

Abstract
Previous studies have reported that activation of nicotinic ACh receptors on cultured pig retinal ganglion cells (RGCs) has a neuroprotective effect against glutamate-induced excitotoxicity (Wehrwein et al., 2004; Thompson et al., 2006). However, the mechanism linking nAChRs to neuroprotection is unknown. Here we tested the hypothesis that signaling cascades involving p38 MAP kinase and PI3 kinase → Akt are involved in linking activation of nAChRs to neuroprotection in isolated pig RGCs. In ELISA studies, regulation of phosphorylated p38 MAP kinase and Akt were analyzed after inducing excitotoxicity or neuroprotection in the presence and absence of specific inhibitors for p38 MAP kinase and PI3 kinase. ELISA results demonstrated that ACh significantly increased phosphorylated Akt and decreased p38 MAP kinase. Glutamate increased phosphorylated p38 MAP kinase but had no significant effect on phosphorylated Akt. Other ELISA studies using p38 MAP kinase and PI3 kinase inhibitors also supported the hypothesis that ACh up-regulated Bcl-2 levels downstream from PI3 kinase and Akt, whereas glutamate down-regulated Bcl-2 levels downstream from p38 MAP kinase.
RGC survival was subsequently assessed by culturing RGCs in conditions to induce excitotoxicity or neuroprotection in the presence or absence of specific inhibitors of p38 MAP kinase or PI3 kinase. The p38 MAP kinase inhibitor significantly decreased the number of RGCs that died by glutamate-induced excitotoxicity but had no effect on the number of cells that survived due to ACh-induced neuroprotection. PI3 kinase inhibitors significantly decreased cell survival caused by ACh-induced neuroprotection but had no effect on cell death caused by glutamate-induced excitotoxicity. These results demonstrate that glutamate mediates excitotoxicity through the p38 MAP kinase signaling pathway and that ACh provides neuroprotection by stimulating the PI3 kinase → Akt → Bcl-2 signaling pathway and inhibiting the p38 MAP kinase → Bcl-2 pathway.
Keywords: neuroprotection, Akt, p38 MAP kinase, excitotoxicity, retina, retinal ganglion cells
Introduction
Excitotoxicity is neuronal cell death caused by excessive neurotransmitter activity (Olney et al., 1978) and has been linked to various diseases of the central nervous system, such as Huntington's, Parkinson's, amyotrophic lateral sclerosis, AIDS, epileptic seizure and Alzheimer's disease (Choi 1988; Romano et al. 1998; Ozdener 2005; Fan and Raymond 2007; Henshall 2007). In the retina, glutamate-induced excitotoxicity has been shown to play a role in diseases such as retinal ischemia, diabetic retinopathy and glaucoma (Quigley et al. 1995; Lafuente et al. 2001; Casson 2006). Previous excitotoxicity studies have demonstrated that relatively high concentrations of glutamate applied to in vivo and ex vivo rodent models caused prolonged influx of non-specific cations in RGCs through ionotropic glutamate channels (Luo et al. 2001; Kawasaki et al. 2002; Wehrwein et al. 2004). Influx of excessive calcium through these glutamate channels triggers activation of apoptotic intracellular signaling cascades and ultimately leads to calcium-induced cell death (Quigley et al. 1995; Lam 1999).
Protecting neurons from excitotoxic cell death has been a significant focus in neuroscience research. Towards this end, a variety of agents that have neuroprotective effects against trauma, degenerative diseases, or excitotoxicity have been studied (Pearson et al. 2001; Ravina et al. 2003; Ozdemir et al. 2005; Shouten 2007). One of the agents that display neuroprotective properties against excitotoxicity in the CNS is acetylcholine (ACh) (Kaneko et al. 1997; Dajas-Bailador et al. 2000). In the brain, β-amyloid is considered to be the main culprit in the pathogenesis of Alzheimer's disease. However, nicotine displaces β-amyloid from its binding site at nAChRs and triggers several different signaling cascades that ultimately have a neuroprotective effect on cell survival (Shaw et al. 2002, Wang et al. 2007). In other studies, nicotine decreased cell loss in parts of the central nervous system associated with Parkinson's disease (Bordia et al. 2007). In the retina, previous studies demonstrated that activation of nicotinic acetylcholine receptors (nAChRs) on cultured adult pig RGCs led to neuroprotection against glutamate-induced excitotoxicity (Wehrwein et al. 2004; Thompson et al. 2006). These porcine studies demonstrated that total RGC survival decreased by an average of 42 ± 5% when cells were chronically treated with 500 µM glutamate for three days. However, if 5 µM ACh was added to plated cells prior to the glutamate insult, glutamate-induced excitotoxicity was significantly reduced, increasing cell survival to near control untreated conditions. These previous studies supported the hypothesis that 500 µM glutamate induces excitotoxic cell death of adult cultured pig RGCs and that ACh has a neuroprotective effect against this cell death through AChRs.
Other neuroprotective studies in the retina have demonstrated the type of ACh receptors associated with ACh-induced neuroprotection (Thompson et al. 2006). In the pig retina, antagonist studies demonstrated that α-bungarotoxin (α-Bgt) sensitive nAChRs in adult pig RGCs were linked to neuroprotection against glutamate-induced excitotoxicity (Wehrwein et al. 2004). Further analysis demonstrated that neuroprotection of large cultured porcine RGCs was initiated through the activation of α7 nAChRs subunits, whereas neuroprotection of small cultured porcine RGCs was initiated through activation of α4β2 nAChR subunits (Thompson et al. 2006). Muscarinic ACh receptors were not found to play any role in ACh-induced neuroprotection in pig RGCs (Wehrwein et al. 2004).
The mechanism responsible for ACh-induced neuroprotection in RGCs is unknown. In other systems, several signaling pathways have been linked to nicotinic ACh receptor (nAChR) neuroprotection against excitotoxicity-induced apoptosis. In some of these studies, agonist binding to α7 nAChRs activates the PI3 K → Akt → Bcl-2 pathway to enhance cell survival (Kihara et al. 2001; Shaw et al. 2002; Dejean et al. 2006; Dasari et al. 2008). Other studies have suggested that inhibition of mitogen-activated protein kinase (MAPK) pathways involved in apoptosis also results in neuroprotection (Dineley et al. 2001). MAPKs are a family of Ser/Thr protein kinases widely conserved among eukaryotes and are involved in many cellular programs such as cell proliferation, differentiation, survival movement and cell death. MAPK signaling cascades are organized into a three-tiered hierarchy. In this hierarchy, a surface stimulus activates MAPKKK to phosphorylate and activate MAPKK, which in turn phosphorylates and activates MAPK. There are three major MAPK pathways identified in mammals. These include MAPK/ERK, SAPK/JNK and p38 MAPK. Activation of the MAPK/ERK pathway has been associated with growth, differentiation, development and cell survival, whereas activation of p38 MAPK and SAPK/JNK pathways have been associated in inflammation, apoptosis, growth and differentiation (Mielke and Herdegen, 2000; Pearson et al. 2001; Zhuang and Schnellmann 2006; Bode and Dong 2007; Borsello and Forloni 2007; Thornton and Rincon 2009).
The objectives of the current study are to demonstrate that excessive glutamate leads to apoptosis in cultured adult pig RGCs and to identify which signaling pathways are involved in glutamate-induced excitotoxicity and ACh-induced neuroprotection using ELISA techniques and pharmacology. Results presented here will support the hypothesis that chronic exposure to a relatively high level of glutamate leads to apoptosis in pig RGCs, that a MAP kinase pathway is involved in glutamate-induced excitotoxicity and that two different signaling cascades are involved in ACh-induced neuroprotection. These results contribute to understanding the mechanism involved with these processes.
Methods
Animal Model
Adult porcine eyes were obtained immediately after slaughter from a local slaughterhouse (Pease Slaughterhouse, Scotts, MI) and kept on ice during transport to Western Michigan University for immediate dissociation and panning. Animal protocols were in accordance with IACUC.
Dissociation and Isolation
Pure RGCs were isolated from adult porcine eyes using a modified version of a two-step panning technique previously described by Wehrwein et al. (2004) and Thompson et al. (2006). Briefly, within 1 hour of eye removal, retinas were removed from the eyes. Isolated retinas were enzymatically treated with papain (27 µl/mg) for 20 minutes at 37°C in modified CO2-independent medium (Gibco), containing 4 mM glutamine, 5% fetal bovine serum (FBS), 5% antibiotic/antimyotic (Gibco) and 4 mM HEPES. After 20 minutes, the enzymatically treated tissue was inactivated with fresh 37°C CO2-independent medium and 1mg/ml DNase. Retinal tissue was subsequently dissociated by gentle trituration and transferred onto Petri dishes that were treated with goat anti-rabbit IgG antibody (Jackson ImmunoResearch; 0.5 mg in 48 ml of 20 mM Tris buffer) for 1 hour at 37°C to eliminate non-specific binding. This represented the first step in the panning procedure.
After an hour, the cells were transferred to other Petri dishes coated with mouse anti-rat Thy 1.1 antibody (BD Biosciences; 10 µg in 10 ml PBS containing zero calcium and zero magnesium) bound to goat anti-mouse IgM (Jackson ImmunoResearch; 0.3 mg in 48 ml of 20 mM Tris buffer). Thy 1.1 antigen is a ganglion cell-specific marker in the retina (Barnstable and Drager, 1984). Cells remained on the IgM/Thy 1.1 plates for 1 hour at 37°C, after which the supernatant was discarded. This represented the second step in the panning procedure and acted to separate the RGCs from other discarded retinal tissue (Wehrwein et al., 2004). The isolated RGCs bound to Thy 1.1 were released using 0.25% trypsin for 10 minutes at 37°C. Trypsin activity was stopped using 1 mg/ml soybean trypsin inhibitor and cells were strained. Cell density of the dissociated RGCs was calculated using a hemocytometer and cells were plated at a density of 1 × 105 cells/ml in modified CO2-independent medium for pharmacology studies or processed for ELISA studies.
ELISA procedure
ELISA techniques were used in this study to quantitatively measure the degree of up- or down-regulation of various phosphorylated enzymes in potential cell signaling cascades linked to excitotoxicity and neuroprotection. ELISAs were chosen to quantify protein content as they are offer a high degree of sensitivity to determine the absolute amounts of antigens in unknown samples (pg/ml) and provide an accurate measurement of the functional proteins in RGCs at the time of treatment.
After plating cells, agonists were added to RGCs to stimulate specific receptors. In each set of experiments, four conditions were kept constant: 1) cells were left untreated, 2) cells were treated with 5 µM ACh, 3) cells were treated with 500 µM glutamate, or 4) cells were treated with 5 µM ACh one hour prior to adding 500 µM glutamate. For experiments including specific inhibitors of the cell signaling enzymes, RGCs were incubated under the following conditions: 1) treated with 10 nM wortmannin, 2) treated with 10 nM LY 294002, or 3) treated with 10 nM SB 203580 for 30 minutes prior to ACh application and 2 1/2 hours before glutamate. RGCs were subsequently incubated for various times to determine peak phosphorylation of the various enzymes analyzed. After incubation, isolated pig RGCs were removed from large Petri dishes, washed with PBS and spun gently into a pellet. The cell pellet was lysed using a cell extraction buffer containing the following: 10 mM Tris, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM sodium pyrophosphate tetrabasic anhydrous, 2 mM sodium orthovanadate, 1% Triton X-100, 10% glycerol, 0.1% sodium dodecyl sulfate, 0.5% deoxycholate, 1 mM phenylmethanesulfonyl fluoride. Lysed cells were vortexed at 10-minute intervals and the cell extracts were transferred to microcentrifuge tubes and centrifuged at 13,000 rpm for 10 minutes at 4°C. Clear lysate was transferred to clean microcentrifuge tubes and kept at −80° C until the protein was assayed.
Each ELISA kit was purchased from Biosource International (Invitrogen) and came with a precoated 96-well plate containing a monoclonal antibody raised against the specific protein to be assayed. ELISA kits were designed to detect and quantify the level of phosphorylated proteins at specific residue sites. The specific residue sites detected by antibodies in each ELISA kits include: Akt[pS473], p38[pTpY180/182], ERK 1/2[pTpY185/187], JNK1/2[pTpY183/185], p53[pS15], Bcl-2[pS70] and NF-κBp65. For normalizing the protein contents of the samples, a total ELISA kit for each protein (Invitrogen) was purchased and used to calculate the total protein present in each sample. Total ELISA kits are independent of phosphorylation states. The percent phosphorylation of each protein was calculated under each experimental condition. All ELISA experiments were repeated a minimum of 7 times with similar results.
ELISA’s were performed according to the manufacturer’s instructions. Absorbance was measured on a PowerWave 200 microplate scanning spectrophotometer. For each assay, a standard curve was calculated from known protein standard concentrations that came in each kit. This standard curve was used to calculate unknown protein concentrations. The assay detected concentrations as low as 2.0 pg/ml of protein.
Pharmacological studies
In pharmacological studies, cells were plated at a density of 1 × 105 cells/ml in culture medium containing 5% fetal bovine serum (FBS), 5% antibiotic/antimyotic (Gibco), 4 mM HEPES, 4 mM glutamine, 0.3µg/ml NGF, 1 µg/ml transferrin, and 6 µg/ml insulin. Cells were allowed to settle for 2 hours in pretreated culture wells. After two hours, fresh modified CO2-independent replaced old medium. The first column of each 24-welled culture dish always contained untreated RGCs. The second column of each culture dish contained isolated RGCs treated for three days with 500 µM L-glutamate to induce excitotoxicity, the third column contained cells treated with 5 µM ACh, and the fourth column of wells contained cells were treated with 5 µM ACh for an hour before addition of 500 µM L-glutamate to induce neuroprotection against excitotoxicity. Cell survival in these four control columns of cells was compared to cell survival in the remaining columns of each 24-well culture dish containing RGCs treated with appropriate agonists and/or antagonists for glutamate or ACh receptors, and/or membrane permeable signaling pathway inhibitors. In studies using specific cell signaling inhibitors, the remaining columns of cells were treated with: 1) 10 nM wortmannin, 2) 10 nM LY 294002, or 3) SB 203580 for 30 minutes prior to ACh and subsequent glutamate application.
All culture supplements, agonists and inhibitors were obtained from Sigma unless noted otherwise. The nicotinic ACh antagonist α-Bgt, p38 MAP kinase inhibitor SB 203580, PI3 kinase inhibitors wortmannin and LY 294002 were obtained from Tocris. Concentrations of specific agents used in this study were determined from dose-response experiments. The minimal dosage that provided a maximum effect was used throughout the study. Once the optimal dosage was obtained, time-course studies were performed to determine the minimal time that elicited maximal effects. As a result of these time-course studies, the majority of inhibitors were applied to cultured cells for 30 minutes before application of ACh and/or glutamate. The one exception to this concerned α-Bgt, which required an hour to produce maximal effects (Wehrwein et al. 2004). Time-course studies using ACh or nicotine demonstrated that ACh and nicotine also needed to be applied to cells 1 hour before glutamate to produce maximal neuroprotective effects (Thompson et al. 2006).
Previous experiments from this lab have demonstrated that 3 days of chronic incubation in glutamate elicited a maximal excitotoxic effect on isolated panned pig RGCs (Wehrwein et al. 2004). After culturing for 3 days, pharmacologically treated and untreated RGCs were loaded with 2 µM of membrane permeable calcein for 1 hour to label viable cells. In the present study, a viable RGC was defined as a RGC body labeled with calcein-AM containing a process extending at least two cell diameters from the cell body (Bozyczko-Coyne et al. 1993). Background fluorescence levels were inherently low with this assay technique because the calcein dye is virtually non-fluorescent before interacting with cells.
To quantify RGC survival and death, five different sections of fluorescent cells from each well were imaged using a Hamamatsu XC-77 CCD camera, captured using a Metamorph Imaging system (Universal Imaging), and counted using ImagePro software (Media Cybernetics, Inc.). Microscopy was performed on a Nikon Diaphot epifluorescent research microscope illuminated by a 100-W mercury arc lamp with an excitation filter EX 510–590, dichroic mirror DM 580 and barrier filter BA590. The photographed sections from each culture well were consistently taken from the same location in each well. The number of cells from these five different sections were counted and averaged. This represented an "N" of one. The number of living RGCs in pharmacologically treated wells were compared to the number of living cells counted in the wells containing untreated RGCs to obtain a percent change from control. To minimize variation, the majority of results were listed as normalized values instead of listing the number of RGCs per well per condition. Although all RGCs were originally plated at the same concentrations, the amount of naturally occurring cell death that occurs during culture (Wehrwein et al. 2004), varied slightly between different experiments due to the variation in aspects of the dissociation process, such as tritration of the retina using Pasteur pipettes, the small variations in the time between eye removal and culturing, etc. Normalizing all pharmacologically treated RGC counts to the control untreated RGC counts in the same experiment eliminated this variation. A minimum of 12 animals were used to generate each pharmacological result. From each animal, cells were plated onto a minimum of 4 individual wells for each agent tested.
Identification of Apoptotic Cells
Apoptosis of RGCs was analyzed by in situ terminal deoxynucleotidyl transferase-mediated dUTP nick-ending labeling (TUNEL) using the ApoTag Red In Situ Apoptosis Detection Kit (Chemicon) according to the manufacturer’s instructions. RGCs were cultured for different amounts of time (0, 1, 3, 5 and 7 days). Labeled RGCs were visualized with the Nikon Diaphot epifluorescent research microscope using filters for rhodamine fluorescence. As in the pharmacological studies, five different sections of fluorescent cells from each culture well were recorded by the Hamamatsu XC-77 CCD camera and captured using the Metamorph Imaging system. The number of TUNEL positive cells were counted for each time point and compared to the number of TUNEL positive RGCs counted under control untreated conditions and normalized.
In control experiments, DNAse positive controls were used to ensure that the apoptotic technique labeled DNA fragments. For these studies, RGCs cultured for 0, 1, 3, 5, and 7 days were pretreated in a buffer containing 30 mM Trizma base, 4 mM MgCl2, and 0.1 mM dithiothreitol at room temperature for 5 minutes. After pretreatment, 1 µg/ml DNAse (Sigma D7291) was applied to cultured RGCs for 10 minutes at room temperature, followed by rinses with distilled water. Following these rinses, RGCs were subjected to the same TUNEL procedure as described above and consistent positive staining was revealed. In negative controls, RGCs were cultured for 0, 1, 3, 5 and 7 days and subjected to the same TUNEL protocol described above minus the addition of TdT. Under these conditions, no TUNEL staining occurred.
In some experiments, RGCs were counterstained with Hoechst 33342 to visual DNA fragmentation following TUNEL staining. Hoechst 33342 (10 µg/ml) was applied to cells already TUNEL stained for 30 minutes at 37°C and washed in Tris buffer before viewed under UV fluorescence.
Statistical Analysis
Statistical analysis was performed on all normalized data using Kruskal-Wallis non-parametric analysis of variance (ANOVA) with post hoc multiple comparisons (Dunn’s test). For data that was not normalized, statistical analysis was performed using ANOVA followed by a Tukey post hoc multiple comparison test. P<0.05 was considered statistically significant for all tests.
Results
Previous studies from this lab have demonstrated that relatively high concentrations of chronic glutamate exposure to adult cultured pig retinal ganglion cells (500 µM for 3 days) significantly decreased cell survival (Wehrwein et al. 2004; Thompson et al. 2006). Figure 1 demonstrates that cell death under these conditions was due to apoptosis. The panel of cells shown in figure 1A represent adult pig RGCs cultured under untreated conditions for three days. Both large and small RGCs have been labeled with 2 µM calcein to visualize living healthy cells (Wehrwein et al. 2004). To determine if glutamate-induced cell loss was a result of apoptosis, isolated RGCs were cultured at 1 × 105 cells/ml for 0, 1, 3, 5 and 7 days under untreated conditions and with 500 µM glutamate using TUNEL labeling.
Figure 1.
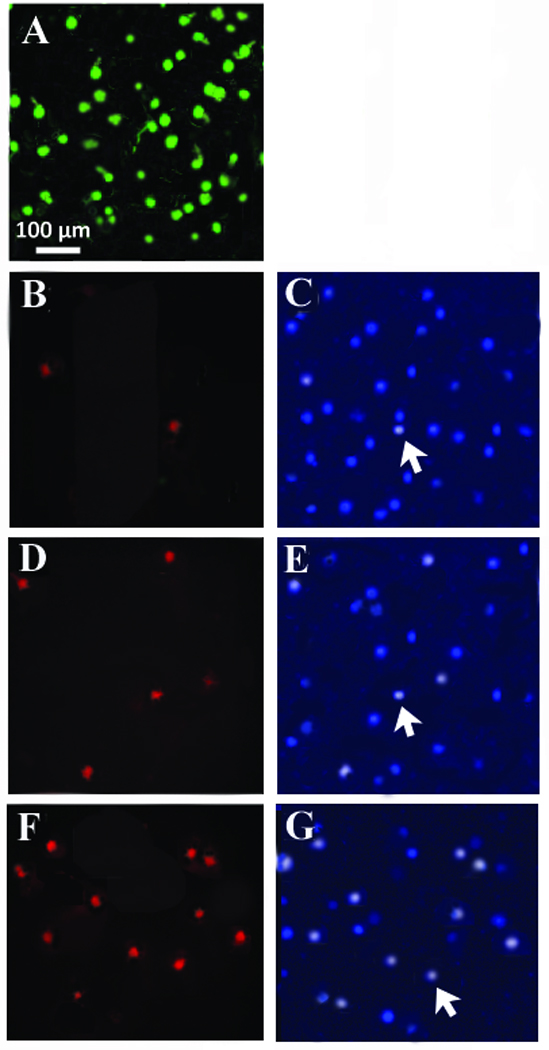
Cultured adult pig RGCs. (1A) Isolated pig RGCs were plated at 1 × 105 cells/ml and cultured for 3 days under control conditions. After 3 days, cells were labeled with calcein AM to label living cells and visualized. (1B). Isolated RGCs were plated and processed for TUNEL staining immediately after plating (Time 0), and counterstained with Hoescht (1C). (1D) TUNEL staining after 1 day in culture with 500 µM glutamate and the corresponding Hoescht stain (1E). (1F) TUNEL staining obtained after 3 days in culture with 500 µM glutamate and the corresponding Hoescht stain (1G). N’s = 7 for each time point with similar results.
The images shown in fig. 1B and 1C were obtained from dissociated RGCs that were plated onto culture dishes and immediately processed for apoptotic and DNA staining (Time 0). Fig. 1B demonstrates that few cells were positive for TUNEL staining at this point and fig. 1C represents the Hoechst stain that corresponds to the same time point. The Hoechst stain labels DNA and will fluoresce brightly if DNA fragmentation and condensation occurs. Fig. 1D and 1E represent TUNEL stained cells after RGCs were cultured in 500 µM glutamate for 1 day (fig. 1D) and the corresponding Hoechst stain (fig. 1E). Fig. 1F and 1G represent TUNEL stained cells after RGCs were cultured in 500 µM glutamate for 3 days (fig. 1F) and the corresponding Hoechst stain (fig. 1G). TUNEL positive cells were observed in all plates, however untreated plates had significantly fewer TUNEL-positive profiles than plates containing RGCs treated with glutamate. In addition, the percentage of TUNEL-positive cells gradually increased for 3 days and then remained relatively constant for the remainder of the week (figure 2), which corresponds with the excitotoxic effects of glutamate on cultured RGCs (Wehrwein et al. 2004).
Figure 2.

Bar graphs summarizing the mean percent of apoptotic positive cells counted at different time points. Mean values were obtained by repeating experiments 7 different times. Error bars represent S.E. * represents significance from untreated conditions (P <0.05).
A number of previous studies have looked at the cell signaling pathways involved in excitotoxicity and neuroprotection (Mielke and Herdegen, 2000; Dineley et al. 2001; Kihara et al. 2001; Pearson et al. 2001; Shaw et al. 2002; Dejean et al. 2006; Zhuang and Schnellmann 2006; Bode and Dong 2007; Borsello and Forloni 2007; Dasari et al. 2008; Thornton and Rincon 2009). In this study, we analyzed up- and down-regulation of several enzymes in these cell signaling pathways to determine if they play a role in glutamate-induced excitotoxicity or ACh-induced neuroprotection in pig RGCs.
Peak Akt phosphorylation
Preliminary studies from this lab suggested that Akt phosphorylation levels changed if RGCs were cultured in ACh. To determine when peak phosphorylation of Akt occurred in cultured pig RGCs, cells were harvested for ELISA processing after cultured in 5 µM ACh or in 500 µM glutamate for various amounts of time (Fig. 3). Under untreated conditions (Time 0), less than 20% of the total Akt in pig RGCs was phosphorylated. When cells were cultured in 5 µM ACh, total phosphorylated Akt levels significantly increased in a time dependent manner up to 12 hours and then steadily decreased. After incubating cells in ACh for 12 hours, 61 ± 4% of total Akt was phosphorylated compared to the levels of phosphorylated Akt measured under untreated conditions. These percentages represented a change of total phosphorylated Akt from 0.29 ng/ml of lysate measured under untreated conditions to 1.01 ng/ml of lysate after 12 hours (Table 1). Although ACh significantly increased Akt phosphorylation after 12 hours, 500 µM glutamate was found to have no significant effect on Akt phosphorylation levels at any time point (Fig. 3, Table 1). These results demonstrated that Akt phosphorylation significantly increased in the presence of ACh. Therefore, Akt may be involved in ACh-induced neuroprotection. On the other hand, glutamate's excitotoxic effect does not appear to be mediated through Akt phosphorylation.
Figure 3.

Peak phosphorylation of Akt. The line graphs in figure 3 represent the percent of total Akt phosphorylation measured using ELISA techniques when adult isolated RGCs were cultured in 5 µM ACh (circles) or 500 µM glutamate (boxes) for various amounts of time before processing cells for lysate formation. * represents a significant difference from untreated conditions (P <0.05). Data were obtained by repeating experiments 7 different times. For each experiment, 12 pig retinas from were used. Error bars represent S.E.
Table 1.
Phosphorylation of enzymes involved in excitotoxicity and neuroprotection. Values listed represent the total amount of phosphorylated enzyme measured from pig RGCs cultured under untreated conditions, in the presence of 5 µM ACh and in the presence of 500 µM glutamate.
| Phosphorylated enzyme | Untreated | ACh | Glutamate | Time to Peak Phosphorylation |
|---|---|---|---|---|
| Akt | .29 (ng/ml lysate) (+/−0.09) |
1.01 (ng/ml)# (+/− 0.14) |
.21 (ng/ml) (+/− 0.06) |
12 hrs |
| p38 | .39 (+/− 0.06) |
.19#^ (+/− 0.04) |
1.17# (+/− 0.2) |
12 hrs |
| JNK | .22 (+/− 0.06) |
.26 (+/− 0.04) |
.21 (+/− 0.04) |
N/A |
| ERK | .38 (+/− 0.08) |
.42 (+/− 0.09) |
.39 (+/− 0.09) |
N/A |
| p53 | .42 (+/− 0.04) |
.36 (+/− 0.02) |
.48 (+/− 0.04) |
N/A |
| Bcl2 | .08 (+/− 0.05) |
.68# (+/− 0.10) |
----* | 20 hrs |
+/− represent standard error.
indicated no detectable level was measured,
represents significance from untreated conditions,
represents significance from glutamate treated and N/A means not applicable.
Significance was determined using an ANOVA followed by a Tukey post hoc multiple comparison test. P < 0.05 was considered statistically significant.
It is possible that different signaling pathways have different time-courses of activation. As a result, ELISA time-course studies were performed on all enzymes examined. The results of these studies demonstrated that peak phosphorylation occurred for all enzymes between 12 and 20 hours under our culture conditions. The time corresponding to peak phosphorylation for each enzyme is listed in Table 1. Cells were harvested at times that corresponded to peak phosphorylation for each analyzed protein.
Phosphorylation of Akt and p38 MAP kinase
Figure 4 represents how phosphorylated Akt (Fig. 4A) and p38 MAP kinase (Fig. 4B) levels change in pig RGCs when excitotoxicity was induced using glutamate or when ACh induced neuroprotection. As represented by the bar graphs in figure 4A, Akt phosphorylation significantly increased from control untreated conditions if ACh was applied alone or if ACh was applied prior to glutamate by a 3-fold increase. When statistical comparisons between groups was analyzed, the increase of phosphorylated Akt due to ACh or ACh and glutamate application was significantly reduced in the presence of 10 nM α-bungarotoxin (α-Bgt) (fig. 4A). On the other hand, glutamate had no significant effect on Akt phosphorylation compared to untreated conditions. These results support the hypothesis that ACh's neuroprotection is mediated through nAChRs and involves Akt.
Figure 4.

Phosphorylation of Akt and p38 MAP kinase. Each bar represents the mean percent of total Akt (fig. 4A) or p38 MAP kinase (fig. 4B) phosphorylation measured using ELISA techniques after isolated RGCs were cultured under various pharmacological conditions for 12 hours. * represents a significant difference from untreated conditions, # represents a significant difference from ACh alone or ACh with glutamate conditions, and the solid circle represents a significant difference from RGCs treated with glutamate, or alpha-Bgt/ACh/glutamate (P <0.05). Data were obtained by repeating experiments 7–12 times. Error bars represent S.E.
Figure 4B demonstrates that p38 MAP kinase phosphorylation significantly decreased when cells were cultured in 5 µM ACh, and significantly increased with RGCs were incubated in 500 µM glutamate. Under untreated conditions, 24 ± 6% of total p38 MAP kinase was phosphorylated after cells were cultured for 12 hours. However, levels of p38 MAP kinase significantly decreased after incubating cells in 5 µM ACh. On the other hand, if cells were cultured in 500 µM glutamate, the percent of phosphorylated p38 MAP kinase significantly increased, representing more than a 2-fold increase in phosphorylated p38 MAP kinase. In other experiments, ACh was applied before glutamate and the amount of phosphorylated p38 MAP kinase was measured. Statistical analysis between groups indicated that the levels of phosphorylated p38 MAP kinase measured under these conditions was significantly less than levels obtained when RGCs were cultured in glutamate only (fig. 4B). Under these conditions, ACh acted to reduce glutamate’s effect on p38 MAP kinase. To verify that the reduction of glutamate’s effect on p38 MAP kinase levels involved ACh receptor activation, inhibition studies using α-Bgt before ACh and glutamate were performed. When α-Bgt was used to block ACh’s effect , phosphorylated p38 MAP kinase levels increased back to levels associated with glutamate alone. Statistical comparisons between groups demonstrated that p38 MAP kinase levels associated with RGCs treated with α-Bgt/ACh/glutamate were significantly different from cells treated with ACh and glutamate, but were not significantly different from phosphorylated p38 MAP kinase levels measured from glutamate exposure alone. These results support the hypothesis that both ACh and glutamate regulate phosphorylation of p38 MAP kinase and that p38 MAP kinase may be involved in the apoptotic pathway induced by 500 µM glutamate. Phosphorylation of p38 MAP kinase is significantly up-regulated by glutamate, suggesting its involvement in the excitotoxic pathway, and it is significantly down-regulated by ACh, increasing the effectiveness of the neuroprotective pathway. Results using α-Bgt also supports ACh's involvement in neuroprotection.
Inhibition of p38 MAP kinase and PI3 kinase
Phosphorylated Akt levels significantly increased after ACh application, which supports the hypothesis that PI3 kinase → Akt is involved in ACh-induced neuroprotection. Likewise, phosphorylated p38 MAP kinase levels significantly increased after glutamate application, thereby supporting a second hypothesis that a MAPKKK→ MAPKK→ p38 MAP kinase pathway is involved in glutamate-induced excitotoxicity. If these hypotheses are correct, inhibiting the enzymes in these pathways should block the effects of ACh and glutamate respectively. Figure 5 summarizes the ELISA results obtained when PI3 kinase and p38 MAP kinase inhibitors were applied to cells before application of ACh, glutamate or ACh and glutamate. As previously shown, under untreated conditions, the percent of total Akt phosphorylation significantly increased from untreated conditions if cells were incubated with ACh. This increase was not significantly blocked if cells were pretreated with the p38 MAP kinase inhibitor, SB 203580 (10 nM) before addition of ACh or ACh and glutamate. However, phosphorylated Akt levels were significantly reduced from ACh-induced levels if inhibitors of PI3 kinase, wortmannin or LY 294002 (10 nM), were added to cells prior to ACh application or prior to ACh and glutamate application (Fig. 5A). Comparison studies between groups demonstrate that the percent of Akt phosphorylation measured from RGCs treated with wortmannin or LY 294002 before ACh and glutamate, were significantly different from the amount of Akt phosphorylation measured when cells were incubated in ACh alone or ACh and glutamate.
Figure 5.

Inhibition of signaling enzymes involved in glutamate-induced excitotoxicity and ACh-induced neuroprotection. Each bar represents the mean percent of total Akt (fig. 5A) or p38 MAP kinase phosphorylation (fig. 5B) measured using ELISA techniques after isolated RGCs were cultured for 12 hours under various pharmacological conditions. * represents a significant difference from untreated conditions, # represents a significant difference from all other conditions in fig. 5B. The solid circle also represents a significant different from all other conditions in fig. 5B. (P <0.05). Results were obtained by repeating experiments 7–12 times. Error bars represent S.E.
As previously demonstrated in fig. 4, glutamate alone significantly increased the percent of total p38 MAP kinase phosphorylation. Addition of the p38 MAP kinase inhibitor, SB 203580, prior to glutamate application, significantly decreased the percent of total p38 MAP kinase phosphorylation to near untreated levels (Fig. 5B, left bars). However, the PI3 kinase inhibitors, wortmannin and LY 294002, had no significant effect on glutamate’s effect on p38 MAP kinase levels (Fig. 5B, left bars). These results support the hypothesis that p38 MAP kinase is likely involved in glutamate-induced excitotoxicity and in the absence of ACh, the PI3 → Akt cascade does not likely contribute to glutamate’s excitotoxic effect.
However, when ACh is present, p38 MAP kinase levels significantly decrease. The right bars in fig. 5B summarize the p38 MAP kinase levels recorded in the presence of ACh and glutamate and when SB 203580, wortmannin or LY 294002 are applied before ACh and glutamate. Phosphorylated p38 MAP kinase levels measured in the presence of ACh and glutamate were significantly reduced from levels obtained when p38 MAP kinase levels were measured from cells incubated in glutamate alone. There was also a significance difference between groups when the cells were treated with SB/ACh/Glut and statistically compared to cells cultured under untreated control conditions. On the other hand, when wortmannin or LY 294002 was applied before ACh and glutamate, comparison analysis between groups demonstrated a significant increase in p38 MAP kinase levels compared to results obtained when ACh and glutamate were applied without PI3 kinase inhibitors, but no significant difference compared to cells incubated in glutamate alone (Fig. 5B). Results obtained using these pharmacological inhibitors support the hypothesis that the PI3 K → Akt pathway is involved in ACh-induced neuroprotection and p38 MAP kinase is involved in glutamate-mediated excitotoxicity.
Additional enzymes involved in excitotoxic and neuroprotective pathways
To determine if other enzymes are involved in excitotoxic and neuroprotective pathways in pig RGCs, the protein content of additional enzymes was determined using ELISA kits and the results were listed in Table 1. Phosphorylated JNK, ERK, or p53 protein content did not vary significantly from control in the presence of ACh or glutamate. However, phosphorylation of Bcl-2 increased significantly from control in the presence of ACh. In the presence of glutamate, Bcl-2 levels dropped to a level that could not be detected. Taken together, these results suggest that JNK, ERK, and p53, enzymes thought to contribute to apoptotic pathways in other systems (Kim et al., 2008, Liu et al., 2008), are not likely to be involved in the excitotoxic or neuroprotective pathways in pig RGCs. However, Bcl-2, proposed to be a downstream target of Akt in cell survival pathways, is significantly up-regulated by ACh and down-regulated by glutamate (Table 1).
To support the hypothesis that Bcl-2 is a downstream target for both ACh and glutamate, SB 203580 was used in ELISA studies to inhibit p38 MAP kinase activity and wortmannin was used to block PI3 kinase activity. The ELISA results based on these experiments are summarized in figure 6. These results were not normalized to total Bcl-2 levels as the phosphorylated Bcl-2 measured in the presence of glutamate was below the detection capabilities of the ELISA (2 pg/ml). However, based on the phosphyorylated Bcl-2 levels measured, SB blocked glutamate’s decrease of phosphorylated Bcl-2. In addition, wortmannin significantly blocked the ACh-induced increase of phosphorylated Bcl-2 as well as the ACh/glutamate increase of Bcl-2. Statistical analysis demonstrated that the ACh/glutamate levels of Bcl-2 was significantly different from Bcl-2 levels obtained when cells were incubated in ACh alone or when cells were incubated in SB/ACh/glut. Taken together, these ELISA results suggest that Bcl-2 is a downstream target to Akt as well as p38 MAP kinase. Akt results in up-regulation of Bcl-2, whereas activation of p38 MAP kinase results in down-regulation of Bcl-2.
Figure 6.

Bcl-2 phosphorylation by Akt and p38 MAP kinase. Each bar represents the mean Bcl-2 phosphorylation measured using ELISA techniques after isolated RGCs were cultured for 20 hours under various pharmacological conditions. The solid circle represents a significant difference from all conditions except when RGCs were cultured in ACh/glutamate or SB 203580/ACh/glutamate. The star represents a significant difference from RGCs treated with ACh, ACh/glutamate or SB 203580/ACh/glutamate. # represents a significant difference from all other pharmacologically treated groups. ^ indicates that Bcl-2 phosphorylation levels were below detection levels when RGCs were incubated in 500 µM glutamate for 20 hours. Results were obtained by repeating experiments 7 times. Error bars represent S.E.
Effect of cell signaling inhibitors on cell survival
Results from ELISA studies can only demonstrate a change in levels of phosphorylated enzymes. To determine if changes in enzyme levels correspond to changes in cell survival, the pharmacological inhibitors of cell signaling enzymes used in ELISA studies were also used on cultured pig RGC to determine their effect on cell survival. In Figure 7, pharmacological inhibitors were used prior to inducing excitotoxicity or neuroprotection in cultured pig RGCs. Cell survival was accessed after 3 days in culture and compared to survival of cultured cells under untreated conditions. In previous studies, the viability of cultured RGCs after 3 days have been reported (Wehrwein et al., 2004). In figure 7A, images of RGCs cultured for 3 days under some of the various pharmacological conditions are illustrated. The bar graphs shown in figure 7B (left bar graphs) quantify and summarize the excitotoxic effect of glutamate and the neuroprotective effect of ACh on survival of cultured pig RGCs. If RGCs were treated with 500 µM glutamate alone, total cell survival significantly decreased compared to total cell survival obtained under untreated conditions. However, if 5 µM ACh was applied to cultured pig RGCs an hour before the glutamate insult, cell survival increased to near untreated control values.
Figure 7.

Inhibitors of PI3 kinase and p38 MAP kinase affect RGC survival. Figure 7A represents images of calcein AM labeled RGCs after being cultured for 3 days under various pharmacological conditions. In figure 7B, each bar represents the mean percent survival of adult isolated RGCs counted after cultured for 3 days in the presence of of agonists +/or antagonists. * represents a significant difference from untreated conditions. The solid circle represents a significant difference from glutamate treated RGCs. (P <0.05). Error bars represent S.E.
The middle bars in figure 7B represent the summarized effect of the p38 MAP kinase inhibitor, SB 203580, on glutamate-induced excitotoxicity and ACh-induced neuroprotection. If glutamate triggers a MAPKKK → MAPKK → p38 MAP kinase pathway, p38 MAP kinase inhibitor should block glutamate-induced excitotoxicity. When 10 nM SB 203580 was applied to cells before glutamate, cell survival significantly increased compared to cells cultured in glutamate alone. SB 203580, however, had no significant effect on cell survival if it was added before ACh, or if it was added before ACh and glutamate to induce neuroprotection (fig. 7B, middle bars). These results support the hypothesis that p38 MAP kinase is involved in glutamate-induced excitotoxicity but not in ACh-induced neuroprotection.
ELISA results demonstrated that ACh mediated up-regulation of Akt suggesting that PI3 kinase and Akt may be involved in neuroprotection. If ACh triggers a PI3 K → Akt → Bcl-2 survival pathway, PI3 kinase inhibitors should block ACh-induced neuroprotection against glutamate-induced excitotoxicity and lead to increased cell death. The right group of bars in figure 7B represents the summarized effect of wortmannin on ACh-induced neuroprotection in cultured RGCs. When cells were pretreated with wortmannin (fig. 7B, right bars) or LY 294002 (not shown) before addition of ACh and glutamate, the normal ACh-induced neuroprotective effect against glutamate-induced excitotoxicity was eliminated. The results obtained from these cell culture studies support the hypotheses generated as a result of the previous ELISA studies. ACh acts as a neuroprotective agent by increasing the PI3-Akt survival pathway and simultaneously inhibits the excitotoxic p38-MAP kinase pathway.
Discussion
In this study, we provide evidence that 500 µM glutamate mediates excitotoxic cell death in isolated pig retinal ganglion cells through the p38 MAP kinase signaling pathway and that relatively low concentrations of ACh (5 µM) provides neuroprotection against glutamate insult by activating the PI3 K → Akt → Bcl-2 signaling pathway and inhibiting p38 MAP kinase → Bcl-2 pathway. These conclusions were based on a combination of ELISA and pharmacological results obtained from cultured cell survival studies.
Neuroprotection evidence
ELISA studies demonstrated that phosphorylated Akt protein content was up-regulated by approximately 300% when cells were incubated in relatively low concentrations of ACh. This increase occurred whether cells were treated with 5 µM ACh alone or if ACh was applied to cells before glutamate insult. ACh also significantly increased levels of Bcl-2 to support the hypothesis that ACh activates the PI3 kinase → Akt → Bcl-2 signaling pathway. Bcl-2 has been identified as a downstream effector of the PI3 kinase → Akt survival pathway. It has been reported that Akt activation leads to the overexpression of Bcl-2 and nicotine can activate Akt via PI3 kinase thereby inducing Bcl-2 and Bcl-x upregulation. (Berra et al. 1998; Matsuzaki et al., 1999: Kihara et al. 2001; Manabe and Lipton 2003; Mai et al. 2003). The results presented in this study support these conclusions and suggest that ACh induces activation of Akt survival pathway and likely involves the anti-apoptotic form of Bcl-2.
While some studies propose that the PI3 K→ Akt→ Bcl-2 pathway mediates cell survival (Henshall et al. 2002) other studies promote PI3 K→ Akt→ NF-κB as the pathway of choice for cell survival (Shimamura et al. 2003; Wang et al. 2007), It is possible that they are both simultaneously activated by Akt, as NF-κB is a nuclear transcription regulator with a specific motif for Bcl-2 transcription (Viatour et al. 2003) and Bcl-2 governs mitochondrial outer membrane permeabilization (Dejean et al., 2006). ELISA studies were not performed to measure phosphorylated NF-κB in cultured pig RGCs , as phosphorylation of NF-κB is not a reliable measurement of NF-κB activity since translocation of NF-κB is required for this transcription factor’s activation. As the procedure to address translocation of NF-κB to RGC nucleus is beyond the scope of this study, the effect of ACh on NF-κB was not addressed. Therefore, it is possible and likely that pAkt phosphorylates other downstream targets besides Bcl-2 during ACh-induced neuroprotection.
ELISA studies also demonstrated that ACh significantly decreased total phosphorylated p38 MAP kinase, which is likely to be involved in the glutamate excitotoxic pathway. In addition, when ACh was applied with glutamate, the levels of p38 MAP kinase measured were significantly less than the levels of p38 MAP kinase recorded when glutamate was applied alone. The possibility that ACh leads to inhibition of p38 MAP kinase was also supported by pharmacology experiments using PI3 kinase inhibitors on cultured pig RGCs. In the presence of two different PI3 kinase inhibitors, ACh's neuroprotection against glutamate-induced excitotoxicity was eliminated. These results suggest that ACh is working on multiple signaling enzymes to enhance cell survival. Therefore, it is likely that ACh acts as a neuroprotective agent by increasing the PI3-Akt survival pathway and simultaneously inhibits the excitotoxic p38-MAP kinase → Bcl-2 pathway (figure 8). When ACh was not present, however, the Akt-induced survival pathway is not activated and inhibition of p38 MAP kinase does not occur.
Figure 8.

Proposed schematic of ACh-induced neuroprotection. Activation of nAChRs triggers cell survival through PI3 kinase → Akt → Bcl-2 and inhibition of the apoptotic p38 MAP kinase → Bcl-2 pathway.
Glutamate-induced excitotoxicity evidence
ELISA and pharmacology studies demonstrated that p38 MAP kinase was involved in glutamate-induced excitotoxicity. ELISA results demonstrated that total phosphorylated p38 MAP kinase protein content increased approximately 2-fold when glutamate was applied to cultured RGCs, while there was no significant effect on phosphorylated Akt protein content. This suggests that p38 MAP kinase, not Akt, is involved in glutamate-induced excitotoxicity.
Glutamate was also found to decrease levels of phosphorylated Bcl-2 to below detection levels, suggesting that Bcl-2 is also a downstream target for p38 MAP kinase. This was substantiated when a significant difference was found in phosphorylated Bcl-2 levels between RGCs cultured in ACh/Glut and those cultured in SB 203580/ACh/Glut. Several other studies have demonstrated that the anti-apoptotic role of Bcl-2 can be regulated by phosphorylation from p38 MAP kinase (De Chiara et al., 2006; Ranawat and Bansal, 2008; Markou et al., 2009). In fact, Bcl-2 phosphorylation by activated p38 MAP kinase is a key event in the early induction of apoptosis under conditions of cellular stress (De Chaira et al., 2006).
The hypothesis that p38 MAP kinase is involved in glutamate-induced excitotoxicity was also supported by pharmacology studies using cultured pig RGCs. If cultured cells were treated with SB 203580 before glutamate insult, glutamate's excitotoxic effect was eliminated. Like all MAP kinases, p38 MAP kinase is activated by dual kinases termed the MAP kinase kinases (MKKs) (Zarubin and Han,2005). Therefore, the likely signaling cascade activated by excessive glutamate in isolated pig RGCs is: MAPKKK → MAPKK → p38 MAP kinase.
In our current study, phosphorylation of ERK, JNK, and p53 in the presence of ACh or glutamate did not vary significantly from untreated levels (Table 1). These MAPK enzymes do not appear to have a role in excitotoxicity or neuroprotection in pig RGCs and support previous research that demonstrates these enzymes do not play an active role in excitotoxicity or neuroprotection in this tissue type. However, other studies have demonstrated these molecules do have a role in RGC survival (Park et al. 2004; Pernet et al. 2005; Zhou et al. 2005). It may be that the different results obtained in these other studies is due to the type of receptor that initiates the second messenger signaling cascades or due to the type of stimulus the cells encounter. In cultured pig RGC system, previous studies have demonstrated that ACh-induced neuroprotection is mediated through activation of nACh receptors (Thompson et al. 2006). In other studies, different receptors are activated by different stimuli to trigger signaling cascades. It is likely that varied stimuli will result in varied responses depending on the pathways triggered. Another issue that can influence experimental outcome is the experimental model used. This pharmacology study was conducted on a primary culture of isolated pig RGCs and therefore only provides a crude model of the behavior of cells in an intact retina. A change in the RGC’s extracellular environment may also be a factor influencing the results from this study and could explain why there are different results recorded from several other studies where signaling pathways in retinal excitotoxicity and neuroprotection have been implicated. Further studies to address this issue are required and are currently underway to demonstrate that ACh plays a neuroprotective role in the retina in vivo.
Previous studies have demonstrated that the link between glutamate receptor activation and signaling cascade initiation is calcium permeation through glutamate channels (Lam et al. 1999; Quigley et al., 1995). What is the likely link between nAChR activation and the neuroprotective pathways identified in this study? One possibility is that PI3 kinase physically associates with nAChRs. When ACh binds to the nAChRs, PI3 kinase is activated and the signaling cascade is triggered. The other likely scenario involves calcium. Calcium as well as sodium ions permeate nAChR channels (Burnashev 1998). It has been proposed that calcium influx through activated alpha-7 nAChRs could be involved in activation of the neuroprotective pathway (Kihara et al. 2001). Previous studies from this lab have demonstrated that large RGCs contain alpha-7 nAChR subunits (Thompson et al.,2006). Therefore, calcium may be the link between receptor activation and the second messenger system cascade. In fact, calcium influx has been found to activate PI3 kinase in other systems (Liu et al. 2007). Figure 8 summarizes a possible scenario linking nAChR activation to signaling messenger cascades leading to neuroprotection in pig RGCs. In this scenario, ACh binds to nAChRs and activate nAChR channels, which allows influx of calcium and sodium. Calcium would then bind to calcium-dependent enzymes to trigger the PI3 → Akt survival pathway and Bcl-2. At the same time, one of the enzymes in the PI3 → Akt survival pathway may inhibit p38 MAP kinase to inhibit apoptosis induced cell death.
This raises an interesting dilemma. Calcium permeation through glutamate receptor channels can trigger apoptosis, whereas calcium permeation through nACh receptor channels leads to neuroprotection. Both types of channels are found on RGCs. How can calcium trigger both events? One possibility is that enzymes involved in excitotoxicity and neuroprotection are compartmentalized. However, it is more likely that this dilemma has to do with the concentration of calcium entering the cells. To induce apoptosis, relatively high concentrations of glutamate need to be applied to isolated pig RGCs, leading to relatively high concentrations of calcium entering the cells and activating the p38 MAP kinase pathway. On the other hand, relatively low levels of calcium are likely to permeate RGCs in the presence of 5 µM ACh to activate the PI3 K – Akt pathway to initiate the neuroprotective pathway and inhibit p38 MAP kinase. Another scenario involves previous immunocytochemical studies demonstrating that two different nAChRs are localized on large and small RGCs (Thompson et al. 2006). In this immunocytochemical study, large RGCs were found to contain only α7 nAChR subunits whereas small RGCs were found to contain only α4β2 nAChR subunits. Activation of both nAChRs resulted in ACh-induced neuroprotection, but the mechanism of that neuroprotection may be different depending on the receptor activated. For instance, we may find that activation of α7 nAChR leads to ACh-induced neuroprotection through activation of the PI3 K – Akt pathway, whereas activation of α4β2 nAChRs leads to ACh-induced neuroprotection through inhibition of p38MAP kinase. The results presented in this study do not address this issue as total RGCs were used for ELISA and pharmacological studies. However, two different mechanisms occurring in two different types of RGCs could reconcile how low calcium permeability through ACh receptors can offset the excitotoxic damage caused by high calcium permeation via glutamate receptors.
Physiological Implications
Could ACh have a neuroprotective role in the retina under physiological conditions? RGCs contain nAChRs and receive cholinergic input from a well-described population of amacrine cells, known as starburst amacrine cells (Famiglietti 1983; Masland,1988). It is therefore possible that one of ACh's functions in the retina is to provide a constant level of neuroprotection to RGCs. To date, it is unknown if ACh release from starburst amacrine cells changes due to glaucoma, diabetic retinopathy or retinal ischemia. Future studies are needed to determine if ACh functions under physiological conditions to provide neuroprotection to RGCs and if ACh release from amacrine cells change during neurodegenerative diseases. A loss of ACh-induced neuroprotection may help explain the progressive nature of retinal neurodegenerative disorders. Information such as this could lead to therapeutic treatments for retinal excitotoxic conditions that would ultimately introduce ACh or nicotine into the eye to stimulate neuroprotection and stop the progression of certain diseases.
Acknowledgments
The authors thank Dr. John Spitsbergen for culturing and ELISA consultation. This work was funded by a Midwest Eye Grant and a grant from NIH/NEI #EY017314 awarded to C.L. Linn.
Footnotes
There is no conflict of interest in this study.
Contributor Information
C.O. Asomugha, Western Michigan University, Kalamazoo, MI
D.M. Linn, Grand Valley State University, Allendale, MI
C.L. Linn, Western Michigan University, Kalamazoo, MI
References
- Barnstable CJ, Drager UC. Thy-1 antigen: a ganglion cell specific marker in rodent retina. Neurosci. 1984;11:847–855. doi: 10.1016/0306-4522(84)90195-7. [DOI] [PubMed] [Google Scholar]
- Berra E, Diaz-Meco MT, Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the Phosphoinositide 3-kinase/Akt pathway. J. of Biol. Chem. 1998;273:10792–10797. doi: 10.1074/jbc.273.17.10792. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong Z. The functional contrariety of JNK. Mol. Carcinogen. 2007;46:591–598. doi: 10.1002/mc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordia T, Grady SR, McIntosh JM, Quik M. Nigrostriatal damage preferentially decreases a subpopulation of alpha6beta2 nAChRs in mouse, monkey, and Parkinson's disease striatum. Mol. Pharm. 2007;72:52–61. doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]
- Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr. Pharm. Design. 2007;13:1875–1876. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, McKenna BW, Connors TJ, Neff NT. A rapid fluorometric assay to measure neuronal survival in vitro. J. Neurosci. Methods. 1993;50:205–216. doi: 10.1016/0165-0270(93)90009-g. [DOI] [PubMed] [Google Scholar]
- Burnashev N. Calcium permeability of ligand-gated channels. Cell calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- Casson RJ. Possible role of excitotoxicity in the pathogenesis of glaucoma. Clin. and Exper. Ophthal. 2006;34:54–63. doi: 10.1111/j.1442-9071.2006.01146.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and disease of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Lima PA, Wonnacott S. The alpha7 nicotinic receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca2+ dependent mechanism. Neuropharm. 2000;39:2799–2807. doi: 10.1016/s0028-3908(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Dasari VR, Veeravalli KK, Saving KL, Gujrati M, Fassett D, Klopfenstein JD, Dinh DH, Rao JS. Neuroprotection by cord blood stem cells against glutamate-induced apoptosis is mediated by Akt pathway. Neurobiol. Disorders. 2008;32:486–498. doi: 10.1016/j.nbd.2008.09.005. [DOI] [PubMed] [Google Scholar]
- De Chaiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F. Bcl-2 phosphorylation of p38 MAPK: identification of target sites and biologic consequences. J. Biol. Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochimica et Biophysica Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha 7 nicotinic acetylcholine receptors: In vitro and in vivo mechanism related to Alzheimer’s disease. J. Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog. in Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Famiglietti EV., Jr Starburst amacrine cells and cholinergic neurons: mirror-symmetric on and off amacrine cells of rabbit retina. Brain Res. 1983;14(261):138–144. doi: 10.1016/0006-8993(83)91293-3. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, Simon RP. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J. Neurosci. 2002;22:8458–8465. doi: 10.1523/JNEUROSCI.22-19-08458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC. Apotposis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Transactions. 2007;35:421–423. doi: 10.1042/BST0350421. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Maeda T, Kume T, Kochiyama H, Akaike A, Shimohama S, Kimura J. Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via alpha7-neuronal receptors and neuronal CNS receptors. Brain Res. 1997;765:135–140. doi: 10.1016/s0006-8993(97)00556-8. [DOI] [PubMed] [Google Scholar]
- Kawasaki A, Han MH, Wei JY, Hirata K, Otori Y, Barnstable CJ. Protective effect of arachidonic acid on glutamate neurotoxicity in rat retinal ganglion cells. Invest. Ophthalmology in Vis. Sci. 2002;43:1835–1842. [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. of Biol. Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Kim YS, Park GB, Lee HK, Song H, Choi IH, Lee WJ, Hur DY. Cross-linking of B7-Hi on EBV-transformed B cells induces apoptosis through reactive oxygen species production, JNK signaling activation, and fasL expression. J. of Immunol. 2008;181:6158–6169. doi: 10.4049/jimmunol.181.9.6158. [DOI] [PubMed] [Google Scholar]
- Lafuente MP, Villegas-Perez PM, Sobrado-Calvo P, Garcia-Aviles A, Miralles de Imperial J, Vidal-Sanz M. Neuroprotective effects of alpha (2)-selective adrenergic agonists against ischemia-induced retinal ganglion cell death. Invest. Ophthal. in Vis. Sci. 2001;42:2074–2084. [PubMed] [Google Scholar]
- Lam TT, Abler AS, Kwong JMK, Tso MOM. N-methyl-D-aspartate (NMDA)-induced apoptosis in rat retina. Invest. Ophthal. in Vis. Sci. 1999;40:2391–2397. [PubMed] [Google Scholar]
- Liu J, Mao W, Ding B, Liang CS. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Amer. J. of Phys. Heart Circulation Phys. 2008;295:H1956–H1965. doi: 10.1152/ajpheart.00407.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZM, Chen GG, Vlantis AC, Tse GM, Shum CK, van Hasselt CA. Calcium-mediated activation of PI3 K and p53 leads to apoptosis in thyroid carcinoma cells. Cell and Mol. Life Sci. 2007;64:1428–1436. doi: 10.1007/s00018-007-7107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Heidinger V, Picaud S, Lambrou G, Dreyfus H, Sahel J, Hicks D. Selective excitotoxic degeneration of adult pig retinal ganglion cells in vitro. Invest. Ophthal. in Vis. Sci. 2001;42:1096–1106. [PubMed] [Google Scholar]
- Mai H, May WS, Gao F, Jin Z, Deng X. A functional role for nicotine in Bcl-2 phosphorylation and suppression of apoptosis. J. of Biol. Chem. 2003;278:1886–1891. doi: 10.1074/jbc.M209044200. [DOI] [PubMed] [Google Scholar]
- Manabe S, Lipton SA. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest. Ophthal. in Vis. Sci. 2003;44:385–392. doi: 10.1167/iovs.02-0187. [DOI] [PubMed] [Google Scholar]
- Markou T, Dowling AA, Kelly T, Lazou A. Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic Res. 2009;43:809–816. doi: 10.1080/10715760903071649. [DOI] [PubMed] [Google Scholar]
- Masland RH. Amacrine cells. Trends in Neurosci. 1988;11:405–411. doi: 10.1016/0166-2236(88)90078-1. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 1999;75:2037–2046. 1999. [PubMed] [Google Scholar]
- Mielke K, Herdegen T. JNK and p38 stress kinases - degenerative effectors of signal-transduction-cascades in the nervous system. Prog. in Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Olney JW, de Guabareff T. Glutamate neurotoxicity and Huntington’s chorea. Nature. 1978;271:557–559. doi: 10.1038/271557a0. [DOI] [PubMed] [Google Scholar]
- Ozdemir D, Tugyan K, Uysal N, Sonmez U, Sonmez A, Acikgoz O, Ozdemir N, Duman M, Ozkan H. Protective effect of melatonin against head trauma-induced hippocampal damage and spatial memory deficits in immature rats. Neurosci. Let. 2005;385:234–239. doi: 10.1016/j.neulet.2005.05.055. [DOI] [PubMed] [Google Scholar]
- Ozdener H. Molecular mechanisms of HIV-1 associated neurodegeneration. J. of Biosci. 2005;30:391–405. doi: 10.1007/BF02703676. [DOI] [PubMed] [Google Scholar]
- Park EM, Joh TH, Volpe BT, Chu CK, Song G, Cho S. A neuroprotective role of extracellular signal-regulated kinase in N-acetyl-O methyldopamine-treated hippocampal neurons after exposure to in vitro and in vivo ischemia. Neurosci. 2004;123:147–154. doi: 10.1016/j.neuroscience.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein kinase pathways: regulation and physiological functions. Endocr. Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Pernet V, Hauswirth WW, Di Polo A. Extracellular signal-regulated kinase ½ mediates survival, but not axon regeneration, of adult injured central nervous system neurons in vivo. J. of Neurochem. 2005;93:72–83. doi: 10.1111/j.1471-4159.2005.03002.x. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest. Ophthal. in Vis. Sci. 1995;36:774–786. [PubMed] [Google Scholar]
- Ranawat P, Bansal MP. Decreased glutathione levels potentiate the apoptotic efficacy of selenium: possible involvement of p38 and JNK MAPKs—in vitro studies. Mol. Cell. Biochem. 2008;309:21–32. doi: 10.1007/s11010-007-9639-7. [DOI] [PubMed] [Google Scholar]
- Ravina BM, Fagan SC, Hart RG, Hovinga CA, Murphy DD, Dawson TM, Marler JR. Neuroprotective agents for clinical trials in Parkinson's disease: A systematic assessment. Neurol. 2003;60:1234–1240. doi: 10.1212/01.wnl.0000058760.13152.1a. [DOI] [PubMed] [Google Scholar]
- Romano C, Chen Q, Olney JW. The intact (ex vivo) retina as a model system for the study of excitotoxicity. Prog. in Ret. Eye Res. 1998;17:465–483. doi: 10.1016/s1350-9462(98)00008-1. [DOI] [PubMed] [Google Scholar]
- Shaw S, Bencherif M, Marrero MB. Janus kinase 2, an early target of alpha7 nicotinic acetylcholine receptor-mediated neuroprotection against Aβ-(1–42) amyloid. J. of Biol. Chem. 2002;277:44920–44924. doi: 10.1074/jbc.M204610200. [DOI] [PubMed] [Google Scholar]
- Shimamura H, Terada Y, Okado T, Tanaka H, Inoshita S, Sasaki S. The PI3-kinase-Akt pathway promotes mesangial cell survival and inhibits apoptosis in vitro via NF-kappa B and Bad. J. Amer. Soc. of Nephr. 14:1427–1434. doi: 10.1097/01.asn.0000066140.99610.32. [DOI] [PubMed] [Google Scholar]
- Shouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr. Opin. in Crit. Care. 2007;13:134–142. doi: 10.1097/MCC.0b013e3280895d5c. [DOI] [PubMed] [Google Scholar]
- Thompson SA, Smith O, Linn DM, Linn CL. Acetylcholine neuroprotection against glutamate-induced excitotoxicity in adult pig retinal ganglion cells is partially mediated through α4 nAChRs. Exp. Eye Res. 2006;83:1135–1145. doi: 10.1016/j.exer.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int. J. of Biol. Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Bentires-Alj M, Chariot A, Deregowski V, deLeval L, Merville MP, Bours V. NF-kB2/p100 induces Bcl-2 expression. Leuk. 2003;17:1349–1356. doi: 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang Z, Chen J, Wu J. Apoptosis induced by NO via phosphorylation of p38 MAPK that stimulates NF-kappaB, p53 and caspase-3 activation in rabbit articular chondrocytes. Cell Biol. Inter. 2007;31:1027–1035. doi: 10.1016/j.cellbi.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Wehrwein E, Thompson SA, Coulibaly SF, Linn DM, Linn CL. Acetylcholine protects isolated adult pig retinal ganglion cells from glutamate induced excitotoxicity. Invest. Ophthal. in Vis. Sci. 2004;45:1531–1543. doi: 10.1167/iovs.03-0406. [DOI] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. The J. of Pharm. and Exp. Therap. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Pernet V, Hauswirth WW, DiPolo A. Activation of the extracellular signal-regulated kinase ½ pathway by AAV gene transfer protects retinal ganglion cells in glaucoma. Mol. Therap. 2005;12:402–412. doi: 10.1016/j.ymthe.2005.04.004. [DOI] [PubMed] [Google Scholar]