;
- BA
= Brodmann area;
- CDR
= Clinical Dementia Rating;
- FH+
= parental family history of Alzheimer disease;
- FH−
= no family history of Alzheimer disease;
- FHm
= maternal family history of Alzheimer disease;
- FHp
= paternal family history of Alzheimer disease;
- GMV
= gray matter volume;
- mtDNA
= mitochondrial DNA;
- TICV
= total intracranial volume.
Alzheimer disease (AD) is the most common neurodegenerative disease. Complex genetic and environmental mechanisms contribute to late-onset AD.1,2 One consistently identified risk factor for AD is family history of dementia.3 Normal individuals with a first-degree relative with AD are at a 4- to 10-fold higher risk of developing AD compared with individuals with no family history.4 Moreover, maternal transmission of AD is significantly more frequent than paternal transmission.5 However, it is still unknown how familial transmission of AD might act biologically to increase risk of late-onset AD.
Epidemiology data indicate that healthy children with a maternal family history of AD may have AD-like disease phenotypes, which are heritable traits found in unaffected family members at a higher rate than in the general population. For example, nondemented offspring of AD-affected mothers perform less well on cognitive testing than offspring of AD-affected fathers.6 A recent brain imaging study found an association between history of maternal AD and AD-like cerebral glucose consumption in cognitively healthy subjects.7 PET scans from subjects with mothers affected with late-onset AD showed reduced metabolic rates for glucose in AD-vulnerable brain regions. Maternal history of AD may also predispose normal individuals to progressive metabolic reductions in AD-vulnerable brain regions.8 Moreover, a recent fMRI study found that both apoE4 and family history affect memory performance in cognitively healthy individuals.9 Although the biologic mechanisms through which a family history of AD influences risk remain unclear, characterizing brain imaging endophenotypes in target populations may be a useful step in identification of risk of developing AD.
Along with PET and fMRI, volumetric MRI is a useful marker of risk and disease progression in AD,10 and MRI traits are informative endophenotypes for basic and clinical studies of AD.11 Voxel-based morphometry (VBM) is a method of processing MRI that can identify small changes in brain volume associated with disease, as well as characterize genetic risk variants responsible for these brain structure changes. We and others have used VBM to characterize regional changes in volume in early AD compared with elderly subjects without dementia.12 In addition, we have reported that the presence of an apoE4 allele in older adults without dementia is associated with imaging markers of risk of AD, namely gray and white matter changes in the medial temporal cortex.13 To date, no studies have assessed whether there are brain volume differences in vivo in cognitively healthy individuals with a family history of late-onset AD. In the present study, we used VBM to test whether cognitively healthy people with a family history of AD have less gray matter volume (GMV), an endophenotype for late-onset AD, than individuals with no family history of AD, and whether these decreases in GMV are different in subjects with a maternal family history.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Kansas University Medical Center Human Subjects Committee. All participants provided informed consent according to institutional guidelines.
Demographics.
Subjects without dementia, aged 60 years and older, were enrolled in the University of Kansas Brain Aging Project. Participants were recruited from a referral-based memory clinic and by media appeals. All subjects received a standard diagnostic evaluation that included medical (history, physical, and laboratory), neuropsychological, and MRI examinations. The absence of cognitive and functional decline was determined by a board-certified neurologist with specialized training in the evaluation of dementia using the Clinical Dementia Rating (CDR).14 All participants included in this analysis had a CDR rating of zero, indicating no dementia. Persons with neurologic disease other than AD, history of ischemic heart disease, history of significant mental illness, diabetes mellitus, or other systemic illness that might impair completion of the study15 were not eligible for the study. All data presented in this study are cross-sectional.
Subjects completed thorough family history examinations with a nurse clinician. A family history of dementia included at least 1 first-degree relative whose dementia onset was between ages 60 and 80 years, and was taken using a standard family history form filled out by a nurse clinician. Participants self-reported names, dates of birth, age at death, age at onset of disease, and clinical information of affected and unaffected family members. Subjects were not included if both of their parents had not lived to the age at risk of late-onset AD (i.e., 60 years) or if both parents had late-onset AD. Only subjects with a positive family history with a single parent affected with AD (FH+) were included in the present study. These subjects were divided into maternal (FHm; only the mother was affected with AD) and paternal (FHp; only the father was affected with AD) family history groups and compared with subjects without a family history of dementia (FH−). Of 67 individuals with complete family history data, 16 cognitively intact individuals had a maternal history of late-onset AD, 8 had a paternal history of AD, and 43 had no parental history of AD.
ApoE genotypes were determined by restriction enzyme isotyping. For these 67 subjects, 21 carried an apoE4 allele (ɛ2/ɛ4, n = 2; ɛ4/ɛ3, n = 17; ɛ4/ɛ4, n = 2) and 46 did not (ɛ2/ɛ3, n = 10; ɛ3/ɛ3, n = 36).
Cognitive measures.
A trained psychometrician administered a psychometric battery including standard measures of memory, language, executive function, and visuospatial ability (tests described in detail elsewhere16). The Mini-Mental State Examination17 was administered as a measure of global cognition. All cognitive performance scores were converted to Z scores based on the mean and SD of a larger cohort of individuals without dementia, described previously.16 The mean of each participant's Z scores was determined to create an index of global cognitive performance.
Imaging.
Structural MRI data were obtained using a Siemens 3.0-T Allegra MRI Scanner. High-resolution T1-weighted anatomic images were acquired (magnetization-prepared rapid gradient echo; 1 × 1 × 1 mm3 voxels, repetition time = 2,500, echo time = 4.38 milliseconds, inversion time = 1,100 milliseconds, field of view = 256 × 256, flip angle = 8 degrees) and processed for voxel-based analysis. Every scan was checked for image artifacts and gross anatomic abnormalities. Data analysis for 67 subjects was performed using the VBM5 toolbox (http://dbm.neuro.uni-jena.de), an extension of the SPM5 algorithms (Wellcome Department of Cognitive Neurology, London, UK) running under Matlab 7.1 (The MathWorks, Natick, MA) on Linux. Processing for VBM has been detailed elsewhere12; in short, we used unified segmentation with hidden Markov random field, priors, nonlinear modulation, saved images with affine registration only, and smoothed at 10-mm full-width at half-maximum gaussian kernel. Total gray matter, white matter, and total intracranial volume (TICV) were computed in cubic centimeters using the normalized tissue maps of each study participant. Modulated outputted images are corrected for nonlinear warping, effectively globally scaling data for TICV. Thus, TICV is not included in the statistics as a global scalar.
Demographic statistical analyses.
SPSS 16.0 (SPSS Inc., Chicago, IL) was used for all statistical analysis outside of imaging space. Continuous demographic and imaging variables were compared between the family history groups using analysis of variance. The χ2 test was used to compare categorical variables between groups.
VBM statistics.
We used VBM to examine regional brain volume differences between groups. We used a full-factorial model (a 3-sample t test), and included age, gender, and apoE4 carrier status as confounding variables. First, we examined whether there were significant GMV differences between FH− and FH+ (FHm and FHp combined) groups. Second, we examined whether there were parent gender effects on GMV by comparing the 3 groups (FH− vs FHp vs FHm). For all analyses, results were considered significant at p < 0.05 after correction for multiple comparisons (family-wise error) according to the small volume correction, with clusters exceeding an extent threshold of 100 voxels and Z > 3.0. To focus our analyses on AD-related regions, we used a masking image of regional GMVs from a previous study. It included regions significantly decreased in subjects with early AD compared with elderly subjects without dementia,12 namely the medial temporal lobe (hippocampus and parahippocampal gyrus), superior, inferior, and middle temporal gyrus (Brodmann area [BA] 20/21/22/37), right insula (BA 13), left cingulate (BA 31), bilateral middle frontal gyrus (BA 6/9), bilateral fusiform gyrus (BA 37/BA 19), and inferior frontal cortex (BA 45/46). The mask was then applied to the full volume of data for each contrast. Voxels are reported with reference to the Montreal Neurological Institute (MNI) standard space within SPM5.18 As a secondary analysis, we compared only the apoE-negative family history groups (FH− vs FHp vs FHm) to verify that decreased volume in the FHm group was not driven solely by a presence of an apoE ɛ4 allele.
RESULTS
Demographic statistical analysis.
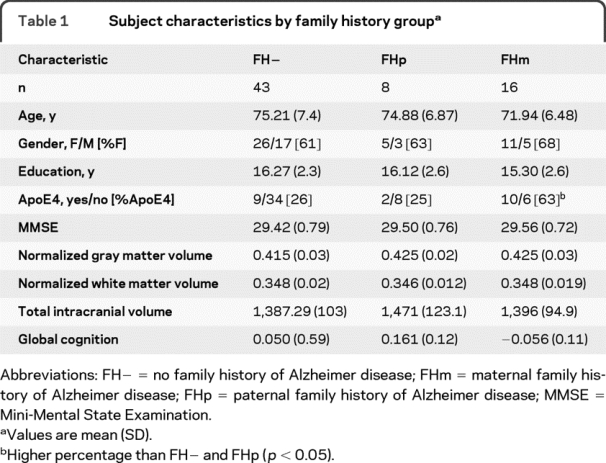
The mean age of the cohort (n = 67) was 74.4 (SD 7.2) years, with no significant difference in age among FH− (n = 43), FHp (n = 8), and FHm (n = 16). Groups were similar for gender distribution, years of education, and global cognition. There were no significant differences in normalized whole brain volume or gray or white matter volume. The FHm group had a significantly greater number of apoE4 alleles than the FHp and FH− groups (table 1).
Table 1 Subject characteristics by family history group
Imaging measures.
As compared with FH− subjects, FH+ subjects had significantly decreased GMV in the precuneus, middle frontal gyrus, inferior frontal gyrus, and superior frontal gyrus (table 2). Furthermore, comparison of the 3 groups (FH− vs FHp vs FHm) revealed that the prefrontal cortex decreases in volume were driven by the FHm group (table 3, figure 1, and figure 2, A and B). More specifically, FHm subjects had significantly smaller right inferior frontal, middle frontal gyri, and left insula GMV when compared with the FH− group. FHm subjects also had significantly less gray matter in the right lingual gyrus, right inferior frontal gyrus, and right middle frontal gyrus when compared with FHp subjects (table 3 and figure 1).
Table 2 Brain regions showing significant gray matter volume reductions in FH+ subjects compared with FH− subjects from global voxel-based morphometry analysis, controlling for age, gender, apoE4, and total intracranial volume
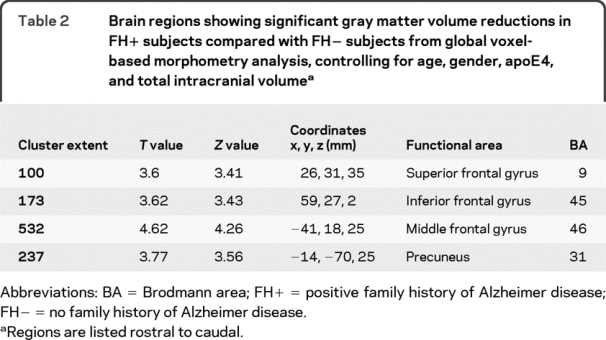
Table 3 Brain regions showing significant gray matter volume reductions in FHm subjects compared with FHp subjects and FH− from global voxel-based morphometry analysis, controlling for age, gender, apoE4, and total intracranial volume


Figure 1 Statistical parametric maps showing gray matter volume reductions in normal FH+ subjects compared with FH− subjects
The first 2 rows display maps from subjects with a maternal family history of Alzheimer disease (FHm) as compared with subjects with no family history (FH−) (A) and subjects with a paternal family history (FHp) (B). Row C shows gray matter volume (GMV) reductions in apoE4-negative FHm subjects compared with apoE4-negative FH− subjects. Anatomic location and description of brain regions for A and B are in table 3. Statistical parametric maps showing GMV reductions in normal FHp subjects as compared with FH− subjects are in row D. Anatomic location and description of brain regions are in table e-1. Areas of gray matter volume decrease are represented on purple–to–yellow, blue–to–light blue, dark orange–to–yellow, and green–to–light green color-coded scales for the 4 contrasts, reflecting Z scores between 2 and 5 for the upper contrast and between 2 and 4 for the lower 3 contrasts. Areas of gray matter volume decrease are displayed on a standardized spatially normalized MRI.

Figure 2 Selected volume parameter estimates from voxel-based morphometry analyses
The graphs show the corresponding parameter estimates for key regions identified in figure 1, representing group differences in family history–related gray matter volumes. Error bars are ±1 SEM. Gray matter volume (GMV) was reduced in those with a maternal family history of Alzheimer disease (FHm) vs those with a paternal family history (FHp) and no family history (FH−) in the inferior frontal gyrus (A), middle frontal gyrus (B), and precuneus (C). The FHp group demonstrated reduced GMV compared with the FH− group but not FHm in the inferior precuneus (D). See tables 3 and e-1 for statistics.
As compared with the FH− group, the FHp group had regional decreases in GMV in the superior frontal gyrus, precuneus (figure 2D), left, middle frontal gyrus, and right fusiform gyrus (table e-1 on the Neurology® Web site at www.neurology.org and figure 1D). However, there were no GMV regional decreases in the FHp group compared with the FHm group.
There were a few GMV reductions in the FH− group compared with the FH+ group in the left insula and left inferior frontal gyrus (table e-2). These regions were not significant in the individual contrasts, which tested whether there were regional GMV decreases in subjects with a paternal family history or a maternal family history compared with subjects with no family history (FHp > FH− and FHm > FH−).
When the analysis was restricted to the apoE4 noncarriers, the FHm group still showed GMV reductions compared with the FH− group in areas overlapping with the larger analysis, namely the bilateral middle frontal gyrus and right inferior frontal gyrus, with additional decreases in the precuneus, bilateral superior frontal gyrus, and middle temporal gyrus (figure 1C). The apoE ɛ4-negative FHm group still showed GMV reductions compared with the FHp group in the same regions as above in the left inferior frontal gyrus, lingual gyrus, and middle frontal gyrus. Additional clusters that reached significance were in the precuneus (figure 2C) and right inferior frontal gyrus.
DISCUSSION
We used VBM techniques to show that cognitively normal individuals with a parent with AD, especially a mother, have reduced GMV in AD-vulnerable brain regions compared with cognitively normal individuals with no family history. More specifically, individuals with a maternal family history of AD had greater GMV reductions in the prefrontal cortices (BA 45, 46, and 47) and the precuneus. These effects remained significant after accounting for potential risk factors for late-onset AD, such as age, gender, and apoE4 genotype. Although we found more frontal than temporal regional GMV decreases in the FHm group, these findings complement and extends reports of an AD endophenotype in FHm but not FHp cognitively normal individuals.7,8,19
Healthy aging is typically associated with some brain atrophy, increases in MRI white matter signal intensity, and decreases in cognitive functioning.20 Advancements in neuroimaging have improved our ability to distinguish visible markers of early transition to AD apart from normal aging. A recent report from the Alzheimer's Disease Neuroimaging Initiative showed that participants with mild cognitive impairment who later converted to AD had lower regional GMV in a number of brain regions at baseline, including temporal, parietal, and frontal cortices.21 GMV reductions in the earliest stages of AD are most often reported in the hippocampus, as well as the precuneus, a region involved in visuospatial processes.22,23 VBM is a sensitive method for identifying regional gray matter atrophy in progressing AD subjects.24 In this study, we used VBM methods and report decreased GMV in subjects with a maternal family history in regions previously shown to be affected in subjects with early AD.12 Our data are consistent with volumetric studies characterizing regions vulnerable to atrophy in the earliest stages of AD.23,25
We report atrophy in the prefrontal cortex of FHm individuals compared with both FHp and FH− groups, which complements and extends PET data showing prefrontal hypometabolism in FHm individuals over time.8 We found that subjects in the paternal family history group had reduced GMV in the prefrontal cortex and precuneus compared with FH− subjects. The paternal group, however, did not have any decreases in GMV compared with the maternal family history group, which would support data showing that FHm subjects have a stronger imaging endophenotype of AD7,8 and that there is a higher mother-to-father ratio among affected parents of subjects with AD.26 We will need to complete a longitudinal examination of our subjects to determine whether the presently reported GMV deficits predispose FHm individuals to additional atrophy in AD-vulnerable regions. It will also be crucial for future studies to analyze whether maternal family history of AD influences disease severity, rate of cognitive decline, or age at onset in subjects with a diagnosis of AD.
Accumulating literature suggests that having an AD-affected father or mother increases one's risk of late-onset, sporadic AD, although the risk is greater when the mother is the affected parent. For example, in a cohort of individuals with AD and a positive family history, the mother was more likely to be the affected parent than the father.26 Both PET and neuropsychological data suggest that FHm status has a greater impact on brain physiology and cognitive function than FHp status.6,7 Gender-specific inheritance phenomena are associated with several genetic paradigms, such as X-linked inheritance, sex-specific imprinting, and mitochondrial DNA (mtDNA) transmission. Currently, there are several lines of evidence supporting a role for mtDNA transmission in AD. Mitochondrial DNA encodes catalytic sites for the enzyme cytochrome oxidase, and cytochrome oxidase activity is reduced in AD.27 Neuronal nuclear genes influencing mitochondrial energy metabolism are underexpressed in AD, particularly in regions like the precuneus.28 Brains pathologically diagnosed with AD have significantly more abundant low-level heteroplasmic mutations in the mtDNA d-loop region,29 and higher levels of the 5-kd “common” mtDNA deletion compared with control brains.30,31 Studies of cytoplasmic hybrids (cybrids) in neuronal cell lines have demonstrated that cell lines expressing AD subject platelet mtDNA have lower mtDNA-related cytochrome oxidase activity, supporting the possibility that mtDNA might differ between AD and control subjects.32,33 Furthermore, AD-related alterations in mtDNA content are neuroanatomically specific to the hippocampus, frontal, and temporal cortices.34
While GMV differences in regions of AD-related atrophy were observed in FH+ participants, we did not find significant GMV changes in the hippocampus, a region most commonly associated with AD-related volumetric changes. AD-related hippocampal atrophy is typically associated with clinically evident cognitive changes (i.e., memory loss); however, our participants did not have dementia or evidence of cognitive or functional decline. There were no significant differences between any groups in our standardized global cognitive measure, consistent with the cross-sectional and longitudinal PET studies of family history. Larger hippocampal volume35 and hippocampal neuronal cells36 are associated with preserved cognition and function in individuals with the presence of high AD neuropathologic burden. So while these individuals may be genetically at risk of AD and demonstrate atrophy in AD-vulnerable regions, protective or compensatory mechanisms may be playing a role maintaining normal cognitive function. This is further suggested by the larger regional volumes in the left insula and prefrontal cortex in FH+ compared with FH− groups. It will be important to replicate these findings in larger community-based samples and longitudinal assessments of the relationship of family with hippocampal atrophy.
It is interesting that our FHm group had significantly more E4 allele carriers than the FHp and FH− groups, similar to another report of higher apoE ɛ4 frequency in an FHm group.37 We and others have found reduced hippocampal GMV in cognitively normal elderly subjects with an apoE4 allele.13,38,39 In the current study, we both controlled for apoE4 and analyzed apoE4 groups separately, which demonstrated that the present findings were not accounted for by apoE4. Excluding FH+ subjects whose parents had dementia only after age 80 years may artifactually reduce the total number of FHm-eligible subjects, and thus we may have selectively enriched for apoE ɛ4 alleles in the FHm group. Moreover, the apoE4 genotype is often overrepresented in subjects who have a family member with AD, and this family history might lead participants to seek involvement in memory studies such as ours.
Our study is limited by a lack of neuropathologic confirmation of parental AD, and it is possible that parents who developed dementia by history may have had another neurodegenerative disorder or a nondegenerative cause of dementia. If the relatives of our subjects did not have AD, it would have likely reduced our ability to detect group differences. family history questionnaires such as ours, however, have been shown to agree with neuropathologic findings.40 Moreover, it is possible that there is a censoring bias in assigning family history groups to individuals whose parents died at an earlier age. Additionally, our study is limited by a cross-sectional design, and further longitudinal evaluations will be important to confirm these findings. The small sample size may have limited our power to resolve volumetric group differences, in particular in the FHp group. Despite these limitations, the regional specificity of our findings in AD-vulnerable regions observed in individuals without dementia and with a maternal family history of AD complement and extend reports of cerebral metabolic differences in subjects with a maternal family history.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by Robyn Honea and Eric Vidoni.
ACKNOWLEDGMENT
The authors thank members of the Kansas University Brain Aging Project team, especially Amith Harsha, Pat Laubinger, Phyllis Switzer, Diane Cunningham, and George Thomas for their assistance with data collection and study as well as helpful suggestions. The authors also thank the research participants of the Kansas University Brain Aging Project for their generosity of time and spirit, which makes this research possible.
DISCLOSURE
Dr. Honea reports no disclosures. Dr. Swerdlow has served on speakers' bureaus for Pfizer Inc and Accera, Inc.; and received a speaker honorarium from Medivation, Inc. Dr. Vidoni receives research support from the Foundation for Physical Therapy. Ms. Goodwin reports no disclosures. Dr. Burns has served on a scientific advisory board for the American Academy of Physician Education; has received royalties from publishing Early Diagnosis and Treatment of Mild Cognitive Impairment (Wiley Press, 2008) and Dementia: An Atlas of Investigation and Diagnosis (Clinical Publishing, Oxford, England, 2007); serves on speakers' bureaus for Pfizer Inc and Novartis; has served as a consultant for Medacorp Consulting and Johnson County Clinical Trials; receives research support from Elan Corporation, Danone, and the Dana Foundation; and has served as an expert witness in legal proceedings regarding competency.
Supplementary Material
Address correspondence and reprint requests to Dr. Robyn A. Honea, University of Kansas School of Medicine, Department of Neurology, 2100 West 36th Ave., Suite 110, Kansas City, KS 66160 rhonea@kumc.edu or jburns2@kumc.edu
Supplemental data at www.neurology.org
Study funding: Supported by grants R03 AG026374, R21 AG029615, and R01 AG022407 from the National Institutes of Aging and K23NS058252 from the National Institute on Neurological Disorders and Stroke. The University of Kansas General Clinical Research Center (M01RR023940) provided essential space, expertise, and nursing support. The Hoglund Brain Imaging Center is supported by grant C76 HF00201.
Disclosure: Author disclosures are provided at the end of the article.
Received July 9, 2009. Accepted in final form October 21, 2009.
REFERENCES
- 1.Reiman EM. Linking brain imaging and genomics in the study of Alzheimer's disease and aging. Ann NY Acad Sci 2007;1097:94–113. [DOI] [PubMed] [Google Scholar]
- 2.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 3.Breitner JC, Silverman JM, Mohs RC, Davis KL. Familial aggregation in Alzheimer's disease: comparison of risk among relatives of early-and late-onset cases, and among male and female relatives in successive generations. Neurology 1988;38:207–212. [DOI] [PubMed] [Google Scholar]
- 4.Cupples LA, Farrer LA, Sadovnick AD, et al. Estimating risk curves for first-degree relatives of patients with Alzheimer's disease: the REVEAL study. Genet Med 2004;6:192–196. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Tortosa E, Barquero MS, Baron M, et al. Variability of age at onset in siblings with familial Alzheimer disease. Arch Neurol 2007;64:1743–1748. [DOI] [PubMed] [Google Scholar]
- 6.Wolf PA, Beiser A, Au R, et al. Parental occurrence of dementia linked to lower cognitive function in the Framingham Offspring Study. Neurology 2005;64:A267–A268. [Google Scholar]
- 7.Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer's disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci USA 2007;104:19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosconi L, Mistur R, Switalski R, et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 2009;72:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu G, McLaren DG, Ries ML, et al. The influence of parental history of Alzheimer's disease and apolipoprotein E epsilon4 on the BOLD signal during recognition memory. Brain 2009;132:383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 2004;62:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuenco KT, Green RC, Zhang J, et al. Magnetic resonance imaging traits in siblings discordant for Alzheimer disease. J Neuroimaging 2008;18:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honea RA, Thomas GP, Harsha A, et al. Cardiorespiratory fitness and preserved medial temporal lobe volume in Alzheimer's Disease. Alzheimer Dis Assoc Disord 2009;23:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honea RA, Vidoni E, Harsha A, Burns JM. Impact of APOE on the healthy aging brain: a voxel-based MRI and DTI study. J Alzheimers Dis Epub 2009 Jul 7. [DOI] [PMC free article] [PubMed]
- 14.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 15.Burns JM, Donnelly JE, Anderson HS, et al. Cardiorespiratory fitness and brain atrophy in early Alzheimer's disease. Neurology 2008;71:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burns JM, Donnelly JE, Anderson HS, et al. Peripheral insulin and brain structure in early Alzheimer disease. Neurology 2007;69:1094–1104. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. Mini-Mental State: A practical method for grading the cognitive state of patients for the clinicians. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 18.Honea RA, Meyer-Lindenberg A, Hobbs KB, et al. Is gray matter volume an intermediate phenotype for schizophrenia? A voxel-based morphometry study of patients with schizophrenia and their healthy siblings. Biol Psychiatry 2008;63:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SC, Schmitz TW, Trivedi MA, et al. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci 2006;26:6069–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramer JH, Mungas D, Reed BR, et al. Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology 2007;21:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misra C, Fan Y, Davatzikos C. Baseline and longitudinal patterns of brain atrophy in MCI patients, and their use in prediction of short-term conversion to AD: results from ADNI. Neuroimage 2009;44:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karas GB, Burton EJ, Rombouts SA, et al. A comprehensive study of gray matter loss in patients with Alzheimer's disease using optimized voxel-based morphometry. Neuroimage 2003;18:895–907. [DOI] [PubMed] [Google Scholar]
- 23.Karas G, Scheltens P, Rombouts S, et al. Precuneus atrophy in early-onset Alzheimer's disease: a morphometric structural MRI study. Neuroradiol 2007;49:967–976. [DOI] [PubMed] [Google Scholar]
- 24.Kinkingnehun S, Sarazin M, Lehericy S, et al. VBM anticipates the rate of progression of Alzheimer disease: a 3-year longitudinal study. Neurology 2008;70:2201–2211. [DOI] [PubMed] [Google Scholar]
- 25.Mosconi L, Sorbi S, de Leon MJ, et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer's disease. J Nucl Med 2006;47:1778–1786. [PubMed] [Google Scholar]
- 26.Edland SD, Silverman JM, Peskind ER, et al. Increased risk of dementia in mothers of Alzheimer's disease cases: evidence for maternal inheritance. Neurology 1996;47:254–256. [DOI] [PubMed] [Google Scholar]
- 27.Parker WD Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology 1990;40:1302–1303. [DOI] [PubMed] [Google Scholar]
- 28.Liang WS, Reiman EM, Valla J, et al. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci USA 2008;105:4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA 2004;101:10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamblet NS, Castora FJ. Elevated levels of the Kearns-Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer's patients. Mutat Res 1997;379:253–262. [DOI] [PubMed] [Google Scholar]
- 31.Corral-Debrinski M, Horton T, Lott MT, et al. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994;23:471–476. [DOI] [PubMed] [Google Scholar]
- 32.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res 2007;85:3416–3428. [DOI] [PubMed] [Google Scholar]
- 33.Swerdlow RH, Parks JK, Cassarino DS, et al. Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology 1997;49:918–925. [DOI] [PubMed] [Google Scholar]
- 34.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 2001;21:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erten-Lyons D, Woltjer RL, Dodge H, et al. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 2009;72:354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iacono D, Markesbery WR, Gross M, et al. The Nun Study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology 2009;73:665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duara R, Barker WW, Lopez-Alberola R, et al. Alzheimer's disease: interaction of apolipoprotein E genotype, family history of dementia, gender, education, ethnicity, and age of onset. Neurology 1996;46:1575–1579. [DOI] [PubMed] [Google Scholar]
- 38.Reiman EM, Uecker A, Caselli RJ, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer's disease. Ann Neurol 1998;44:288–291. [DOI] [PubMed] [Google Scholar]
- 39.Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the ɛ4 allele for apolipoprotein E. N Engl J Med 1996;334:752–758. [DOI] [PubMed] [Google Scholar]
- 40.Kawas C, Segal J, Stewart WF, et al. A validation study of the Dementia Questionnaire. Arch Neurol 1994;51:901–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.