

Abstract
Pharmacological activation of group II metabotropic glutamate receptors (mGluR2/3) inhibits cocaine self-administration and reinstatement of drug-seeking behavior, suggesting a possible use of mGluR2/3 agonists in the treatment of cocaine dependence. In the present study, we investigated whether elevation of the endogenous mGluR2/3 ligand N-acetyl-aspartatylglutamate (NAAG) levels with the N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) inhibitor 2-PMPA attenuates cocaine self-administration and cocaine-induced reinstatement of drug seeking. NAALADase is a NAAG degradation enzyme that hydrolyzes NAAG to N-acetylaspartate and glutamate. Systemic administration of 2-PMPA (10–100 mg/kg, i.p.) inhibited intravenous self-administration maintained by low unit doses of cocaine and cocaine (but not sucrose)-induced reinstatement of drug-seeking behavior. Microinjections of 2-PMPA (3–5 μg/side) or NAAG (3–5 μg/side) into the nucleus accumbens (NAc), but not into the dorsal striatum, also inhibited cocaine-induced reinstatement, an effect that was blocked by intra-NAc injection of LY341495, a selective mGluR2/3 antagonist. In vivo microdialysis demonstrated that 2-PMPA (10–100 mg/kg, i.p.) produced a dose-dependent reduction in both extracellular DA and glutamate, an effect that was also blocked by LY341495. Finally, pretreatment with 2-PMPA partially attenuated cocaine-enhanced extracellular NAc DA, while completely blocking cocaine-enhanced extracellular NAc glutamate in rats during reinstatement testing. Intra-NAc perfusion of LY341495 blocked 2-PMPA-induced reductions in cocaine-enhanced extracellular NAc glutamate, but not DA. These findings suggest that 2-PMPA is effective in attenuating cocaine-induced reinstatement of drug-seeking behavior, likely by attenuating cocaine-induced increases in NAc DA and glutamate via presynaptic mGluR2/3s.
Keywords: NAAG, 2-PMPA, cocaine, dopamine, glutamate, reinstatement, relapse
Cocaine addiction is characterized by high rates of relapse to drug use after abstinence. Despite advances in understanding the neurobiology of addiction, no effective anti-relapse pharmacotherapies exist. Since cocaine priming significantly increases extracellular dopamine (DA) and glutamate in the nucleus accumbens (NAc) and group II metabotropic glutamate receptors (mGluR2/3) negatively control DA and glutamate release (Anderson and Pierce, 2005; Kalivas, 2004), it has been proposed that mGluR2/3 agonists may be effective in attenuating cocaine-induced relapse (Xi et al., 2002). LY379268 is a recently well-characterized, systemically effective mGluR2/3 agonist (Imre, 2007). It is reported that LY379268 significantly inhibits intravenous cocaine self-administration, cocaine-induced reinstatement, and incubation of cocaine craving in rats or non-human primates (Baptista et al., 2004; Adewale et al., 2006; Peters and Kalivas, 2006; Lu et al., 2007). However, LY379268, at doses that inhibit cocaine reward or relapse, also inhibits food-taking or food-seeking behavior and incubation of sucrose craving (Peters and Kalivas, 2006; Liechti et al, 2007; Uejima et al., 2007). These data suggest that LY379268 may have unwanted side-effects in the treatment of drug addiction. In addition, recent studies suggest that the pharmacological effects of LY379268 may be mediated predominantly by activation of the mGluR2 subtype, because mGluR2, but not mGluR3, deletion abolishes the pharmacological action of LY379268 and other mGluR2/3 agonists (LY354740, LY404039) (Linden et al., 2006; Fell et al., 2008; Woolley et al., 2008). Further, the selective mGluR2 positive allosteric modulator LY487379 produces putatively antipsychotic effects similar to LY379268 in animal models (Johnson et al., 2003; Galici et al., 2005), and mGluR2 deletion produces enhanced locomotor, conditioned place preference, and NAc DA responses to cocaine (Morishima et al., 2005). These findings raise the question of whether or not the mGluR3 subtype is also involved in the pharmacological actions of cocaine or of mGluR2/3 agonists.
N-acetyl-aspartylglutamate (NAAG) has been shown to be an endogenous mGluR3 agonist (Wrobleska et al., 1997). NAAG is co-released with glutamate or other neurotransmitters and functionally prevents excessive neurotransmitter release by activation of presynaptic mGluR3s (Neale et al., 2000, 2005). NAAG is inactivated by the enzyme N-acetylated-α-linked-acidic dipepetidase (NAALADase), which hydrolyzes NAAG to N-acetyl-aspartate and glutamate (Tsukamoto et al., 2007). Selective inhibition of NAALADase by 2-PMPA or GPI-5693 (Jackson et al., 1996; Tsukamoto et al., 2007) significantly elevates brain NAAG levels (Slusher et al., 1999; Nagel et al., 2006) and inhibits cocaine-induced behavioral sensitization (Shippenberg et al, 2000), cocaine-induced conditioned place preference (CPP) (Slusher et al, 2001) and cocaine-kindled seizures (Witkin et al., 2002), suggesting involvement of the endogenous NAAG-NAALADase signaling pathway in cocaine addiction (Zhou et al., 2005). However, it remains unclear whether 2-PMPA or NAAG has similar putative therapeutic efficacy to LY379268 in attenuating cocaine self-administration or cocaine-induced relapse.
In the present study, we first studied the effects of the selective NAALADase inhibitor 2-PMPA on intravenous cocaine self-administration maintained by different doses of cocaine and on cocaine- or sucrose-induced reinstatement of reward-seeking behavior, and then investigated the effects of 2-PMPA or NAAG microinjections into the NAc or dorsal striatum on cocaine-induced reinstatement to determine locus of action in the brain. Finally, to further assess whether DA- and/or glutamate-related mechanisms underlie the pharmacological effects of mGluR2/3 agonists in animal models of drug addiction, in vivo microdialysis was used to study the effects of 2-PMPA on basal or cocaine-enhanced NAc DA and glutamate in rats during reinstatement testing in the presence or absence of LY341495, a selective mGluR2/3 antagonist.
MATERIALS AND METHODS
Animals
Male Long-Evans rats (Charles River Laboratories, Raleigh, NC, USA) weighing 250 to 300 g were used. Rats were housed individually in a climate-controlled room on a reversed light-dark cycle (lights on at 7:00 PM, lights off at 7:00 AM) with free access to food and water. The animal facility was fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the U.S. National Academy of Sciences, and were approved by the Animal Care and Use Committee of the National Institute on Drug Abuse of the U.S. National Institutes of Health.
Experiment 1: Intravenous Cocaine Self-Administration
Single-dose cocaine self-administration
Intravenous (i.v.) catheterization surgery and cocaine self-administration protocols were as previously described (Xi et al., 2006a, 2006b). Briefly, i.v. catheterization was performed under sodium pentobarbital (60 mg/kg, i.p.) anesthesia with aseptic surgical technique. After 7 days recovery from surgery, animals were placed into standard operant chambers from Med Associates Inc. (Saint Albans, VT, USA) for cocaine self-administration (0.5 mg/kg/infusion) under FR2 reinforcement. To avoid cocaine overdose during the self-administration period, each animal was limited to a maximum of 50 cocaine injections per 3hr session. After stable cocaine-maintained responding was achieved (i.e., less than 10% variability in mean inter-response interval and less than 10% variability in mean active lever presses for at least 3 consecutive days), each rat randomly received 1 of 3 doses of either 2-PMPA (10, 30, 100 mg/kg, i.p.) or vehicle (50 mM HEPES, 1 ml, i.p.) 30 min prior to the test session. Animals then received an additional 5–7 days of self-administration of cocaine alone until the baseline response rate was re-established prior to testing the next dose of 2-PMPA. The order of testing for the various doses of drug or vehicle was counterbalanced according to a Latin square design.
Multiple-dose cocaine self-administration training
To determine whether the pharmacological action of 2-PMPA was also cocaine dose-dependent, we further studied the effects of 2-PMPA on cocaine self-administration maintained by a full range of cocaine doses (0.031, 0.063, 0.125, 0.25, 0.5 mg/kg/infusion) in a single session. The session consisted of five sequential 20-min components, each preceded by a 20-min time-out period for changing cocaine dose. The infusion volumes and durations in each component were identical except that cocaine concentrations for corresponding unit cocaine doses differed. There was a 30-min extinction period (0 mg/kg cocaine) before daily cocaine self-administration training. Training continued until stable cocaine-maintained responding was achieved (i.e., a minimum of 10 mg/kg cocaine intake within a session, less than 10% variation in total number of cocaine injections for 3 consecutive days, and at least 5-fold higher maximal response rates compared to those maintained during extinction). Then, each rat randomly received either vehicle or 1 of 2 doses of 2-PMPA (10, 30 mg/kg, i.p.) 30 min prior to the test session. Animals then received an additional 5–7 days of self-administration of cocaine alone until the baseline response rate was re-established prior to testing the next dose of 2-PMPA. The order of testing for the various doses of drug or vehicle was counterbalanced according to a Latin square design.
Experiment 2: Cocaine-induced reinstatement of drug-seeking behavior
Surgery and general procedures for cocaine self-administration prior to behavioral extinction were the same as described above.
Extinction
After stable cocaine self-administration was established, animals were exposed to extinction conditions, during which cocaine was replaced by saline, and the cocaine-associated cue-light and tone were turned off. Active lever pressing led only to saline infusion. Daily 3 h extinction sessions for each rat continued until that rat lever-pressed less than 10 times per 3 h session for at least 3 consecutive days. After the animals met the established extinction criteria, they were divided into 14 dose groups (6–14 rats per group) for reinstatement testing.
Effects of 2-PMPA on cocaine-induced reinstatement
Four groups of rats were used to observe the effects of systemic administration of 2-PMPA on cocaine-induced relapse. On the reinstatement test day, each group of animals received either vehicle (50 mM HEPES, 1 ml, i.p.) or 1 dose of 2-PMPA (10, 30, 100 mg/kg, i.p.), 30 min after 2-PMPA administration, all rats were given a priming injection of cocaine (10 mg/kg, i.p.) immediately before the initiation of reinstatement testing. During the reinstatement test, the conditions were identical to those in extinction sessions. Cocaine-induced active-lever presses (reinstatement) were recorded, although these did not lead to either cocaine infusions or presentation of the conditioned cue-light and tone. Reinstatement test sessions lasted 3 h.
Effects of microinjection of 2-PMPA, NAAG or LY341495 into the NAc or dorsal striatum on cocaine-induced reinstatement
To determine the loci of action and the receptor mechanism of 2-PMPA in the rat brain, three additional groups of rats were also surgically implanted with intracranial guide cannulae (20 gauge, 14 mm; Plastics One, Roanoke, VA, USA) into the NAc (AP +1.7 mm, ML ±2.0 mm, DV −5.0 mm, 6° angle from vertical) or dorsal striatum (AP 0.48 mm, ML ±2.5 mm, DV −3.0 mm, 6° angle from vertical), according to the atlas of Paxinos and Watson (1986) before cocaine self-administration training. After the extinction criteria was established, each group of rats randomly received either intra-NAc microinjections of 2-PMPA (0, 3, 5 μg/0.5μl/side), NAAG (0, 3, 5 μg/0.5μl/side) or intra-dorsal striatum microinjections of 2-PMPA in the absence or presence of LY341495 (5 μg/0.5μl/side). Thirty min after microinjections, all rats were given a priming injection of cocaine (10 mg/kg, i.p.) immediately before the initiation of reinstatement testing. The other protocols for cocaine-induced reinstatement were the same as described above.
After the microinjection experiments were completed, rats were anesthetized with a high dose of pentobarbital (>100mg/kg i.p.) and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were removed and placed in 10% formalin for histological verification of microinjection locations.
Effects of 2-PMPA on sucrose-triggered reinstatement of sucrose-seeking behavior
The procedures for oral sucrose self-administration, extinction, and reinstatement testing were identical to the procedures for cocaine self-administration, extinction, and reinstatement testing except for the following: 1) no surgery was performed on the animals in the sucrose experiment; 2) active lever presses led to delivery of 0.1 ml of 5% sucrose solution into a liquid food tray on the operant chamber wall; and 3) reinstatement was triggered initially by 3 “free” sucrose deliveries. The effects of 2-PMPA (30, 100 mg/kg, i.p., 30 min prior to sucrose priming) on sucrose-triggered reinstatement were evaluated.
Experiment 3: In vivo microdialysis with HPLC
In vivo microdialysis protocols were as reported previously (Xi et al., 2006a). Briefly, rats were anesthetized with sodium pentobarbital, and guide cannulae (20 gauge, Plastics One, Roanoke, VA) were surgically implanted into the NAc (AP +1.6 mm, ML ± 2.0 mm, DV −4.0 mm, 6° from vertical), according to the rat brain atlas of Paxinos and Watson (1998). The guide cannulae were fixed to the skull with 4 stainless steel jeweler’s screws (Small Parts Inc., Miami Lakes, FL, USA) and dental acrylic. After 7 days of recovery from surgery, rats were divided into two groups. One group of rats (drug naïve rats) were used directly for in vivo microdialysis, while another group of rats were trained for cocaine self-administration first and then used for in vivo microdialysis beginning at 24 hrs after the last cocaine self-administration. Microdialysis probes were inserted into the NAc 12 hr before the onset of microdialysis to minimize damage-induced neurotransmitter release. Microdialysis samples were collected every 20 min into 10 μl 0.5 M perchloric acid to prevent DA degradation. After collection, samples were frozen at −80°C. Dialysate DA and glutamate were measured using high pressure liquid chromatography (HPLC) with electrochemical and flourometric detection, respectively, as reported previously (Xi et al., 2006a). DA and glutamate values were quantified with external standard curves (DA 0.1–1.0 nM; glutamate 10–1000 nM). The limits of detection for DA and glutamate were 0.01–10 nM and 1 nM-10 μM, respectively.
Effects of 2-PMPA or LY341495 on basal or cocaine-enhanced extracellular DA and glutamate in the NAc
To determine the neurochemical mechanisms underlying the antagonism of 2-PMPA on cocaine-induced reinstatement of drug seeking, we further observed the effects of 2-PMPA (0, 30, 100 mg/kg, i.p.) and/or LY341495 (1 mg/kg, i.p.) on basal extracellular DA and glutamate, and then observed the effects of 2-PMPA pretreatment on cocaine-enhanced NAc DA and glutamate in rats during reinstatement test.
After microdialysis experiments were completed, rats were anesthetized with a high dose of pentobarbital (>100mg/kg i.p.) and perfused transcardially with 0.9% saline followed by 10% formalin. Brains were removed and placed in 10% formalin for histological verification of microdialysis probe locations in rat brain.
Drugs
Cocaine HCl was provided by the National Institute on Drug Abuse (NIDA, Baltimore, MD) and dissolved in physiological saline. 2-PMPA (2-(phosphonomethyl)pentanedioic acid) was provided by Guilford Pharmaceuticals Inc. (Baltimore, MD, USA). LY341495 was purchased from Tocris Bioscience (Ellisville, MO, USA). 2-PMPA was dissolved in 0.5 M HEPES buffer (vehicle) purchased from MP Biomedicals, Inc (Solon, Ohio, USA) for sustemic (i.p.) administration or artificial cerebrospinal fluid (aCSF) for intracranial microinjections or in vivo microdialysis. The pretreatment time (30 min prior to cocaine) of 2-PMPA was chosen on the basis of our preliminary pilot studies and an in vivo microdialysis finding that a significant reduction in extracellular DA and glutamate occurs at 20 min after 2-PMPA administration.
Data analyses
All data are presented as means (± S.E.M.). One-way analysis of variance (ANOVA) was used to analyze the effects of 2-PMPA or NAAG on cocaine self-administration or cocaine-induced reinstatement of drug-seeking behavior. Two-way ANOVA with repeated measures were used to analyze the effects of 2-PMPA on cocaine-enhanced NAc DA and glutamate. Individual group comparisons were carried out using the Bonferroni procedure.
Results
2-PMPA inhibited cocaine-induced reinstatement
Figure 1A illustrates that systemic administration of 2-PMPA (10, 30, 100 mg/kg, i.p.) failed to alter cocaine self-administration maintained by 0.5 mg/kg cocaine (F3,28=0.27, p=NS). Figure 1B illustrates that 2-PMPA, at 10 or 30 mg/kg, significantly and dose-dependently inhibited cocaine self-administration maintained by lower doses of cocaine (0.06, 0.125, 0.25 mg/kg/infusion). Two-way ANOVA with repeated measures over cocaine dose revealed a significant treatment (vehicle vs. 2-PMPA) main effect (F2,10=41.81, p<0.001), significant time (cocaine dose) main effect (F4,20=15.75, p<0.001) and a significant treatment × time interaction (F8,40=5.52, p<0.001). Individual group comparisons revealed a statistical significant reduction in cocaine self-administration maintained by 0.06, 0.125 or 0.25 mg/kg/infusion cocaine after 10 mg/kg (t=7.61, p<0.001) or 30 mg/kg (t=12.86, p<0.001) 2-PMPA, when compared to the vehicle control group. Figure 1C illustrates that 2-PMPA (10, 30, 100 mg/kg) significantly inhibited cocaine-induced reinstatement of drug-seeking behavior (F3,51 = 4.19, p=0.01). Individual group comparisons revealed a statistically significant reduction in the total number of active lever presses after 30 mg/kg (t=6.07, p=0.001) and 100 mg/kg (t=3.33, p<0.05) 2-PMPA, when compared to the vehicle treatment group. Fig. 1D illustrates that systemic administration of 2-PMPA (30, 100 mg/kg, i.p.) failed to alter sucrose-induced reinstatement of sucrose-seeking behavior (F2,23=0.16, p=NS). In addition, 2-PMPA also failed to inhibit oral sucrose self-administration under FR2 reinforcement (data not shown).
Figure 1.

Effects of 2-PMPA (10, 30, 100 mg/kg, i.p.) on intravenous cocaine self-administration (A, B) and cocaine- or sucrose-triggered reinstatement of reward-seeking behavior (C, D) in rats. *p<0.05, **p<0.01, ***p<0.001, compared to the vehicle treatment group.
Intra-NAc microinjections of 2-PMPA or NAAG inhibit cocaine-induced reinstatement
Figure 2A illustrates that microinjections of 2-PMPA (5, 10 μg/μl/side) into the dorsal striatum failed to alter cocaine-induced reinstatement (F2,18=0.27, p=NS). However, when microinjected into the NAc, 2-PMPA dose-dependently inhibited cocaine-induced reinstatement of drug-seeking behavior (Fig. 2B, F4,43=2.97, p<0.05). Individual group comparisons revealed a significant reduction in drug-seeking after 10 μg/side (t=4.40, p<0.05), but not 5 μg/side (t=1.50, p=NS) 2-PMPA. The reduction in cocaine-seeking produced by 10 μg/side 2-PMPA was blocked by pretreatment with 10 μg/side of LY341495, a selective mGluR2/3 antagonist. LY341495 alone had no effect on cocaine-induced reinstatement (Fig. 2B). Figure 2C illustrates that microinjection of the same doses of NAAG into the NAc also significantly inhibited cocaine-induced reinstatement (F3,35=10.78, p<0.001). Individual group comparisons reveals a significant reduction in cocaine-seeking behavior after 5 μg/side (t=6.14, p<0.001) or 10 μg/side (t=6.83, p<0.001) NAAG. The reduction in cocaine-seeking produced by 10 μg/side NAAG was also blocked by the pretreatment with 10 μg/side LY341495.
Figure 2.

Microinjections of 2-PMPA into the dorsal striatum (A) failed to effect reinstatement, while microinjections of 2-PMPA (B) or NAAG (C) into the NAc dose-dependently inhibited cocaine-primed reinstatement. Intra-NAc pretreatment with LY341495 blocked the 2-PMPA- or NAAG-induced inhibition of cocaine-primed reinstatement. Intra-NAc LY341495 alone had no effect on cocaine-induced reinstatement. *p<0.05, ***p<0.01, compared to the vehicle treatment group.
2-PMPA inhibits NAc DA or glutamate release by activation of mGluR2/3 receptors
Figure 3A illustrates that systemic administration of the same doses (10, 30, 100 mg/kg) of 2-PMPA dose-dependently lowered extracellular NAc DA. Two-way ANOVA with repeated measurements over time revealed statistically significant treatment (2-PMPA vs. vehicle) main effect (F4,30=5.32, p<0.01), time main effect (F14,56=3.43, p<0.05), and treatment × time interactions (F56,420=1.46, p<0.05). Individual group comparisons revealed a significant reduction in NAc DA after 30 mg/kg (t=3.65, p<0.05) or 100 mg/kg (t=4.28, p<0.05), but not 10 mg/kg (t=1.23, p=NS) 2-PMPA. The reduction in extracellular DA produced by 30 mg/kg 2-PMPA was completely blocked by pretreatment with 1 mg/kg LY341495. Figure 3B illustrates that the changes in area under curve (AUC) after 2-PMPA administration, indicating that 2-PMPA dose-dependently lowered extracellular DA in the NAc, an effect that was blocked by LY341495 (F4,29=6.9, p<0.001).
Figure 3.

Effects of 2-PMPA on basal levels of extracellular DA and glutamate in the NAc. Systemic administration of 2-PMPA (10, 30, 100 mg/kg, i.p.) alone dose-dependently lowered extracellular DA (A, B) and glutamate (C, D). The effect produced by 30 mg/kg 2-PMPA was blocked by LY341495 (1 mg/kg, i.p.). *p<0.05, compared to baseline (before 2-PMPA) in each treatment group (A, C) or to the vehicle treatment group (B, D).
Figure 3C illustrates that 2-PMPA modestly lowered extracellular glutamate in the NAc. Two-way ANOVA with repeated measures over time revealed a statistically significant treatment main effect (F3,24=2.56, p<0.05), time main effect (F11,33=4.48, p<0.001), and treatment × time interaction (F33,264=1.61, p<0.05). Individual group comparisons revealed a significant reduction in extracellular glutamate after 30 mg/kg (t=3.51, p<0.05) and 100 mg/kg (t=3.67, p<0.05), but not 10 mg/kg (t=1.14, p=NS) 2-PMPA. Figure 3D illustrates the changes in extracellular glutamate (AUC) after 2-PMPA, indicating that systemic administration of 2-PMPA dose-dependently lowered extracellular glutamate in the NAc, an effect that was blocked by pretreatment with 1 mg/kg LY341495 (F3,30=4.87, p<0.001).
2-PMPA inhibited cocaine-induced increases in NAc DA and glutamate
Figure 4A illustrates the effects of 2-PMPA (10–100 mg/kg, i.p., 40 min prior to cocaine) on cocaine-enhanced extracellular DA in the NAc measured during reinstatement testing. Since high doses of 2-PMPA significantly lowered basal levels of extracellular DA, we renormalized cocaine-induced changes in DA over the new baseline, i.e., the mean values of the two 20-min dialysis samples immediately before cocaine administration, to analyze 2-PMPA’s pharmacological action in cocaine-enhanced NAc DA. The two dotted lines indicate the changes in basal DA after 2-PMPA administration. Two-way ANOVA with repeated measurement over time did not reveal a statistically significant treatment main effect (F3,31=1.48, p=NS), but revealed a statistically significant time main effect (F7,217=50.11, p<0.001) and treatment × time interaction (F21,217=1.62, p<0.05). Individual group comparisons indicate that cocaine-induced increases in DA were significantly decreased after 100 mg/kg 2-PMPA at 20 min (t=3.34, p<0.001) and 40 min (t=2.73, p<0.05), when compared to the vehicle treatment group. Figure 4B shows the changes in the area under curve (AUC) in cocaine-enhanced DA, indicating that 2-PMPA produced a dose-dependent reduction in cocaine-induced increase in DA (F3,31=3.61, p<0.05).
Figure 4.

Effects of 2-PMPA on cocaine-enhanced extracellular DA and glutamate in the NAc. Pretreatment with 2-PMPA (10, 30, 100 mg/kg, i.p., 40 min prior to cocaine) dose-dependently attenuated cocaine-enhanced NAc DA (A, B) and glutamate (C, D). *p<0.05, **p<0.01, ***p<0.001, compared to baseline (before cocaine) in each treatment group. #p<0.05, ##p<0.01, compared to the vehicle treatment group.
Figure 4C illustrates the effects of 2-PMPA on cocaine-induced increases in NAc glutamate measured during the reinstatement testing. Since high doses of 2-PMPA also lowered basal levels of extracellular glutamate, we renormalized cocaine-induced changes in glutamate over the new baseline, i.e., the mean values of the two 20-min dialysis samples immediately before cocaine administration, to analyze 2-PMPA’s pharmacological action in cocaine-enhanced NAc glutamate. The two dotted lines indicate the changes in basal glutamate after 2-PMPA administration. Two-way ANOVA with repeated measurement over time did not reveal a statistically treatment main effect (F3,28=1.19, p=NS), but revealed a significant time main effect (F7,196=4.99, p<0.01) and treatment × time interaction (F21, 196=1.79, p<0.05). Individual group comparisons revealed a statistically significant reduction in cocaine-induced increases in NAc glutamate after 30 mg/kg at 20 min (t=2.89, p<0.05) and 40 min (t=4.06, p<0.05) or after 100 mg/kg 2-PMPA at 20 min (t=3.59, p<0.05) and 40 min (t=4.86, p<0.01), when compared to the vehicle treatment group. Figure 4D illustrates the changes in cocaine-induced increases in NAc glutamate (AUC data), indicating that 2-PMPA dose-dependently inhibited NAc glutamate response to cocaine priming (F3,27=4.19, p<0.05).
Intra-NAc LY341495 blocks the antagonism by 2-PMPA of cocaine-enhanced NAc glutamate
Fig. 5A illustrates that intra-NAc local perfusion of LY341495 (1–300 μM) had no significant effect on NAc DA (F1,12=0.53, p=NS). Fig. 5B illustrates that intra-NAc local perfusion of LY341495 (300 μM) failed to block 2-PMPA-induced reduction in cocaine-enhanced NAc DA. Two-way ANOVA with repeated measures revealed a statistically significant treatment (vehicle vs. 2-PMPA) main effect (F2,22 = 4.13, p<0.05), time main effect (F7,154=36.81, p<0.001) and treatment x time interaction (F14,154 =4.58, p<0.001). Individual group comparisons revealed a significant reduction in cocaine-enhanced DA after 100 mg/kg 2-PMPA administration in the presence (t=2.63, p<0.05) or absence (t=2.03, p<0.05) of intra-NAc LY341495 perfusion, compared to the vehicle control group.
Figure 5.

Effects of local intra-NAc administration of LY341495 on basal and cocaine-enhanced extracellular NAc DA and glutamate. Intra-NAc administration of LY341494 neither altered basal extracellular NAc DA (A) nor attenuated 2-PMPA-induced reduction in cocaine-enhanced ANc DA (B). In contrast, intra-NAc administration of LY341495 dose-dependently elevated NAc glutamate (C) and blocked 2-PMPA-induced reduction in cocaine-enhanced NAc glutamate (D). *p<0.05, **p<0.01, ***p<0.001, compared to baseline (before cocaine) in each treatment group. #p<0.05, ##p<0.01, ###p<0.001, compared to vehicle treatment group.
Fig. 5C illustrates that intra-NAc perfusion of LY341495 (1–300 μM) dose-dependently elevated extracellular glutamate in the NAc (F1,11=6.90, p<0.05). Fig. 5B illustrates that intra-NAc local perfusion of LY341995 (300 μM) significantly blocked 2-PMPA-induced reduction in cocaine-enhanced NAc glutamate. Two-way ANOVA with repeated measures over time revealed a significant treatment main effect (F2,21=3.63, p<0.05), time main effect (F7,147=5.92, p<0.001), and treatment x time interaction (F14,147=1.84, p<0.05). Individual group comparisons revealed a significant reduction in cocaine-enhanced glutamate only in the absence of intra-NAc LY341495 (t=2.61, p<0.05), when compared to the vehicle control group.
Histology
Figure 6 illustrates the loci of intracranial microinjections of 2-PMPA or NAAG and the placement of microdialysis probes in rat brain. Microinjector tips aimed at the NAc were found to be within the NAc shell and core (Fig. 6A). Microinjection tips aimed at the dorsal striatum were found to be in the dorsal striatum, near the border of the corpus callosum (Fig. 6B). Fig. 6C shows the locations of microdialysis probes in the NAc. The membrane portions of the probes were primarily in the NAc core. Some were at the interface between the core and shell, and a few were in the shell.
Figure 6.
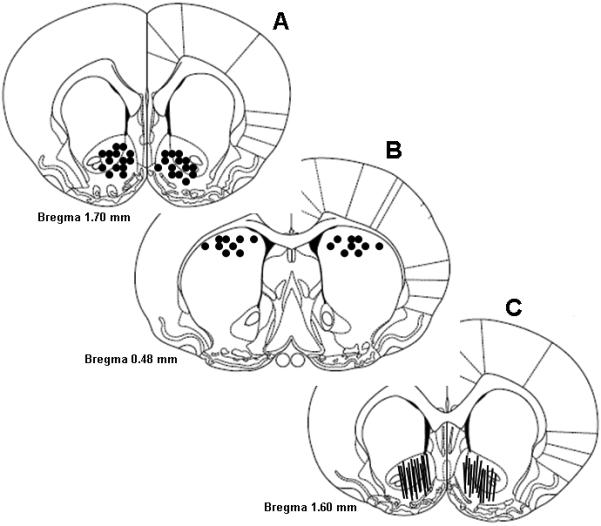
Schematic reconstructions of positions of intracranial microinjections or microdialysis probes in rat brain, indicating that the tips of microinjection guide cannula were located in the NAc (A) or the dorsal striatum (B), and that active microdialysis membranes tended to span the length of the core and shell compartments of the NAc (C).
Discussion
The major findings of the present study include: 1) systemic administration of the NAALADase inhibitor 2-PMPA significantly inhibited intravenous cocaine self-administration maintained by low unit doses of cocaine and inhibited cocaine-induced reinstatement of drug-seeking behavior. In contrast, 2-PMPA had no effect on non-drug (sucrose)-induced reinstatement of sucrose-seeking behavior; 2) microinjection of 2-PMPA or NAAG into the NAc, but not the dorsal striatum, also inhibited cocaine-induced reinstatement, an effect that was blocked by intra-NAc LY341495, a selective mGluR2/3 antagonist; 3) 2-PMPA dose-dependently lowered both extracellular DA and glutamate in the NAc by a mGluR2/3-dependent mechanism;, 4) pretreatment with 2-PMPA partially attenuated cocaine-enhanced extracellular NAc DA, while completely blocking cocaine-enhanced extracellular NAc glutamate in rats during reinstatement testing; and finally, 5) intra-NAc local perfusion of LY341495 failed to alter 2-PMPA-induced reduction in cocaine-enhanced NAc DA, but blocked 2-PMPA-induced reduction in cocaine-enhanced NAc glutamate. Intra-NAc LY341495 alone also failed to alter basal extracellular NAc DA, but dose-dependently increased extracellular NAc glutamate. These data suggest that the NAAG-mGluR2/3-glutamate signaling system in the NAc plays an important role in cocaine-induced relapse to drug-seeking behavior. Overall, these findings are consistent with previous studies demonstrating that 2-PMPA inhibits repeated cocaine-induced locomotor sensitization (Shippenberg et al., 2000), cocaine-induced CPP (Slusher et al., 2001) and high dose cocaine-kindled seizures (Witkin et al., 2002).
An important finding of the present study is that 2-PMPA only inhibits intravenous cocaine self-administration maintained by lower doses (0.03, 0.0625, 0.125, 0.25 mg/kg/infusion), but not the higher dose (0.5 mg/kg/infusion), of cocaine, suggesting that the pharmacological action of 2-PMPA in animal models of addiction is cocaine dose-dependent. The higher cumulative doses (0.5 mg/kg/infusion × 50 maximal infusions = 25 mg/kg) of cocaine may overcome 2-PMPA’s pharmacological effect. This is supported by our recent findings that 2-PMPA dose-dependently inhibits cocaine self-administration under progressive-ratio reinforcement (Xi et al., 2009), cocaine-enhanced brain-stimulation reward (Xi et al., 2009) and cocaine-induced reinstatement of drug-seeking behavior (present study), in which much lower doses (2–10 mg/kg) of cocaine were used.
Another important finding of the present study is that systemic or local intra-NAc (but not intra-dorsal striatum) administration of 2-PMPA significantly inhibited cocaine-induced reinstatement of drug-seeking behavior, suggesting an important role for the NAc in cocaine-induced reinstatement. Since intra-NAc local administration of 2-PMPA or NAAG dose-dependently inhibited cocaine-induced reinstatement, while co-administration of the selective mGluR2/3 antagonist LY341495 blocked the actions of 2-PMPA or NAAG on reinstatement, we suggest that a NAAG-mGluR2/3 mechanism may underlie the antagonism by 2-PMPA of cocaine-triggered relapse. That is, 2-PMPA inhibits NAALADase activity, causing an increase in brain NAAG levels that subsequently activates mGluR2/3s and inhibits cocaine-induced reinstatement of drug-seeking behavior. We note that systemic administration of 2-PMPA did not produce a typical pharmacological dose-response relationship. That is, higher doses of 2-PMPA appeared to be less effective than lower doses in attenuating cocaine-induced reinstatement of drug-seeking behavior. The mechanisms for the loss of the pharmacological action at high doses are unclear. First, it is likely that a high dose of 2-PMPA may produce non-selective binding to other functional proteins. Slushner et al (1999) reported that 2-PMPA, at 10 μM, was inactive in over 100 different receptor, transporter, ion channel and enzyme assays. Second, a high level of brain NAAG produced by high doses of 2-PMPA may act on other receptor signaling systems, which may attenuate the pharmacological action of NAAG on mGlu2/3 receptors.
A third important finding of the present work is that systemic administration of 2-PMPA dose-dependently lowered extracellular NAc DA and glutamate. Pretreatment with 2-PMPA only partially attenuated cocaine-enhanced extracellular NAc DA but completely blocked cocaine-enhanced extracellular NAc glutamate, suggesting an important role of NAc glutamate in mediating the pharmacological action of 2-PMPA. The importance of glutamate in the reinstatement of cocaine-seeking behavior has been extensively studied and established during the past decade (Kalivas, 2004; Knackstedt and Kalivas, 2009). It has been reported that there is a significant reduction in basal levels of extracellular glutamate in rats after cocaine self-administration and an enhanced glutamate release in the NAc during reinstatement of cocaine seeking (Baker et al., 2003; McFarland et al., 2003; Xi et al., 2006a). Congruently, normalization of decreased extracellular glutamate or blockade of NAc AMPA receptors blocks cocaine-induced reinstatement (Cornish and Kalivas, 2000; Di Ciana and Everitt, 2001; Baker et al., 2003; Xi et al., 2006a). Consistent with these findings, the present finding that 2-PMPA dose-dependently inhibited cocaine-enhanced NAc glutamate supports a critical role for NAc glutamate in cocaine-triggered reinstatement and the anti-relapse effects produced by 2-PMPA or NAAG. This is congruent with previous reports that increased glutamate tone on presynaptic mGluR2/3 by either blocking presynaptic cannabinoid CB1 receptors with AM251 (Xi et al., 2006a) or facilitating cystine-glutamate antiporter activity with N-acetylcystine (Baker et al., 2003; Moran et al., 2005) inhibits both cocaine-induced increases in NAc glutamate and reinstatement of drug-seeking behavior (Knackstedt and Kalivas, 2009).
The mechanisms by which 2-PMPA inhibits basal and cocaine-enhanced extracellular glutamate are not yet fully understood. An important observation in the present study is that the mGluR2/3 antagonist LY341495 almost completely blocked 2-PMPA-induced reductions in both basal and cocaine-enhanced extracellular glutamate (Figs. 3 and 5), suggesting that the pharmacological effects of 2-PMPA on glutamate may be mediated predominantly by activation of brain mGluR2/3 secondary to an increase in brain NAAG levels after 2-PMPA administration. This is consistent with a previous report that another NAALADase inhibitor, ZJ-43, inhibits traumatic brain injury-induced increases in glutamate release in the hippocampus by activation of brain mGluR3s (Zhong et al., 2006). Previous studies have shown that cocaine-induced increases in NAc glutamate are derived predominantly from presynaptic vesicular tetrodotoxin-sensitive sources (McFarland et al., 2003; Xi et al., 2006a), while mGluR2/3 may inhibit both Ca++-dependent vesicular and tetrodotoxin-insensitive nonvesicular NAc glutamate release (Xi et al., 2002). These data suggest that 2-PMPA-induced reduction in basal extracellular glutamate may be mediated by inhibiting both vesicular and nonvesicular glutamate release, while 2-PMPA-induced reduction in cocaine-enhanced NAc glutamate may be mediated predominantly by inhibiting presynaptic vesicular release. We note that inhibition of NAALADase by 2-PMPA causes a reduction in nonvesicular glutamate release from NAAG hydrolysis, which may also contribute to the reduction in basal extracellular NAc glutamate after 2-PMPA administration. Given that LY341495 almost completely blocked 2-PMPA-induced reductions in NAc glutamate (Fig. 3), we suggest that an increased NAAG-mGluR2/3 mechanism after 2-PMPA administration may play a critical role in lowering extracellular. Of course, we can not exclude such a possibility that that NAAG hydrolysis may occur inside the cell or synapse. Thus, 2-PMPA-induced changes in glutamate release deriving from NAAG hydrolysis may be difficult to assay using present in vivo brain microdialysis techniques.
In addition to NAc glutamate, cocaine priming also significantly increases extracellular NAc DA, suggesting an important role for NAc DA in cocaine-induced relapse (Shalev et al., 2002; Anderson and Pierce, 2005). This is supported by the evidence that systemic administration of DA reuptake inhibitors (De Vries et al., 1999) or intra-NAc infusions of DA (Cornish and Kalivas, 2000) evokes reinstatement of cocaine seeking. Administration of D2-like DA receptor agonists (Self et al., 1996; Khroyan et al., 2000; De Vries et al., 2002) reinstates cocaine seeking, while administration of D2-like DA receptor antagonists attenuates cocaine-induced reinstatement (Khroyan et al., 2000; Vorel et al., 2002; Xi et al., 2006b). In the present study, we also found that 2-PMPA dose-dependently lowered extracellular NAc DA, and that pretreatment with 2-PMPA partially attenuated cocaine-enhanced NAc DA, suggesting that a DA-dependent mechanism may contribute to the therapeutic effects of 2-PMPA on cocaine-primed relapse. We note that systemic administration of LY341495 blocked 2-PMPA-induced reductions in NAc DA, while intra-NAc local perfusion of LY341495 failed to block 2-PMPA-induced reductions in cocaine-enhanced NAc DA, suggesting that mGluR2/3 may be located predominantly on DA cell bodies and dendrites or on other cells outside the NAc, but not on DA terminals within the NAc. Another possibility for this finding is that a non-mGluR2/3 mechanism may underlie the action of 2-PMPA on cocaine-enhanced NAc DA.
Early studies suggested that NAAG is a selective mGluR3 agonist (Wroblewska et al., 1997). However, further studies indicate that NAAG may also bind to mGluR2 with similar affinity in CHO cells expressing rat mGluR2s (Cartmell et al., 1998). These data suggest that NAAG may be an endogenous mGluR2/3 agonist with only somewhat higher selectivity for mGluR3s over mGluR2s. This is in contrast to LY379268, which has 2-fold higher selectivity for mGluR2s over mGluR3s (Collado et al., 2002). Our NAAG-mGluR2/3 mechanism is further supported by our finding that pretreatment with the selective mGluR2/3 antagonist LY3414952 significantly blocked 2-PMPA- or NAAG-induced inhibition of cocaine-induced reinstatement, and 2-PMPA-induced reduction in extracellular NAc glutamate. However, these findings appear to conflict with two recent reports suggesting that NAAG may not be a selective mGluR3 agonist and that glutamate in unpurified NAAG may actually underlie the electrophysiological actions of NAAG observed in in vitro cells expressing human or rat mGluR3s or mGluR2s (Fricker et al., 2009; Chopra et al., 2009). However, this view is not supported by our findings in vivo that intranasal NAAG microinjection (by which drugs may directly enter the brain from the nose), but not similar doses of glutamate, inhibit cocaine-enhanced brain-stimulation reward in rats (Xi et al., 2009). Together, the present data suggest that the potential pharmacotherapeutic effects of 2-PMPA on cocaine-induced relapse to drug-seeking behavior appear to be mediated by activation of NAc mGluR2s and/or mGluR3s.
Regardless of the precise mechanisms underlying their pharmacological actions, 2-PMPA appears to differ significantly from LY379268. LY379268 has been shown to produce broad functional inhibition of multiple neurotransmitter release, of locomotor activity, and of drug and natural reward (Imre, 2007). In addition, repeated use of LY379268 also appears to produce rapid tolerance (Cartmell et al., 2000; Liechti et al., 2007). In contrast, 2-PMPA selectively inhibits cocaine reward and relapse (Xi et al., 2009; present study), but fails to inhibit basal levels of locomotion (Shippenberg et al., 2000), food-induced CPP (Slushner et al., 2001), sucrose-induced reinstatement (present study), basal brain reward functions (Xi et al., 2009), and learning and memory (Slushner et al., 1999). These data suggest that 2-PMPA or other NAALADase inhibitors may have fewer undesirable effects in potential therapeutic use for the treatment of cocaine addiction.
In conclusion, the present study demonstrates that the selective NAALADase inhibitor 2-PMPA significantly inhibits cocaine self-administration and cocaine-induced reinstatement of drug-seeking behavior, but has no effect on sucrose-seeking behavior. The underlying mechanisms may relate to 2-PMPA-induced increases in brain NAAG levels, which subsequently inhibit cocaine-enhanced extracellular glutamate and DA by activation of brain mGluR2/3s. The present findings support the potential use of 2-PMPA or other NAALADase inhibitors in the treatment of cocaine dependence.
Acknowledgement
This research was supported by the Intramural Research Program of the National Institute on Drug Abuse, National Institutes of Health, Department of Health and Human Services.
Footnotes
Disclosure/Conflict of Interest
All authors hereby declare that, except for income received from their respective primary employers, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional services. Co-authors Ajit G. Thomas and Barbara S. Slusher declare that their primary employer is Guilford Pharmaceuticals Inc. Co-author Ashby declares that, in addition to income received from his primary employer, he has received grant support from Guilford Pharmaceuticals Inc. for research on Guilford compounds.
References
- Adewale AS, Platt DM, Spealman RD. Pharmacological stimulation of group ii metabotropic glutamate receptors reduces cocaine self-administration and cocaine-induced reinstatement of drug seeking in squirrel monkeys. J. Pharmacol. Exp. Ther. 2006;318:922–931. doi: 10.1124/jpet.106.105387. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Pierce RC. Cocaine-induced alterations in dopamine receptor signaling: implications for reinforcement and reinstatement. Pharmacol. Ther. 2005;106:389–403. doi: 10.1016/j.pharmthera.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Baptista MA, Martin-Fardon R, Weiss F. Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J. Neurosci. 2004;24:4723–4727. doi: 10.1523/JNEUROSCI.0176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Adam G, Chaboz S, Henningsen R, Kemp JA, Klingelschmidt A, Metzler V, Monsma F, Schaffhauser H, Wichmann J, Mutel V. Characterization of [3H]-(2S,2′R,3′R)-2-(2′,3′-dicarboxy-cyclopropyl)glycine ([3H]-DCG IV) binding to metabotropic mGlu2 receptor-transfected cell membranes. Br. J. Pharmacol. 1998;123:497–504. doi: 10.1038/sj.bjp.0701647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Monn JA, Schoepp DD. Tolerance to the motor impairment, but not to the reversal of PCP-induced motor activities by oral administration of the mGlu2/3 receptor agonist, LY379268. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:39–46. doi: 10.1007/s002109900151. [DOI] [PubMed] [Google Scholar]
- Chopra M, Yao Y, Blake TJ, Hampson DR, Johnson EC. The neuroactive peptide N-acetylaspartylglutamate (NAAG) is not an agonist at the mGluR3 subtype of metabotropic glutamate receptor. J. Pharmacol. Exp. Ther. 2009;330:212–219. doi: 10.1124/jpet.109.152553. [DOI] [PubMed] [Google Scholar]
- Collado I, Pedregal C, Mazón A, Espinosa JF, Blanco-Urgoiti J, Schoepp DD, Wright RA, Johnson BG, Kingston AE. (2S,1′S,2′S,3′R)-2-(2′-carboxy-3′-methylcyclopropyl) glycine is a potent and selective metabotropic group 2 receptor agonist with anxiolytic properties. J. Med. Chem. 2002;45:3619–3629. doi: 10.1021/jm0110486. [DOI] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J. Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries TJ, Schoffelmeer AN, Binnekade R, Raasø H, Vanderschuren LJ. Relapse to cocaine- and heroin-seeking behavior mediated by dopamine D2 receptors is time-dependent and associated with behavioral sensitization. Neuropsychopharmacology. 2002;26:18–26. doi: 10.1016/S0893-133X(01)00293-7. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Schoffelmeer AN, Binnekade R, Vanderschuren LJ. Dopaminergic mechanisms mediating the incentive to seek cocaine and heroin following long-term withdrawal of IV drug self-administration. Psychopharmacology (Berl) 1999;143:254–260. doi: 10.1007/s002130050944. [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- Fell MJ, Svensson KA, Johnson BG, Schoepp DD. Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (-)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid ( LY404039) J. Pharmacol. Exp. Ther. 2008;326:209–217. doi: 10.1124/jpet.108.136861. [DOI] [PubMed] [Google Scholar]
- Fricker AC, Selina Mok MH, de la Flor R, Shah AJ, Woolley M, Dawson LA, Kew JN. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology. 2009;56:1060–1067. doi: 10.1016/j.neuropharm.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Galici R, Echemendia NG, Rodriguez AL, Conn PJ. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J. Pharmacol. Exp. Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- Imre G. The preclinical properties of a novel group II metabotropic glutamate receptor agonist LY379268. CNS Drug Rev. 2007;13:444–464. doi: 10.1111/j.1527-3458.2007.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzanti BA, Trainor DA. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated-α-linked acidic dipeptidase. J. Med. Chem. 1996;39:619–622. doi: 10.1021/jm950801q. [DOI] [PubMed] [Google Scholar]
- Johnson MP, Baez M, Jagdmann GE, Jr., Britton TC, Large TH, Callagaro DO, Tizzano JP, Monn JA, Schoepp DD. Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2- trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J. Med. Chem. 2003;46:3189–3192. doi: 10.1021/jm034015u. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Curr. Opin. Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Khroyan TV, Barrett-Larimore RL, Rowlett JK, Spealman RD. Dopamine D1- and D2-like receptor mechanisms in relapse to cocaine-seeking behavior: effects of selective antagonists and agonists. J. Pharmacol. Exp. Ther. 2000;294:680–687. [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Glutamate and reinstatement. Curr. Opin. Pharmacol. 2009;9:59–64. doi: 10.1016/j.coph.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liechti ME, Lhuillier L, Kaupmann K, Markou A. Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence. J. Neurosci. 2007;27:9077–9085. doi: 10.1523/JNEUROSCI.1766-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden AM, Baez M, Bergeron M, Schoepp DD. Effects of mGlu2 or mGlu3 receptor deletions on mGlu2/3 receptor agonist ( LY354740)-induced brain c-Fos expression: specific roles for mGlu2 in the amygdala and subcortical nuclei, and mGlu3 in the hippocampus. Neuropharmacology. 2006;51:213–228. doi: 10.1016/j.neuropharm.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Lu L, Uejima JL, Gray SM, Bossert JM, Shaham Y. Systemic and central amygdala injections of the mGluR(2/3) agonist LY379268 attenuate the expression of incubation of cocaine craving. Biol. Psychiatry. 2007;61:591–598. doi: 10.1016/j.biopsych.2006.04.011. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J. Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Miyakawa T, Furuyashiki T, Tanaka Y, Mizuma H, Nakanishi S. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4170–4175. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale JH, Bzdega T, Wroblewska B. N-Acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J. Neurochem. 2000;75:443–452. doi: 10.1046/j.1471-4159.2000.0750443.x. [DOI] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Gehl LM, Wroblewska B, Bzdega T. The neurotransmitter N-acetylaspartylglutamate in models of pain, ALS, diabetic neuropathy. CNS injury and schizophrenia. Trends Pharmacol. Sci. 2005;26:477–484. doi: 10.1016/j.tips.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Nagel J, Belozertseva I, Greco S, Kashkin V, Malyshkin A, Jirgensons A, Shekunova E, Eilbacher B, Bespalov A, Danysz W. Effects of NAAG peptidase inhibitor 2-PMPA in model chronic pain - relation to brain concentration. Neuropharmacology. 2006;51:1163–1171. doi: 10.1016/j.neuropharm.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. fourth ed. Academic Press; San Diego, CA: 1998. [Google Scholar]
- Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology. 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- Self DW, Barnhart WJ, Lehman DA, Nestler EJ. Opposite modulation of cocaine-seeking behavior by D1- and D2-like dopamine receptor agonists. Science. 1996;271:1586–1589. doi: 10.1126/science.271.5255.1586. [DOI] [PubMed] [Google Scholar]
- Shalev U, Grimm JW, Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol. Rev. 2002;54:1–42. doi: 10.1124/pr.54.1.1. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Rea W, Slusher BS. Modulation of behavioral sensitization to cocaine by NAALADase inhibition. Synapse. 2000;38:161–166. doi: 10.1002/1098-2396(200011)38:2<161::AID-SYN7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Thomas A, Paul M, Schad CA, Ashby CR., Jr. Expression and acquisition of the conditioned place preference response to cocaine in rats is blocked by selective inhibitors of the enzyme N-acetylated-alpha-linked-acidic dipeptidase (NAALADASE) Synapse. 2001;41:22–28. doi: 10.1002/syn.1056. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC, Tortella FC, Wozniak KM, Yudkoff M, Potter BM, Jackson PF. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999;5:1396–1402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T, Wozniak KM, Slusher BS. Progress in the discovery and development of glutamate carboxypeptidase II inhibitors. Drug Discov. Today. 2007;12:767–776. doi: 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Uejima JL, Bossert JM, Poles GC, Lu L. Systemic and central amygdala injections of the mGluR2/3 agonist LY379268 attenuate the expression of incubation of sucrose craving in rats. Behav Brain Res. 2007;181(2):292–6. doi: 10.1016/j.bbr.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Vorel SR, Ashby CR, Jr., Paul M, Liu X, Hayes R, Hagan JJ, Middlemiss DN, Stemp G, Gardner EL. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J. Neurosci. 2002;22:9595–9603. doi: 10.1523/JNEUROSCI.22-21-09595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Gasior M, Schad C, Zapata A, Shippenberg T, Hartman T, Slusher BS. NAALADase (GCP II) inhibition prevents cocaine-kindled seizures. Neuropharmacology. 2002;43:348–356. doi: 10.1016/s0028-3908(02)00124-7. [DOI] [PubMed] [Google Scholar]
- Woolley ML, Pemberton DJ, Bate S, Corti C, Jones DN. The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology. 2008;196:431–440. doi: 10.1007/s00213-007-0974-x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. NAAG selectively activates mGluR3 receptors in transfected cells. J. Neurochem. 1997;69:174–181. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- Xi Z-X, Baker DA, Shen H, Carson DS, Kalivas PW. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J. Pharmacol. Exp. Ther. 2002;300:162–171. doi: 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- Xi Z-X, Gilbert JG, Peng X-Q, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J. Neurosci. 2006;26:8531–8536. doi: 10.1523/JNEUROSCI.0726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z-X, Kiyatkin M, Li X, Peng X-Q, Wiggins A, Spiller K, Li J, Gardner EL. N-acetylaspartylglutamate (NAAG) inhibits intravenous cocaine self-administration and cocaine-enhanced brain-stimulation reward in rats. Neuropharmacology. 2009 Jun 24; doi: 10.1016/j.neuropharm.2009.06.016. 2009. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z-X, Newman AH, Gilbert JG, Pak AC, Peng X-Q, Ashby CR, Jr., Gitajn L, Gardner EL. The novel dopamine D3 receptor antagonist NGB 2904 inhibits cocaine’s rewarding effects and cocaine-induced reinstatement of drug-seeking behavior in rats. Neuropsychopharmacology. 2006;31:1393–1405. doi: 10.1038/sj.npp.1300912. [DOI] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Van KC, Bzdega T, Smyth A, Zhou J, Kozikowski AP, Jiang J, O’Connor WT, Berman RF, Neale JH, Lyeth BG. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J. Neurochem. 2006;97:1015–1025. doi: 10.1111/j.1471-4159.2006.03786.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat. Rev. Drug Discov. 2005;4:1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]