

Abstract
Background:
THAP1 encodes a transcription factor (THAP1) that harbors an atypical zinc finger domain and regulates cell proliferation. An exon 2 insertion/deletion frameshift mutation in THAP1 is responsible for DYT6 dystonia in Amish-Mennonites. Subsequent screening efforts in familial, mainly early-onset, primary dystonia identified additional THAP1 sequence variants in non-Amish subjects.
Objective:
To examine a large cohort of subjects with mainly adult-onset primary dystonia for sequence variants in THAP1.
Methods:
With high-resolution melting, all 3 THAP1 exons were screened for sequence variants in 1,114 subjects with mainly adult-onset primary dystonia, 96 with unclassified dystonia, and 600 controls (400 neurologically normal and 200 with Parkinson disease). In addition, all 3 THAP1 exons were sequenced in 200 subjects with dystonia and 200 neurologically normal controls.
Results:
Nine unique melting curves were found in 19 subjects from 16 families with primary dystonia and 1 control. Age at dystonia onset ranged from 8 to 69 years (mean 48 years). Sequencing identified 6 novel missense mutations in conserved regions of THAP1 (G9C [cervical, masticatory, arm], D17G [cervical], F132S [laryngeal], I149T [cervical and generalized], A166T [laryngeal], and Q187K [cervical]). One subject with blepharospasm and another with laryngeal dystonia harbored a c.-42C>T variant. A c.57C>T silent variant was found in 1 subject with segmental craniocervical dystonia. An intron 1 variant (c.71+9C>A) was present in 7 subjects with dystonia (7/1,210) but only 1 control (1/600).
Conclusions:
A heterogeneous collection of THAP1 sequence variants is associated with varied anatomical distributions and onset ages of both familial and sporadic primary dystonia.
GLOSSARY
- HRM
= high-resolution melting;
- PD
= Parkinson disease;
- THAP
= thanatos-associated protein;
- UTR
= untranslated region.
THAP1 has joined the dystonia family.1 An exon 2 insertion/deletion frameshift mutation in THAP1 is responsible for DYT6 dystonia in Amish-Mennonites.1 Identification of a different exon 2 mutation (F81L) in a German family suggested that sequence variants in THAP1 may contribute to the development of dystonia in diverse populations.1 In 2 follow-up studies, 11 additional sequence variants were identified in familial, mainly early-onset, primary dystonia.2,3
In most neurology subspecialty practices, the majority of patients with dystonia are adults with largely sporadic focal or segmental involvement. Approximately 8% to 27% of these late-onset probands have at least 1 first-degree relative with dystonia.4–8 These data suggest the possibility that adult-onset primary dystonia is due, in large part, to sequence variants of low penetrance in a distinct collection of genes. In support of this hypothesis, DYT1, DYT5, DYT6, DYT11, and DYT12 dystonia exhibit variable expressivity and incomplete penetrance.9 Late-onset DYT1 dystonia usually manifests as hand-forearm dystonia and rarely generalizes. Similarly, patients with DYT11 may develop late-onset hand-forearm or cervical dystonia. However, screening efforts have shown that DYT1 and DYT11 mutations are only rarely associated with adult-onset sporadic dystonia.10–12
In contrast to DYT1 and DYT11, DYT6 dystonia commonly affects muscles of the head, neck, and larynx, with relatively less limb involvement.13,14 Moreover, in comparison with DYT1, DYT6 dystonia seems to exhibit a later age-of-onset and often remains focal or segmental in distribution.1,2,13,14 Based on these considerations, we sought to identify THAP1 sequence variants in a large cohort of subjects with primary, mainly adult-onset dystonia.
METHODS
Standard protocol approvals, registrations, and patient consents.
All human studies were performed in accordance with institutional review board guidelines, and all subjects gave informed consent.
Subjects.
Subjects with dystonia and neurologically normal controls were acquired from outpatient clinics at participating institutions and support group meetings of the National Spasmodic Dysphonia Association, National Spasmodic Torticollis Association, Benign Essential Blepharospasm Research Foundation, Dystonia/Spasmodic Torticollis, and Dystonia Medical Research Foundation. Subjects acquired at support group meetings were examined by M.S.L. Subjects with Parkinson disease (PD) were recruited from the clinics of M.S.L. and R.F.P. Clinical diagnoses were made by means of history and examination by 1 or more neurologists or neurolaryngologists at each institution. Neurologically normal controls were defined as individuals with no personal or first-degree family history of movement or neurodegenerative disorder. All controls acquired at the University of Tennessee Health Science Center and support group meetings were examined by M.S.L. or R.F.P. Dystonia was classified in accordance with established schemes.15,16 Subjects with known DYT1 dystonia were not recruited into our study.
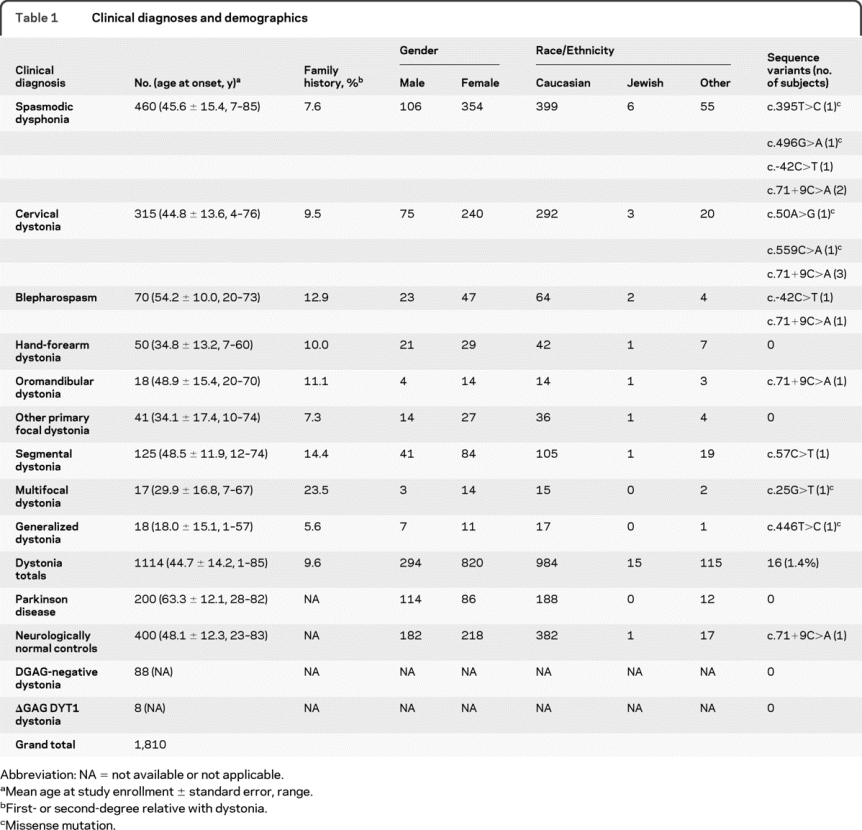
Clinical diagnoses for 1,810 subjects interrogated with high-resolution melting (HRM) appear in table 1. Table 1 does not include family members of probands with THAP1 sequence variants. Of the 400 neurologically normal control subjects, 184 were obtained from Coriell Institute for Medical Research (neurologically normal Caucasian control panels NDPT020 and NDPT024). Demographic information and dystonia distribution were not available for 96 DNA samples received from Athena Diagnostics. The panel from Athena included 8 samples with confirmed DYT1 ΔGAG deletions and 88 ΔGAG-negative samples associated with a clinical diagnosis of dystonia. Either in-person clinical evaluations or detailed telephone interviews were conducted with available family members of probands with THAP1 sequence variants.
Table 1 Clinical diagnoses and demographics
DNA isolation and mutation analysis.
DNA was extracted from peripheral blood leukocytes using a DNA Isolation Kit for Mammalian Blood (Roche, Morristown, NJ). The Oragene™ DNA Self-Collection Kit (Genotek, Kanata, Ontario, Canada) was used to acquire DNA from family members unable to visit one of the participating clinical sites.
DNA quantity and quality were analyzed with a NanoDrop ND-100 spectrophotometer (NanoDrop Technologies, Wilmington, DE), Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen, Carlsbad, CA), and agarose gel electrophoresis. Poor-quality samples were rescued by whole genome amplification with a REPLI-g® Mini Kit (Qiagen, Valencia, CA). Samples that could not be rescued were not used for HRM and do not appear in table 1.
HRM analyses were performed using the LightCycler® 480 Real-Time PCR system and HRM Master Mix (Roche Applied Science, Indianapolis, IN) in accordance with manufacturer instructions and our laboratory protocol.12 With Primer3 (frodo.wi.mit.edu), PCR primers were placed on flanking intronic and untranslated regions (UTRs) to encompass the coding regions of the 3 THAP1 exons (table e-1 on the Neurology® Web site at www.neurology.org). Optimized HRM reactions were performed in 96-well plates using 20 ng of template DNA, 1X HRM Master Mix, 2.5 mM MgCl2, and 200 nM of each primer in a 20-μL reaction volume. Detailed PCR cycling and HRM conditions are presented in the e-Methods.
All samples were run in duplicate. Using LightCycler® 480 Gene Scanning Software (Roche Applied Science), melting curves and difference plots were analyzed by 3 investigators (Y.Z., S.G., and M.S.L.) blinded to phenotype. All samples were unambiguously assigned to genotypes by Gene Scanning software. Then, we sequenced samples with shifted melting curves. For sequencing, 5 μL of the PCR products were cleaned using ExoSAP-IT® (United States Biochemical, Cleveland, OH). Then, 1 to 2 μL of the purified PCR products were sequenced in the forward and reverse directions using a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA). In addition, to evaluate the sensitivity and specificity of HRM, all 3 THAP1 exons were sequenced in 200 neurologically normal controls and 200 subjects with dystonia.
Statistics.
The Fisher exact test was used to evaluate association of the c.71+9C>T sequence variant with dystonia.
In silico analyses.
PMUT and PolyPhen were used to predict the pathologic character of single amino acid mutations. PMUT (mmb2.pcb.ub.es:8080/PMut) is a Web-based tool used for the annotation of pathologic changes in proteins.17 PMUT is based on the use of neural networks trained to detect pathologic missense mutations and has shown a success rate of greater than 80%. PMUT final output includes a pathogenicity prediction along with a confidence index ranging from 0 (low) to 9 (high). PolyPhen (genetics.bwh.harvard.edu/pph) predicts the possible impact of an amino acid substitution on the function of a human protein based on empirical rules applied to the amino acid sequence, phylogenetic profile scores, and calculation of structural parameters.18 PolyPhen output includes position-specific independent counts scores. The Kyte and Doolittle hydrophobicity scale was also used for analysis of individual amino acid changes.19 Protein sequence alignment was performed with ClustalW2 (www.ebi.ac.uk/Tools/clustalw2/).
RESULTS
High-resolution melting.
HRM robustly distinguished sequence variants from control DNA (figure 1). Furthermore, Gene Scanning software clustered sequence variants into discrete groups (figure 1B). Based on follow-up sequencing of samples exhibiting shifted melting curves and sequencing data from 200 neurologically normal controls and 200 subjects with dystonia, HRM showed 100% diagnostic specificity and sensitivity.

Figure 1 Detection of THAP1 sequence variants with high-resolution melting
Normalized and temperature-shifted high-resolution melting curves (A) and difference plots (B) of exon 1 differentiate 4 sequence variants from 1 normal control (each in duplicate).
THAP1 mutations in primary dystonia.
Among 1,210 subjects with dystonia, 200 subjects with PD, and 400 neurologically normal controls, 9 distinct shifted melting curve profiles were identified in 16 individuals with primary dystonia and 1 control (tables 1 and 2). All shifted melting curves were due to single base substitutions (table 2 and figure 2). No deletions or insertions were identified with sequencing. Single base substitutions were found in the 5′UTR (c.-42C>T), exon 1 (c.25G>T, c.50A>G, c.57C>T), intron 1 (c.71+9C>A), and exon 3 (c.395T>C, c.446T>C, c.496G>A, c.559C>A) of THAP1. No sequence variants were localized to exon 2. None of the subjects harboring sequence variants were of Amish descent. Among the 18 affected subjects from 16 families, 14 were female (78%) and 4 were male (22%). Sites of onset included arm (2/18), leg (1/18), cervical (6/18), laryngeal (6/18), upper face (2/18), and jaw (1/18). Dystonia remained focal in 15 of 18 (83%) at most recent anatomical classification. The mean age at onset was 48 years and ranged from 8 to 69 years. All but 2 subjects had late-onset (>20 years) dystonia.
Table 2 Phenotypes, genotypes and in silico analysis of missense mutations
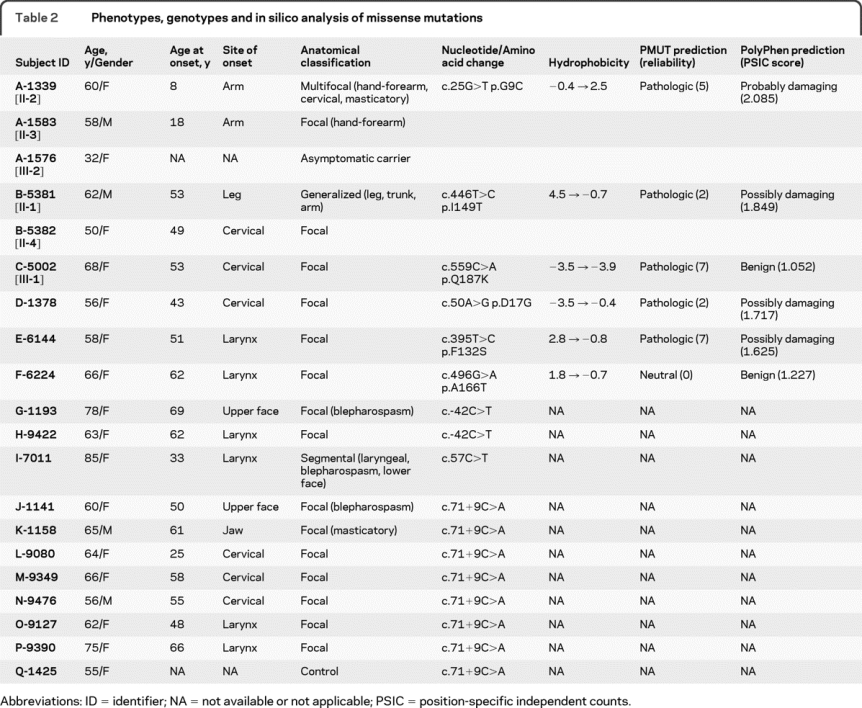

Figure 2 THAP1, THAP1, and localization of mutations in primary dystonia
(A) Genomic structure of THAP1 and contiguous regions (NT 007995), 3-exon transcript (NM 018105), and translated protein (NP 060575) along with the locations of sequence variants. NLS = nuclear localization signal; Pro = proline-rich region; THAP = thanatos-associated protein. (B) Alignment of exon 1 and the 5′ portion of intron 1 in human, mouse, and rat. Sequence variants are highlighted in yellow (missense) or turquoise. Exonic nucleotides, uppercase; intronic nucleotides, lowercase. (C) Alignment of THAP1 homologues with ClustalW2: AVFPMILW, red (small + hydrophobic including aromatic − Y); DE, blue (acidic); RK, magenta (basic); STYHCNGQ, green (hydroxyl + amine + basic + Q). ClustalW2 generates the following symbols denoting the degree of conservation observed in each column: asterisk (*) means that residues in that column are identical in all sequences, period (.) means that conserved substitutions have been observed, and colon (:) means that semiconserved substitutions are observed. Sequence variants are highlighted.
Nine non-Jewish Caucasian subjects from 6 families were found to harbor unique missense mutations in conserved regions of THAP1 (G9C, D17G, F132S, I149T, A166T, and Q187K). Families A, B, and C have a strong family history of dystonia (figure 3). Based on in silico analysis with PMUT, 5 of 6 variants were predicted to be pathologic. Although PMUT classified the A166T variant as neutral, the reliability of this call was 0. PolyPhen predicted that 4 of these 6 variants were either possibly or probably damaging.

Figure 3 Partial pedigrees of multiplex families with missense mutations in THAP1
Genotypes confirmed by sequencing are shown below individual family members.
In family A, the proband, a 61-year-old woman (family A [II-2], figure 3), first noted difficulty writing at age 8 years. Her hand-forearm dystonia persisted into her adult years and has remained task specific. Cervical and jaw-opening masticatory dystonia became manifest at ages 35 and 60 years, respectively. She has shown consistent benefit from injections of botulinum toxin type A for treatment of her cervical and masticatory dystonia. Her mother (deceased) reportedly had childhood-onset unilateral hand tremor and writer's cramp. The proband has 2 sisters and 1 brother. Her brother has writer's cramp. One of the proband's 2 daughters harbors the c.25G>T variant but is asymptomatic at age 32 years. The c.25G>T missense mutation in exon 1 of THAP1 leads to substitution of glycine with a hydrophobic cysteine (G9C) residue in the highly conserved THAP domain of THAP1 (figure 2C).
In family B, the proband, a 63-year-old man (family B [II-1], figure 3), has a 9-year history of slowly progressive left leg and truncal dystonia. On neurologic examination, mild right arm dystonia was noted, particularly during ambulation. The c.446T>C sequence variant found in this subject leads to substitution of a strongly hydrophobic isoleucine residue with a threonine (I149T) in the nuclear localization signal domain of THAP1 (figure 2C). The proband's father was diagnosed with cervical dystonia, allegedly after a neck injury, and died at age 58 years from suicide in the setting of significant disability from long-standing cervical dystonia. The proband also has a 50-year-old sister with cervical dystonia (II-4, figure 3) who harbors the same c.446T>C sequence variant. By report, none of their children exhibit dystonia. However, these and other family members were not accessible for neurologic examination and acquisition of DNA.
In family C, the proband, a 71-year-old woman (family C [III-1], figure 3), had onset of cervical dystonia at age 53 years. Her sequence variant is located at the C terminus of THAP1 (c.559C>A) and leads to replacement of glutamine with a lysine residue (Q187K) in the coiled-coil domain of THAP1. A 60-year-old sister, who was unavailable for examination, reportedly has a diagnosis of reflex sympathetic dystrophy with bilateral limb pain but no confirmed diagnosis of dystonia. The proband's parents are deceased and had no reported signs of dystonia at the time of their deaths. The proband's maternal grandfather had probable cervical dystonia with abnormal neck posturing, reportedly similar to the proband's, although he was never formally diagnosed with dystonia. One of the proband's cousins (family C [III-4], figure 3) is now 51 years old and has had cervical dystonia for approximately 10 years.
Missense mutations were detected in 3 additional subjects in our cohort. Subject D-1378 with a c.50A>G (p.D17G) sequence variant is a 57-year-old woman with cervical dystonia that became apparent at age 43 years. Injections of botulinum toxin type A over a period of 8 years have been associated with moderate control of pain and abnormal posturing. She has 2 brothers who are reportedly normal, although the older brother experienced a prolonged episode of atraumatic wry neck during his college years.
Subject E-6144 with laryngeal dystonia (adductor subtype) harbors a c.395T>C (p.F132S) sequence variant. She has shown consistently excellent results with injections of botulinum toxin type A into her thyroarytenoid muscles. There is no reported dystonia in her family.
Subject F-6224 with a c.496G>A (p.A166T) sequence variant also has adductor laryngeal dystonia. The A166T variant is localized to the coiled-coil domain of THAP1. This subject's dystonia has responded well to injections of botulinum toxin type A. Her maternal grandmother had an undiagnosed late-onset parkinsonism syndrome. She has 2 siblings and 3 children, all neurologically normal by report.
Three additional sequence variants were identified in 11 non-Jewish Caucasian subjects. None of these variants had been reported in available databases (www.hgvs.org/dblist/ccent.html). A c.-42C>T sequence variant in the 5′UTR region of THAP1 was found in 2 female subjects, one with blepharospasm and the other with laryngeal dystonia (adductor subtype). A synonymous exon 1 sequence variant (c.57C>T) was identified in a subject with segmental craniocervical dystonia that included laryngeal dystonia (adductor subtype), blepharospasm, and lower facial dystonia. In 8 non-Jewish Caucasian subjects, a c.71+9C>A sequence variant was found in a conserved region of intron 1, adjacent to the consensus 5′ splice site (figure 2B).
The c.71+9C>A variant was found in cases of blepharospasm, cervical dystonia, masticatory dystonia, and laryngeal dystonia (table 2). In addition, 1 putatively normal control subject harbored this sequence variant. Subject K-1158 had jaw-opening masticatory dystonia that has responded to injections of botulinum toxin type A into the lateral pterygoid muscles. Both subjects with laryngeal dystonia had the adductor subtype.
Control subject (Q-1425) was reexamined and interviewed by M.S.L. after discovery of her THAP1 sequence variant. She then presented the history of 2 seizures, possibly febrile, between ages 6 months and 3 years and 3 possible generalized seizures occurring between ages 20 and 30 years. She was prophylactically treated with phenytoin for 5 years. Over the past 1.5 years, she has experienced trismus, possibly triggered by placement of a molar crown. On examination, spontaneous and volitional masticatory, facial, and lingual movements appeared normal. There is no history of dystonia in her 3 children, 2 siblings, or parents. A 1-tailed Fisher exact test failed to confirm nonrandom associations between phenotype (dystonia, normal) and the presence of the c.71+9C>A intron 1 variant (p = 0.198).
DISCUSSION
In 1997, the DYT6 locus was mapped to chromosome 8 in 2 large Mennonite families with mainly early-onset, predominantly cranial-cervical dystonia.20 With additional family members, the DYT6 gene locus was narrowed to a 23-cM region on chromosome 8q21–22.14 Finally, in 2009, 2 distinct THAP1 mutations were identified in 5 DYT6 families.1 Indicative of a founder effect, 4 Amish-Mennonite families harbored the same F45fs73X protein variant. Another family, of German origin, was found to have a c.241T>C (p.F81L) sequence variant.
In a follow-up study from the same laboratory, 36 non-DYT1 multiplex families in which at least 1 person had nonfocal involvement before age 22 years were screened for THAP1 sequence variants.2 Eight families had novel THAP1 sequence variants.2 Among the 48 affected subjects from 14 families described in their 2 reports, females (n = 29) outnumbered males (n = 19), and all but 3 subjects had childhood or adolescent onset. The median age at onset was 13 years. The spectrum of anatomical involvement was broad and included generalized dystonia (n = 22), segmental dystonia (n = 16), multifocal dystonia (n = 5), and focal dystonia (n = 5). Most subjects had limb onset (33/48). The most common sites of anatomical involvement on examination were arm (92%), cranial (77%), and laryngeal (67%). In another follow-up study, 160 subjects with dystonia (early-onset, generalized, positive family history, facial or laryngeal) were screened for THAP1 sequence variants, and 2 novel truncating deletion mutations were identified in 2 subjects with early-onset generalized dystonia with laryngeal involvement.3 In contrast to previous studies, our cohort mainly consisted of late-onset focal cases with major concentrations of laryngeal (41%, 460/1,114) and cervical (28%, 315/1,114) dystonia, and the majority of sequence variants were identified in these groups of subjects.
THAP1 encodes a 213 residue transcription factor that contains a DNA sequence-specific zinc-dependent THAP domain (1–81aa), a proline-rich region, a nuclear localization signal (146–162aa), and a coiled-coil domain (figure 2).21,22 The THAP domain is highly conserved across species (figure 2). THAP1 plays an important role in transcriptional regulation in the context of cell proliferation and pRb/E2F cell cycle pathways. THAP1 expression is central to endothelial cell proliferation during angiogenesis.23 Other THAP zinc fingers, including human THAP2 and THAP3, share structural homology but do not recognize the same DNA target sequence. THAP1 localizes to promyelocytic leukemia nuclear bodies with the proapoptotic leucine-zipper protein Par-4 and potentiates tumor necrosis factor α–induced apoptosis.24 Three sequence variants identified in our study, along with 8 of 12 previously reported variants, are located in the THAP domain of THAP1. Using a gel-shift assay, the F81L missense mutation in the THAP domain of THAP1 was shown to reduce binding affinity of THAP1 to target DNA.1 Other THAP domain missense variants are likely to produce similar effects on DNA binding. In contrast, the c.57C>T (p.P19P) sequence variant in subject I-7011 could affect pre–messenger RNA splicing accuracy or efficiency leading to altered levels of splice variants.25 Alternatively, the p.P19P variant is simply a rare polymorphism and not causally associated with the appearance of dystonia. The intron 1 (c.71+9C>A) and 5′UTR (c.-42C>T) sequence variants described herein could also exerts effects on splicing fidelity or expression levels. In this regard, both underexpression and overexpression of THAP1 may be deleterious.23
In our study, single amino acid substitutions (p.F132S, p.I149T, p.A166T, and p.Q187K) outside the THAP domain of THAP1 were also found to be associated with 1 case of late-onset generalized dystonia and 4 cases of late-onset focal dystonia (2 cervical dystonia and 2 laryngeal dystonia). In contrast, the 3 previously published variants located in the C-terminal half of THAP1 are predicted to cause protein truncation and were associated with generalized or segmental dystonia.2,3 When combined with data from previous publications, one may speculate that nontruncating sequence variants outside the THAP domain of THAP1 are more likely to be associated with later onset and more restricted anatomical involvement.
Our study has shown that a heterogeneous collection of THAP1 sequence variants may be causally associated with primary dystonia. However, it must be emphasized that the molecular pathogenicity of each variant has not been established. Variable expressivity in age at onset and anatomical distribution was found in individual families. Given that most of our subjects had sporadic dystonia, many THAP1 sequence variants may show low penetrance. Although we found HRM to be an efficient tool for high-throughput screening, HRM does have limitations and may not detect certain homozygous variants or large insertions and deletions.26 Therefore, the frequency of THAP1 sequence variants reported herein may be an underestimate.
AUTHOR CONTRIBUTIONS
M.S.L. designed the study and conducted the statistical analyses.
ACKNOWLEDGMENT
The authors thank C. Lohnes, J. Dennhardt, A. Fitzgerald, E. Heintzen, L. Carpenter, C. Keppel, J. Hartlein, T. Pretorius, A. Strongosky, J. Searcy, H. Lam, and C. Lim for assistance with clinical data collection.
STUDY FUNDING
Supported by the Neuroscience Institute at the University of Tennessee Health Science Center (M.S.L.), Dystonia Medical Research Foundation (M.S.L.), and NIH National Institute of Neurological Disorders and Stroke grant R01NS048458 (M.S.L.). At Washington University School of Medicine, work was supported by NINDS grants P30NS05710 (Neuroscience Blueprint Grant), Clinical Sciences Translation Award RR024992, the American Parkinson's Disease Association (APDA) Advanced Research Center, the Greater St. Louis Chapter of the APDA, the Barnes-Jewish Hospital Foundation (Jack Buck Fund for PD Research and the Elliot H. Stein Family Fund), and the Missouri Chapter of the Dystonia Research Foundation and the Murphy Fund; at Mayo Clinic Florida, work was supported by the NIH NINDS, Morris K. Udall Center of Excellence for Parkinson Disease Research grant P50-NS40256 and NIA R01AG015866 (Z.K.W., R.J.U., and J.A.V.G.), NINDS R01 NS057567-01A2, Pacific Alzheimer's Research Foundation PARF-C06-01 and CR 90052025 Mayo Clinic Jacksonville Research Committee (Z.K.W., R.J.U.), and NIA P01AG017216 (Z.K.W.); and at the Parkinson's & Movement Disorder Institute, work was supported by the Long Beach Memorial Foundation, Orange Coast Memorial Foundation, and the Parkinson's & Movement Disorder Foundation.
DISCLOSURE
Dr. Xiao and Dr. Zhao report no disclosures. Dr. Bastian serves on the Scientific Advisory Board of Olympus Surgical; and received honoraria from Olympus Surgical Education event. Dr. Perlmutter serves on the scientific advisory boards of the American Parkinson Disease Association, Dystonia Medical Research Foundation, MO Chapter of the Dystonia Medical Research Fund, Greater St. Louis Chapter of the APDA; serves as an editorial board member of Neurology; received travel expenses and/or honoraria for lectures or educational activities not sponsored by industry; received honoraria from Parkinson Disease Study Group for grant reviews; received honoraria from Medtronic Inc. for partial fellowship support. Receives research support from NIH [1R01 NS41509 (PI), R01 NS050425 (PI), R01 NS058714 (PI), P30 NS057105 (Project coleader), NIH/NCRR RR024992 (Core leader), R01ES013743 (Coinvestigator), R01 NS039821, R01NS058797 (Coinvestigator); RO1 HD056015 (Coinvestigator), U54 NS065701 (Co-PI and Project leader)]; receives research support from the Huntington Disease Society of American Center of Excellence, Michael J. Fox Foundation, HiQ Foundation, McDonnell Center for Higher Brain Function, Greater St. Louis Chapter of the American Parkinson Disease Association, American Parkinson Disease Association, Bander Foundation for Medical Ethics and Advanced PD Research Center at Washington University; received research support from the BJH Foundation. Dr. Racette received research support from Teva (PI), Eisai (PI), and Solvay (PI); receives research support from Schwarz (PI), Solstice (PI), Eisai (PI), Allergan (PI), and Neurogen (PI); received government research support from NIH [5R01 NS037167-10 (PI-Foroud,T)]; received research support for “Epidemiology of Parkinsonism in Welders” Administrative Supplement to Support College Undergraduate Research (NIEHS) (PI); receives research support from NIH [U10 NS44455 (PI), R01HG02449 (PI-Shoulson, I), R01 ES013743-01A2 (PI), P42 ES04696 (PI-Checkoway, H), K24 NS060825 (PI), R21 ES017504 (PI), K23 NS43351 (PI)]; received research support from BJHF/ICTS [Neuropathology of Chronic Manganese Exposure” (PI)]; and received research support from received research support from the Michael J. Fox Foundation. Dr. Tabbal receives research support from NIH [1R01NS41509 (Coinvestigator), RO1 NS050425 (Coinvestigator)]. Dr. Karimi receives research support from NIH [1R01NS41509 (Coinvestigator), RO1 NS058714 (Coinvestigator)]. Dr. Paniello reports no disclosures. Dr. Wszolek served as a consultant for Bayer Schering Pharmaceuticals; has served as co-Editor-in-Chief of Parkinsonism and Related Disorders, Editor-in-Chief of the Polish Edition of Neurology, and Regional Editor the European Journal of Neurology, serves as an editorial board member of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds patents that may accrue revenue, Mayo 2004-185, 2004-291, and 2007-104; receives royalties from Parkinsonism and Related Disorders (Elsevier), Polish Edition of Neurology (Medycyna Praktyczna), and European Journal of Neurology (Wiley-Blackwell); received research support from Allergan, Inc.; received research support from NIH [P01AG017216-1 (Coinvestigator), R01AG015866-1 (Coinvestigator), P50NS 40256 (Clinical Core Leader); and CIHR P 121849 (Coinvestigator)]; received research support from Mayo Clinic Florida, Research Committee CR program; received research support from PARF, C06-01. Dr. Uitti received research support form Novartis, Medtronic, Inc., Eisai Inc., and Advanced Neuromodulations Systems; receives research support from NIH/NINDS [P50NS 40256 (Coinvestigator)]; serves as an Associate Editor for Neurology; His institution has received annual royalties from the licensing of the technology related to PARK8/LRRK2 greater than the federal threshold for significant financial interest. Dr. Van Gerpen reports no disclosures. Dr. Simon has received research support from the NINDS [1R01NS058988 (PI), U10 NS44482 (PI at BIDMC), NINDS R01 NS037167 (subcontract/Investigator)]; received research support from NINDS [1R03NS053840 (PI)]; and served as a consultant to the Gerson Lehrman Group (GLG) and Link Medicine; received research support from Michael J. Fox, National Parkinson Foundation Center of Excellence Research Grant, and National Parkinson Foundation “Mega-Research Project” Award (Co-PI). Dr. Simon has received honoraria for lectures or consultation from Link Medicine and from UCB Pharma. Dr. Tarsy serves as a consultant for Esai Neuroscience; receives funding from Medtronic and Allergan; receives patient education grants from Allergan, Boehringer Ingelheim, Valeant, Teva Neurosciences; receives research funding from Schwarz, Boehringer Ingelheim, Solvay, Neurogen; and receives foundation support from National Parkinson Foundation. Dr. Hedera reports no disclosures. Dr. Truong served on the speakers' bureau of Allergan; serves on the speakers' bureau for Teva; served as a consultant for Schering Plough; received compensation for activities with Solstice Neurosciences and GlaxoSmithKline, Inc.; received funding for travel from Ipsen, Allergan, and Teva; received research support from UCB Pharma Merz, Ipsen, Schering Plough, and Acadia. Dr. Frei served on the speakers' bureau for Allergan. Dr. Batish is an employee of Athena Diagnostics. Dr. Blitzer serves as an editor for Laryngoscope and the Journal of the American Academy of Otolaryngology - Head and Neck Surgery; served on the scientific advisory boards for Allergan, Inc. and Revance Therapeutics; has received honoraria for activities with Myotech; has received research support from Allergan, Inc., Merz Pharma, and Revance Therapeutics; and has received royalty payments from Xomed/Medtronics. Dr. Pfeiffer serves on the scientific advisory board for the National Parkinson Foundation; served as a consultant for Solvay and Ipsen; serves on the speakers' bureaus of Novartis, GlaxoSmithKline, Boehringer Ingelheim, Teva, and UCB/Schwarz; served on the scientific advisory boards for Kyowa, Solvay, Ipsen, and Theravance; served as a legal consultant for Spriggs & Hollingsworth and Davis Graham & Stubbs as a legal consultant. Dr. Pfeiffer serves as Co-Editor-in-Chief for Parkinsonism and Related Disorders; receives royalty payments from Neurogastroenterology (Butterworth Heinemann, 2008), Parkinson's Disease (CRC Press, 2008, 2009); and Parkinson's Disease and Nonmotor Dysfunction (Humana Press, 2008); has received research support from Kyowa, Novartis, Boehringer Ingelheim, Eisai, UCB/Schwarz, and Santhera. Dr. Gong reports no disclosures. Dr. LeDoux serves on the scientific advisory board for the Dystonia Medical Research Foundation; serves on the editorial board of Parkinsonism and Related Disorders; serves on the speakers' bureau for Boehringer Ingelheim Pharmaceuticals, Inc., and Teva Neuroscience; receives research support from NIH/NINDS [R01NS048458 (PI), 5U01 NS052592 (Site PI)]; the Dystonia Medical Research Foundation, Bachmann-Strauss Dystonia & Parkinson Foundation, Merz Pharma, Xenoport, Boehringer Ingelheim Pharmaceuticals, Inc., and HP Therapeutics Foundation; and receives royalty payments from Animal Models of Movement Disorders (Elsevier, 2005).
Supplementary Material
Address correspondence and reprint requests to Dr. Mark S. LeDoux, University of Tennessee Health Science Center, Department of Neurology, 855 Monroe Ave., Link Building, Suite 415, Memphis, TN 38163 mledoux@utmem.edu.
Editorial, page 192
Supplemental data at www.neurology.org
Disclosure: Author disclosures are provided at the end of the article.
Received May 27, 2009. Accepted in final form September 15, 2009.
REFERENCES
- 1.Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 2009;41:286–288. [DOI] [PubMed] [Google Scholar]
- 2.Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol 2009;8:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Djarmati A, Schneider SA, Lohmann K, et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol 2009;8:447–452. [DOI] [PubMed] [Google Scholar]
- 4.Grandas F, Elston J, Quinn N, Marsden CD. Blepharospasm: a review of 264 patients. J Neurol Neurosurg Psychiatry 1988;51:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duane DD. Spasmodic torticollis: clinical and biologic features and their implications for focal dystonia. Adv Neurol 1988;50:473–492. [PubMed] [Google Scholar]
- 6.Chan J, Brin MF, Fahn S. Idiopathic cervical dystonia: clinical characteristics. Mov Disord 1991;6:119–126. [DOI] [PubMed] [Google Scholar]
- 7.Maniak S, Sieberer M, Hagenah J, Klein C, Vieregge P. Focal and segmental primary dystonia in north-western Germany: a clinico-genetic study. Acta Neurol Scand 2003;107:228–232. [DOI] [PubMed] [Google Scholar]
- 8.Defazio G, Martino D, Aniello MS, et al. A family study on primary blepharospasm. J Neurol Neurosurg Psychiatry 2006;77:252–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bressman SB. Dystonia: phenotypes and genotypes. Rev Neurol (Paris) 2003;159:849–856. [PubMed] [Google Scholar]
- 10.Klein C, Friedman J, Bressman S, et al. Genetic testing for early-onset torsion dystonia (DYT1): introduction of a simple screening method, experiences from testing of a large patient cohort, and ethical aspects. Genet Test 1999;3:323–328. [DOI] [PubMed] [Google Scholar]
- 11.Grundmann K, Laubis-Herrmann U, Dressler D, et al. Lack of mutations in the epsilon-sarcoglycan gene in patients with different subtypes of primary dystonias. Mov Disord 2004;19:1294–1297. [DOI] [PubMed] [Google Scholar]
- 12.Xiao J, Bastian RW, Perlmutter JS, et al. High-throughput mutational analysis of TOR1A in primary dystonia. BMC Med Genet 2009;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozelius LJ. Update on the genetics of primary torsion dystonia loci DYT6, DYT7, and DYT13 and the dystonia-plus locus DYT12. Adv Neurol 2004;94:109–112. [PubMed] [Google Scholar]
- 14.Saunders-Pullman R, Raymond D, Senthil G, et al. Narrowing the DYT6 dystonia region and evidence for locus heterogeneity in the Amish-Mennonites. Am J Med Genet A 2007;143A:2098–2105. [DOI] [PubMed] [Google Scholar]
- 15.Bressman SB. Dystonia genotypes, phenotypes, and classification. Adv Neurol 2004;94:101–107. [PubMed] [Google Scholar]
- 16.Fahn S, Bressman SB, Marsden CD. Classification of dystonia. Adv Neurol 1998;78:1–10. [PubMed] [Google Scholar]
- 17.Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 2005;21:3176–3178. [DOI] [PubMed] [Google Scholar]
- 18.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002;30:3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982;157:105–132. [DOI] [PubMed] [Google Scholar]
- 20.Almasy L, Bressman SB, Raymond D, et al. Idiopathic torsion dystonia linked to chromosome 8 in two Mennonite families. Ann Neurol 1997;42:670–673. [DOI] [PubMed] [Google Scholar]
- 21.Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Girard JP. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc Natl Acad Sci USA 2005;102:6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bessiere D, Lacroix C, Campagne S, et al. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J Biol Chem 2008;283:4352–4363. [DOI] [PubMed] [Google Scholar]
- 23.Cayrol C, Lacroix C, Mathe C, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood 2007;109:584–594. [DOI] [PubMed] [Google Scholar]
- 24.Roussigne M, Cayrol C, Clouaire T, Amalric F, Girard JP. THAP1 is a nuclear proapoptotic factor that links prostate-apoptosis-response-4 (Par-4) to PML nuclear bodies. Oncogene 2003;22:2432–2442. [DOI] [PubMed] [Google Scholar]
- 25.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002;3:285–298. [DOI] [PubMed] [Google Scholar]
- 26.Wittwer CT. High-resolution DNA melting analysis: advancements and limitations. Hum Mutat 2009;30:857–859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.