

Abstract
B cell follicles are specialized microenvironments that support events necessary for humoral immunity 1, 2, 3. Following antigen encounter, activated B cells initially seek T cell help at the follicle-T zone boundary and then move to interfollicular and T-zone distal (outer) regions of the follicle 4, 5, 6, 7, 8, 9, 10. Subsequently, some cells move to the follicle center, become germinal center (GC) B cells and undergo antibody affinity maturation 1, 2, 11. Although germinal ‘centers’ within follicles were described in 1885 12, the molecular cues mediating segregation of B cells between outer and center follicle have remained undefined. Here we establish a role for the orphan G-protein coupled receptor, Epstein Barr Virus-induced molecule-2 (EBI2) 13, in this process. EBI2 is expressed in mature B cells and increases in expression early after activation before being down-regulated in GC B cells. EBI2 deficiency led to a reduction in the early antibody response to a T-dependent antigen. EBI2-deficient B cells failed to move to the outer follicle at day 2 of activation and instead were found in the follicle center, whereas EBI2 over-expression was sufficient to promote B cell localization to the outer follicle. In mixed bone marrow chimeras, EBI2-deficient B cells phenocopied GC B cells in preferentially localizing to the follicle center. When down-regulation of EBI2 in wild-type B cells was antagonized, participation in the GC reaction was impaired. These studies identify an important role for EBI2 in promoting B cell localization in the outer follicle, and show that differential expression of this receptor helps position B cells appropriately for mounting T-dependent antibody responses.
The propensity of B cells to migrate to outer versus center follicle at different stages of the antibody response, together with the established roles of G-protein coupled receptors (GPCRs) in controlling lymphocyte positioning events, led us to ask whether novel GPCRs differentially expressed between early activated and GC B cells may be involved in this subcompartmentalization. These criteria focused our attention on EBI2 (GPR183), a Gαi-coupled orphan receptor 13,14 abundantly expressed in EBV infected and activated human B cells and down-regulated in GC B cells 15,16. To explore the expression pattern of mouse Ebi2 we generated a reporter mouse line carrying the EGFP gene inserted in place of the Ebi2 open reading frame (Suppl. Fig. 1). Analysis of Ebi2GFP/+ mice showed that EBI2 is upregulated during B cell maturation in the bone marrow (BM), and is expressed in mature recirculating B cells in BM, spleen and lymph nodes (Fig. 1a). Expression of the GFP reporter tracked closely with changes in Ebi2 transcript abundance (Fig. 1b). GFP levels were further upregulated after BCR engagement with anti-IgM, or combined anti-IgM and anti-CD40 stimulation (Fig. 1c). To examine expression following receipt of T cell help we employed an adoptive transfer system in which B cells from C57Bl/6 (B6) Ebi2GFP/+ mice were transferred to the coisogenic strain, bm12, that bears a three amino acid difference in the I-Ab MHC class II molecule and contains a high frequency of I-Ab responsive helper T cells 6,17. This approach permits tracking of an activated B cell population in non-transgenic mice at early time points after receiving T cell help. Two days after adoptive transfer of anti-IgM stimulated B cells we found higher GFP expression in B cells transferred to bm12 compared to B6 recipients, indicating that interaction with helper T cells promoted EBI2 upregulation (Fig. 1d). EBI2 expression was maintained in plasma cells but was markedly (∼25-fold) down-regulated in GC B cells (Fig. 1a and b). Most CD4 T cells and a smaller fraction of CD8 T cells also expressed EBI2, though at lower levels than B cells (Fig. 1a and b). In sections, GFP was detectable throughout the follicle and T zone but was almost undetectable within GCs, identified by their expression of GL7, making these structures appear as EBI2-deficient islands in a ‘sea’ of EBI2-expressing cells (Fig. 1e).
Figure 1. Ebi2 upregulation in activated B cells and down-regulation in GCs.
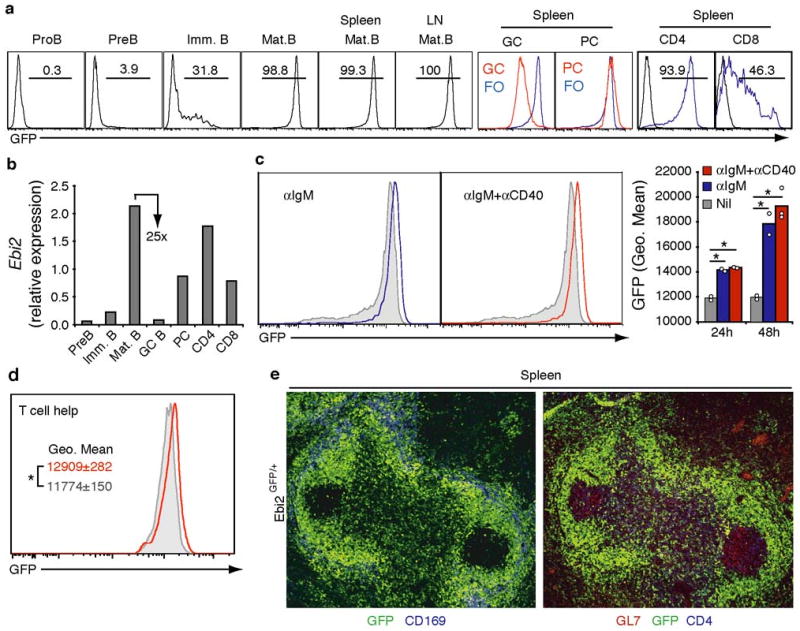
a, Flow cytometric detection of GFP fluorescence in the indicated BM, spleen and lymph node (LN) cell subsets from Ebi2GFP/+ mice. PC, plasma cells. b, Q-PCR analysis of Ebi2 transcript abundance in the indicated cell populations. Expression is shown relative to Hprt1. c and d, Flow cytometric detection of GFP fluorescence in B cells stimulated for one day with anti-IgM or anti-IgM and anti-CD40 (c) or that were stimulated for 1 h with anti-IgM and exposed in vivo for 2 days to T cell help (red) or not provided with T cell help (gray) (d). Numbers in a indicate percent of cells in gate. PC, plasma cells; FO, follicular B cells. Gray histograms in c indicate unstimulated cells. Bar graph in c shows geometric mean Ebi2GFP/+ fluorescence for one and two day cultures and summarizes 3 experiments. e, Immunofluorescence microscopy of fixed spleen tissue from an Ebi2GFP/+ mouse, stained to detect GFP+ cells (green) and CD169+ marginal zone macrophages (blue, left panel) or GL7+ GC B cells and CD4 T cells (red and blue, respectively, right panel).
An initial analysis of lymphoid tissues from EBI2-deficient mice showed the presence of organized follicles and T cell compartments, and the mice had normal numbers of B and T cells (Suppl. Fig. 2 and data not shown). Movement of activated B cells to the follicle-T zone boundary within six hours of BCR stimulation occurred similarly for EBI2-deficient and wild-type cells (Fig. 2a) suggesting that BCR mediated EBI2 induction (Fig. 1) is not required for this CCR7-dependent relocalization event 8. We then asked whether the next stage(s) of activated B cell migration that occur during T-dependent responses, movement to the outer follicle and interfollicular regions, were EBI2-dependent. To permit in situ tracking of activated B cells responding to T cell help, we used the bm12 adoptive transfer approach introduced above. Littermate control B cells that had received anti-IgM stimulation were able to respond to T cell help within bm12 recipients and relocalized to the outer follicle at day 2 (Fig. 2b), as did cotransferred wild-type Igha B cells (Fig. 2b), consistent with earlier studies using Ig-transgenic B cells 6. By contrast, EBI2-deficient B cells were unable to localize to this region and instead favored the central area of the follicle (Fig. 2b). To determine whether upregulation of EBI2 could be sufficient to control B cell localization to the interfollicular and outer follicle regions within lymphoid tissues we transduced B cells with Ebi2-encoding or control retroviruses and transferred them to wild-type recipients. One day later the Ebi2-overexpressing cells, identified by expression of a hCD4 reporter, were situated in interfollicular regions and in the outer follicle (Fig. 2c). This contrasted with the distribution of B cells transduced with the control retrovirus, where the cells distributed uniformly within the follicle (Fig. 2c). Thus EBI2 appears to be both necessary and sufficient to promote positioning of activated B cells in the outer follicle and interfollicular regions. Consistent with an essential role for EBI2 in the correct positioning of B cells during the early phases of T-dependent humoral responses, EBI2-deficient mice mounted a reduced day 7 IgG1 antibody response to nitrophenyl-chicken gamma globulin (NP-CGG) in alum (Fig. 2d). The IgM response was not affected (Fig. 2d).
Figure 2. EBI2 promotes localization of activated B cells in the outer follicle.
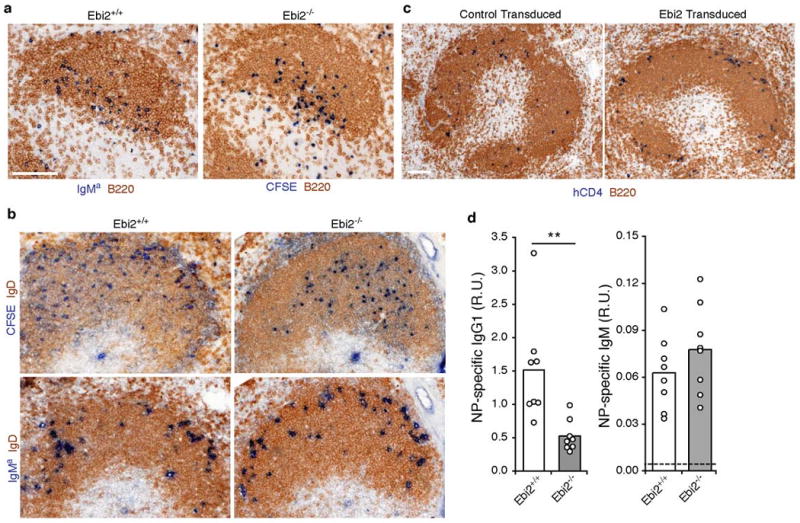
a-c, Immunohistochemical staining of spleen cryosections. a, Distribution of wild-type and EBI2-deficient B cells that had been stimulated with anti-IgM in vitro for 1h, analyzed 6 h after being transferred to wild-type hosts. Ebi2−/− B cells were CFSE labeled prior to transfer. Sections were stained with an antibody to detect CFSE (Ebi2-/-) or co-transferred wild-type Igha (IgMa, Ebi2+/+) B cells (blue) and endogenous B cells (IgD, brown). b, Distribution of anti-IgM treated wild-type and EBI2-deficient B cells (CFSE, blue), and internal control Igha B cells (IgMa, blue), after 2 days exposure to T cell help in bm12 hosts. Upper and lower panels are serial sections. Endogenous B cells were detected with anti-IgD (brown). c, Distribution of B cells transduced with control or Ebi2-expressing retrovirus (hCD4, blue), one day after transfer. Endogenous B cells were detected with anti-IgD (brown). d, Anti-NP IgG1 and IgM serum titers in wild-type and EBI2-deficient mice on day 7 following immunization with NP-CGG in alum. R.U., relative units.
As another approach to examine the role of EBI2 in determining B cell localization, we examined cell distribution in 20:80 mixed BM chimeras that contained a minority of EBI2-deficient cells (20% Ighb EBI2-deficient or littermate control and 80% Igha wild-type). Remarkably, EBI2-deficient B cells selectively localized in a GC-like location at the center of follicles in spleen, lymph nodes and Peyer's patches of the BM chimeras (Fig. 3a). These foci of cells were not GCs as they maintained high expression of IgD and lacked expression of GL7 (Fig. 3a and Supp. Fig. 3). In contrast, EBI2-deficient and wild-type T cells appeared intermingled throughout the T zone (Suppl. Fig. 4). Analysis of 90:10 mixed BM chimeras containing a majority of EBI2-deficient (or littermate control) cells showed that EBI2-deficient B cells colocalized with the CD35+ follicular dendritic cell (FDC) network at the center of follicles whereas the minor population of wild-type B cells in these mice was partially excluded from this area and enriched in interfollicular regions or outer follicles (Fig. 3b). Similar findings were made in lymph nodes and Peyer's patches (Suppl. Fig. 5). It seems possible that in mice where most B cells lack EBI2, there is increased EBI2 ligand availability and wild-type B cells predominate at sites of ligand production. In control mixed BM chimeras reconstituted with a minority (20:80) or majority (90:10) of Ebi2+/+ BM, the two types of wild-type B cells were intermingled in both the follicle center and periphery (Fig. 3a, b and Suppl. Figs. 3 and 5). FDCs are dependent on LTα1β2 for their maintenance 18. To test whether the segregation of wild-type and EBI2-deficient B cells was dependent on FDCs, we treated mixed BM chimeras with LTβR-Ig, a LTα1β2 antagonist 19, for 2-3 weeks. The CD35+ FDC networks were depleted after treatment (Fig. 3c) as expected 19,20. Under these conditions, wild-type and EBI2-deficient cells no longer showed a segregated distribution in splenic B cell areas (Fig. 3c) suggesting a role for FDCs or other LTα1β2-dependent cells in this process. In mice that lack CXCL13, a chemokine made broadly by the follicular stromal cell network 20,21, B cell localization in the outer splenic white-pulp (where follicles would normally be located) is reduced but not completely blocked 22 (Fig. 3d). In CXCL13-deficient mice reconstituted with a mixture of EBI2-deficient and wild-type BM, EBI2-deficient B cells were selectively diminished within the white-pulp cords and instead accumulated in the marginal zone that surrounds the white-pulp (Fig. 3d). These findings provide further evidence for EBI2-mediated attraction of B cells to the outer white-pulp.
Figure 3. EBI2-deficient B cells localize to the follicle center in a LTα1β2- and CXCL13-dependent manner.
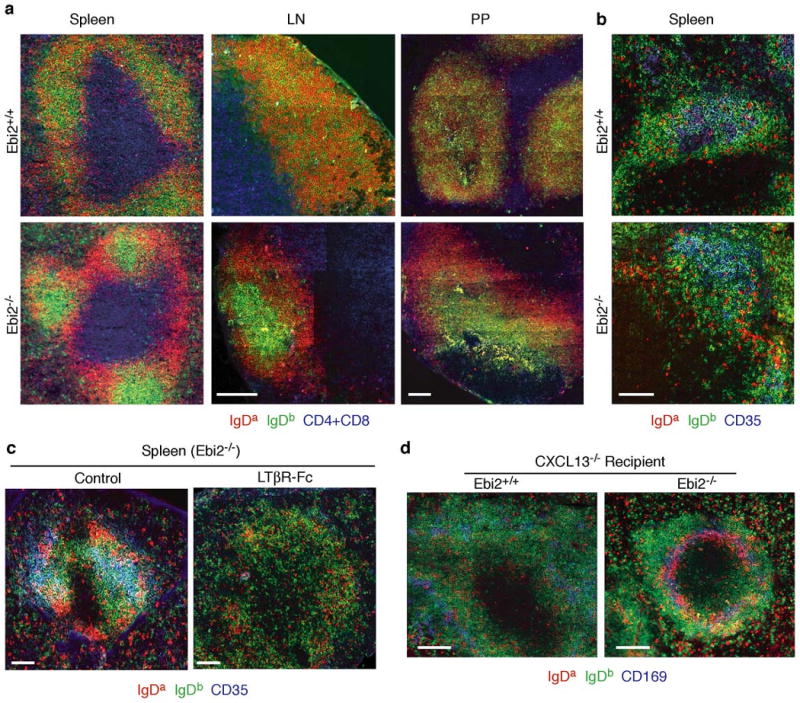
a, Distribution of wild-type and EBI2-deficient B cells in spleen, lymph nodes (LN) and Peyer's patches (PP) of 20:80 mixed BM chimeras (20% Ighb Ebi2+/+ or Ebi2−/− and 80% Igha wild-type). Sections were stained to detect Ebi2+/+ or Ebi2−/− B cells (IgDb, green), Igha control B cells (IgDa, red), and T cells (CD4+CD8, blue). b, Spleen sections from 90:10 mixed BM chimeras (90% Ighb Ebi2+/+ or Ebi2−/− and 10% Igha wild-type) stained to detect B cells as in a and for CD35 to highlight FDC networks (blue). c, Similar analysis to b in control or 3 week LTβR-Fc treated 90:10 Ebi2-/- mixed BM chimeras. d, Distribution of wild-type and EBI2-deficient B cells in CXCL13−/− hosts reconstituted with 70:30 BM mixtures (70% Ighb Ebi2+/+ or Ebi2−/− and 30% Igha wild-type). Spleen sections stained to detect EBI2-deficient B cells (IgDb, green), wild-type B cells (IgDa, red) and marginal zone macrophages (CD169, blue).
The above findings suggested that EBI2 down-regulation in GC B cells may promote their positioning at the follicle center. To directly test the significance of EBI2 down-regulation during GC development, we enforced constitutive EBI2 expression in hen egg lysozyme-specific Ig-transgenic B cells using retroviral gene transduction and then tested their ability to participate in GC and plasma cell responses following short-term adoptive transfer to hen egg lysozyme immunized hosts (Fig. 4a). The frequency of Ebi2 or control vector transduced B cells amongst the GC and plasma cell populations was tracked using the hCD4 reporter. By flow cytometric analysis, Ebi2-transduced cells showed a reduced ability to participate in GC responses compared to vector transduced cells, while participating with wild-type efficiency in the plasma cell response (Fig. 4a and b). Although some Ebi2-transduced cells could take on a GC phenotype (Fig. 4a), the cells were unable to position in GCs (Fig. 4c and Suppl. Fig. 6). Cells transduced with the control vector were readily detected within GCs (Fig. 4c and Suppl. Fig. 6). Ebi2 transduced cells contained Ebi2 transcripts in amounts within two-fold of those present in follicular B cells and at least 25-fold higher than in GC B cells (Suppl. Fig. 6). These experiments suggest that EBI2 down-regulation is necessary for localization of developing GC B cells to the follicle center.
Figure 4. Maintained Ebi2 expression impairs participation in GC response.
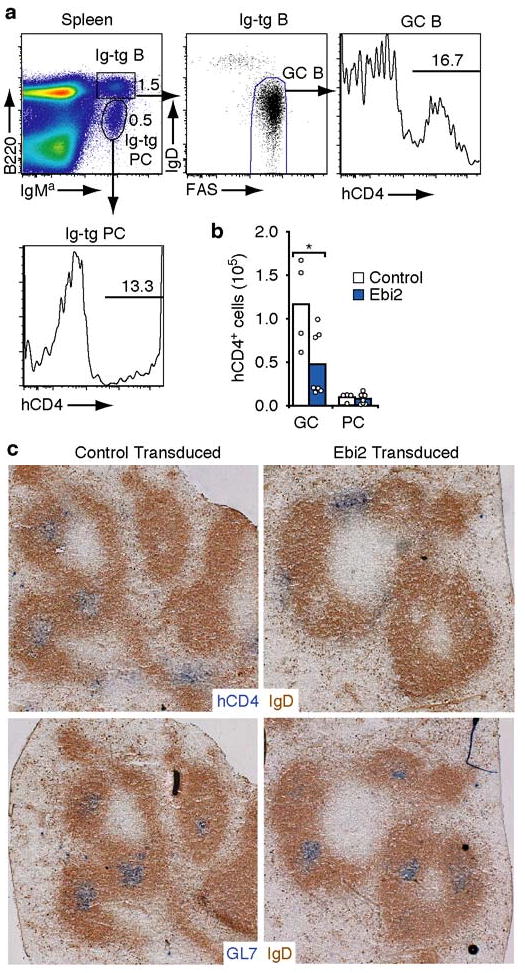
a, Flow cytometric analysis of spleen cells from an immunized mouse receiving Ebi2-transduced Ig-transgenic (IgMa) B cells, 4 days after transfer, showing gating scheme to identify representation of transduced (hCD4+) cells amongst GC B cells (B220+IgMa+IgDloFashi) and plasma cells (PC, B220loIgMa+). Numbers indicate frequency of cells in the indicated gate. b, Number of transduced (hCD4+) B cells having a GC or plasma cell phenotype. c, Distribution of transduced B cells (hCD4+, blue) in sections of spleen from mice receiving control vector or Ebi2-transduced B cells. Endogenous naïve B cells are stained brown (IgD) and GCs are detected in serial sections using GL7 (blue, lower panels).
In summary, we establish that EBI2 is upregulated in B cells after BCR and CD40 engagement and is necessary to promote positioning of activated B cells to interfollicular regions and the outer follicle. Cognate B cells interact with helper T cells and undergo proliferation in these regions 7,10. Defects in these processes are likely responsible for the diminished ability of EBI2-deficient mice to mount an early T-dependent IgG antibody response. Our findings suggest EBI2 is needed within B cells for these events but we do not exclude that it also plays a role in directing activated CD4 helper T cells and possibly other cell types to these regions. GC B cells markedly down-regulate EBI2, a change that appears necessary to favor localization of activated B cells at the follicle center, in association with the antigen-presenting and GC-supportive 23 FDC network. Bcl6, a transcription factor required for GC development, negatively regulates EBI2 expression 15. We suggest that by favoring appropriate niche occupancy, negative regulation of EBI2 represents an important component of the Bcl6 gene expression program directing GC over plasma cell fate. Although the ligand for EBI2 remains undefined, we speculate that it is more concentrated in the outer compared to center follicle as well as in interfollicular regions. We propose that while CXCR5 is sufficient to promote B cell localization in follicles 22,24, cells expressing EBI2 are more strongly attracted to the outer follicle compared to cells lacking this receptor. An additional FDC-derived cue may favor positioning in the center follicle, and when cells lose responsiveness to EBI2 ligands, positioning in response to this cue is dominant. The activity of EBI2 provides a possible explanation for why CXCR5-deficient B cells continue to localize in interfollicular areas and in regions corresponding to the outer follicle 22,24. Our findings may also help explain the niche preferences of certain B cell lymphomas, particularly follicular center lymphoma 25. Indeed, expression array studies demonstrate that EBI2 is down-regulated in follicular and GC lymphomas 16,26. Moreover, it seems possible that the marked EBI2 induction observed early following EBV infection 13 serves as a mechanism used by the virus to promote positioning in niches that favor survival of the infected B cells.
Methods Summary
EGFP was inserted in place of the Ebi2 open reading frame within E14 (129) ES cells using standard procedures and Ebi2GFP/+ mice were backcrossed to C57Bl/6 for six generations. 6-12 week old C57BL/6 were from either the National Cancer Institute or Jackson Laboratories. B6(C)-H2-Ab1bm12/KhEgJ (bm12) mice and B6.Cg-IghaThy1aGPi1a/J (IgMa) mice were from Jackson Laboratories. MD4 mice 27 and CXCL13-deficient mice 22 were from an internal colony. Bone marrow chimeras were generated as described 11 and analyzed after 6-12 weeks. NP-CGG immunizations were performed using 50 μg of NP-CGG (Solid Phase Sciences, USA) in Alum (Accurate Chemical & Scientific Corp., USA). The retroviral construct was made by inserting the mouse EBI2 open reading frame, with a preprolactin-FLAG leader sequence 28 in place of the ATG, into the MSCV2.2 retroviral vector containing cytoplasmic-domain truncated human CD4 as an expression marker downstream of the internal ribosomal entry site 8. B cells were isolated and in some cases labeled with 2.5 μM of 5(and 6)-carboxy-fluorescein diacetate succinimidyl ester (CFSE, Molecular Probes) as described 29. For in vivo analysis of Ebi2 expression, 5-10×106 purified B cells as in 11, were transferred into bm12 experiments. Transduced cells were adoptively transferred one day after spin-infection for transfers to non-immunized hosts or immediately after spin-infection for transfers to immunized hosts. For GC experiments, B6 mice received 105 MD4 B cells and 105 OTII CD4+ T cells at day -1, were intraperitoneally immunized with 50μg HEL-OVA in RIBI adjuvant system (Sigma) at day 0 and received approximately 106 EBI2 or control vector transduced cells at day 1. Mice were analyzed on day 5. Flow cytometry, ELISA, immunohistochemsitry and immunofluorescence microscopy were by standard techniques and are detailed in Full Methods.
Supplementary Material
Acknowledgments
We thank Chris Allen for help with microscopy and discussions, Jesse Green, Betsy Gray and Kazuhiro Suzuki for helpful advice and Jinping An for expert assistance with the mouse colony. We also thank Nigel Killen and Jay Dietrich for help with gene targeting. J.P. is an Associate and J.G.C. an Investigator of the Howard Hughes Medical Institute. This work was supported in part by grants from the NIH.
Footnotes
Author Contributions: J.P.P. and J.G.C. designed and conceptualized the research; J.P.P., L.K. and Y.X. did the experiments. J.P.P., L.M.K. and J.G.C. analyzed the data, prepared the figures and wrote the manuscript.
References
- 1.MacLennan ICM. Germinal Centers. Annu Rev Immunol. 1994;12:117. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 2.Kelsoe G. The germinal center: a crucible for lymphocyte selection. Semin Immunol. 1996;8:179. doi: 10.1006/smim.1996.0022. [DOI] [PubMed] [Google Scholar]
- 3.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu YJ, Oldfield S, MacLennan ICM. Memory B cells in T cell-dependent antibody responses colonize the splenic marginal zones. Eur J Immunol. 1988;18:355. doi: 10.1002/eji.1830180306. [DOI] [PubMed] [Google Scholar]
- 5.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 7.Garside P, et al. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 8.Reif K, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- 9.Pape KA, et al. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J Exp Med. 2003;197:1677. doi: 10.1084/jem.20012065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity. 2009;30:599. doi: 10.1016/j.immuni.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen CD, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 12.Nieuwenhuis P, Opstelten D. Functional anatomy of germinal centers. Am J Anat. 1984;170:421. doi: 10.1002/aja.1001700315. [DOI] [PubMed] [Google Scholar]
- 13.Birkenbach M, et al. Epstein-Barr virus induced genes: first lymphocyte-specific G-protein coupled peptide receptors. J Virol. 1993;67:2209. doi: 10.1128/jvi.67.4.2209-2220.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenkilde MM, et al. Molecular pharmacological phenotyping of EBI2. An orphan seven-transmembrane receptor with constitutive activity. J Biol Chem. 2006;281:13199. doi: 10.1074/jbc.M602245200. [DOI] [PubMed] [Google Scholar]
- 15.Shaffer AL, et al. Signatures of the immune response. Immunity. 2001;15:375. doi: 10.1016/s1074-7613(01)00194-7. [DOI] [PubMed] [Google Scholar]
- 16.Cahir-McFarland ED, et al. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol. 2004;78:4108. doi: 10.1128/JVI.78.8.4108-4119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mengle-Gaw L, et al. Gene conversion between murine class II major histocompatibility complex loci. Functional and molecular evidence from the bm 12 mutant. J Exp Med. 1984;160:1184. doi: 10.1084/jem.160.4.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Ann Rev Immunol. 1999;17:399. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 19.Mackay F, et al. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur J Immunol. 1997;27:2033. doi: 10.1002/eji.1830270830. [DOI] [PubMed] [Google Scholar]
- 20.Ngo VN, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999;189:403. doi: 10.1084/jem.189.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunn MD, et al. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 22.Ansel KM, et al. A chemokine driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 23.Tew JG, et al. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol Rev. 1997;156:39. doi: 10.1111/j.1600-065x.1997.tb00957.x. [DOI] [PubMed] [Google Scholar]
- 24.Forster R, et al. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 25.Lossos IS, Levy R. Higher grade transformation of follicular lymphoma: phenotypic tumor progression associated with diverse genetic lesions. Semin Cancer Biol. 2003;13:191. doi: 10.1016/s1044-579x(03)00015-4. [DOI] [PubMed] [Google Scholar]
- 26.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 27.Goodnow CC, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 28.Ishii K, et al. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J Biol Chem. 1993;268:9780. [PubMed] [Google Scholar]
- 29.Okada T, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 2005;3:e150. doi: 10.1371/journal.pbio.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.