

Abstract
There is increasing recognition that stochastic processes regulate highly predictable patterns of gene expression in developing organisms, but the implications of stochastic gene expression for understanding haploinsufficiency remain largely unexplored. We have used simulations of stochastic gene expression to illustrate that gene copy number and expression deactivation rates are important variables in achieving predictable outcomes. In gene expression systems with non-zero expression deactivation rates, diploid systems had a higher probability of uninterrupted gene expression than haploid systems and were more successful at maintaining gene product above a very low threshold. Systems with relatively rapid expression deactivation rates (unstable gene expression) had more predictable responses to a gradient of inducer than systems with slow or zero expression deactivation rates (stable gene expression), and diploid systems were more predictable than haploid, with or without dosage compensation. We suggest that null mutations of a single allele in a diploid organism could decrease the probability of gene expression and present the hypothesis that some haploinsufficiency syndromes might result from an increased susceptibility to stochastic delays of gene initiation or interruptions of gene expression.
Haploinsufficiency refers to a phenotype associated with the inactivation of a single allele in a diploid organism. A common notion of haploinsufficiency is that the phenotype reflects a requirement for >50% of the diploid level of gene product. For example, a leading medical textbook defines haploinsufficiency as “meaning that a half-normal amount of gene product is insufficient to maintain a normal phenotype (i.e., the process is sensitive to reduced dosage)” (ref. 1, p. 383). This definition assumes that a cell must maintain relatively stable levels of gene product above a half-normal threshold level and precludes gene dosage compensation, i.e., increasing expression from the remaining allele to achieve a steady-state level of product comparable to the diploid state.
Although the steady-state level of many gene products may be relatively stable, there is increasing recognition that aspects of the initial activation of gene expression are probabilistic, or stochastic (2–12). Therefore, the highly predictable patterns of gene expression in multicellular organisms are achieved by a system that has probabilistic features. We have used a minimal model of stochastic gene expression to illustrate that gene copy number and expression off-rates can be critical variables in achieving predictable outcomes. We present the hypothesis that some haploinsufficiency diseases result from an increased susceptibility to stochastic delays of gene initiation or interruptions of gene expression, events that are normally buffered by increased gene copy number and that are relatively insensitive to dosage compensation.
METHODS
The simulations in this analysis are presented in nondimensional form. Temporal properties were scaled relative to the product half time, Tp. The activation rate constant, ka, was chosen such that the average time required for gene activation was one-fourth of the product lifetime (Ta = Tp/4). For example, if the product half-life were 1 h, the time for half-maximal gene activation would be 15 min; if the product half-life were 2 h, the half-time of activation would be 30 min, etc.
Although times, rates, and concentrations have been normalized for the sake of generality, the following actual parameter values were used for simulations. The half-time, Tp, for the first-order degradation of product, P, was 4 h with a corresponding rate constant of kp = 4.8 × 10−5 s−1. Except for models of stable expression (i.e., kd = 0), ka = kd = 2 × 10−4 s−1 for “slow” kinetics or was 10-fold faster at 2 × 10−3 s−1 for “fast” kinetics. The uncompensated single-gene expression rate was Jp = 24 fM/s. P and S concentrations were normalized to their Michaelis–Menten constants (1.0 nM) and were expressed in units of P (uP) and S (uS).
Simulations were performed by using a prerelease version 2.0 of kinecyte biological modeling software (RainTown, Seattle) for the MacOS. kinecyte solves the underlying differential equations by using fourth-order Runge–Kutta integration adapted from Press et al. (32) with a fixed integration step-size of Tp/20 to accommodate stochastic gene expression. Halving the integration step-size to Tp/40 had no significant effect on integration accuracy. For stochastic gene expression, random intervals for active/inactive transitions were based on first-order reaction rate constants and a pseudo-random integer number generator to determine the probability during each integration interval according to an exponentially distributed interval histogram (13).
RESULTS
Stochastic Activation of Stable Genes.
The basic model of gene expression, product accumulation, and product degradation is shown in Fig. 1A. As in similar models of stochastic gene expression (9), we consider a gene to switch randomly between inactive (G) and active (G*) states according to first-order reaction kinetics where ka is the activation rate constant (with a corresponding half-time of activation, Ta = log2/ka) and kd (with corresponding Td) is the deactivation rate constant that determines how long a gene dwells in the active state. When active, each gene expresses a product (P) at a rate Jp. The product is degraded by a first-order process with a rate constant and half-time of kp and Tp, respectively. Although there are many variables that could affect product stability—e.g., mRNA half-life, translation efficiency, product compartmentalization and processing, and protein half-life—we have collapsed the production and degradation kinetics of both RNA and protein into a single product pool and have kept this variable constant so that we can explore the properties of expression kinetics.
Figure 1.

A stochastic model of gene expression. (A) This minimal model of stochastic gene expression kinetics consists of a pool of product, P, and two identical genes (indicated by brackets and the subscript 2). P is degraded according to first-order kinetics with a half time, Tp. (For a first-order process, the rate constant corresponding to a half time, T, is k = log2/T.) Each gene functions independently of the other and can be inactive (G) or active (G*). Each active gene expresses P at a rate Jp (in nM/s, for instance) so that, if two genes are active, then the net rate of P-synthesis is twice Jp. Genes switch spontaneously between active and inactive state according to first-order kinetics with an activation half time of Ta and a deactivation half time of Td (and corresponding rate constants, ka and kd). (B) Both genes (G1 and G2) are initially inactive and then are allowed to activate (as indicated by the black bars at the top) independently and randomly with a half time, Ta, that is 1/4 the half time of product degradation (i.e., Ta = Tp/4). In this simulation, the deactivation rate (kd) was 0 and expression was stable after activation. (C) The deactivation half time was set to match the activation half time used in A (i.e., Td = Ta = Tp/4) so that, in the steady state, each gene is active on average 50% of the time [Ta/(Ta + Td)]. Jp was doubled from its value in B to maintain an average steady-state P level of 0.5. Stochastic activation and deactivation events for each gene (indicated by the intermittent black lines) produced a “noisy” pattern of product accumulation and depletion. To the right, an amplitude histogram (accumulated in the steady state for a period of 250 Tp) shows the dispersion, or variance, of the product accumulated over time, i.e., the expression noise. (D) Activation and deactivation kinetics that were 10× faster than in A (i.e., Ta = Td = Tp/40, or fast-unstable kinetics) created such brief active and inactive periods that the product level changed only slightly. This markedly reduced product variance, as seen in the decreased variance of the amplitude histogram. (E) When a single gene synthesized product with the same kinetics as in C, the dispersion of expression noise increased as seen by comparing the black histogram with the white histogram taken from C. (F) When the same overall synthesis rate was distributed among four genes, the expression noise was reduced compared with that of two genes (C).
If a pair of genes, G1 and G2, representing two alleles of a gene in a diploid cell are initially inactive and then are induced to initiate expression, each gene will initiate independently with an average latency of Ta. In Fig. 1B, the stochastic nature of activation was evident as a randomly staggered activation of G2 first and then G1. In this case, the activation rate constant was set to be 4× the product degradation rate constant (such that Ta = Tp/4), and the deactivation rate constant was set at zero (kd = 0). Because expression would persist continuously after initiation, we refer to this as “stable” expression kinetics. Once activated, such stable genes do not deactivate, and product P accumulates from an initial level of zero to reach a steady-state level of 0.5 uP. In cases where only a single gene actively expressed product the steady level was 0.25 uP because of a 50% decline in total expression rate (simulation not shown).
Unstable Genes Produce Expression Noise.
If the gene deactivation rate constant was greater than zero (kd > 0), gene expression was intermittent, or “unstable,” resulting in random fluctuations in the amount of product. This is evident in Fig. 1C, where deactivation and activation rate constants were set to be equal (i.e., Ta = Td = Tp/4; referred to below as “slow-unstable” expression kinetics). In this simulation, the expression rate Jp was doubled to compensate for the fact that expression was active only half of the time [i.e., the dwell time for active expression is Ta/(Ta + Td)]. In contrast to the stable gene expression modeled in Fig. 1B, product levels randomly fluctuated around a mean value of 0.5 uP with excursions to higher and lower product levels, as seen in the amplitude histogram to the right of the simulation panel. (The range of product in this simulation was from ≈0.25 to 0.75 uP). In a biological context, this would represent the fluctuation around a mean value of the product level in a cell with a variance determined by the relation of the expression kinetics to the product half-life.
Faster Expression Kinetics and Increased Gene Copy Number Reduce Expression Noise.
The variance of product levels can be reduced by accelerating the kinetics of intermittent gene expression relative to the product half-life. In Fig. 1D, activation and deactivation rate constants were increased by a factor of 10 relative to their values in Fig. 1C (Ta = Td = Tp/40; referred to below as “fast-unstable” expression kinetics). Compared with the slow-unstable kinetics (Ta = Td = Tp/4), fast-unstable kinetics maintained the same mean product level but substantially reduced the variance around the mean. (The range of product was from 0.40 to 0.60 uP, or ≈40% of that for slow expression kinetics.) Further acceleration of expression kinetics relative to product degradation kinetics would produce extremely narrow variations in product level that would approach the invariant level of a stable system. Therefore, evolution could achieve relatively stable product levels either by selecting for highly stable expression or by selecting for unstable expression with activation/deactivation rates that were fast relative to the product half-life.
The range of product level was also related to the number of gene units that contributed to the product pool. For example, when the same net expression rate used in the two-gene model of Fig. 1C was distributed to a single gene (Fig. 1E) or to four genes (Fig. 1F), the average product level was identical but the variance of the product level increased with one “big” gene and decreased with four “small” genes. The amplitude histogram for the single-gene case (Fig. 1E, the black histogram compared with the white histogram copied from Fig. 1B) had a range approaching 1.0 uP. The histogram for the four-gene case (Fig. 1F, black histogram) was narrower than for the two-gene case. Therefore, the range of product level caused by random gene activation/deactivation kinetics decreased both as expression kinetics became faster relative to the product half-life and as the number of expression units increased.
Haploinsufficiency and Expression Noise.
An implication of stochastic gene expression, therefore, is that steady-state product levels in a cell would be expected to fluctuate around a mean and that, although loss of one allele would result in a 50% decrease in the average steady state product level, transient excursions significantly below the 50% level could occur. To illustrate this, we modeled a positively autoregulated system in which the product, P, acted as its own expression stimulus such that expression was irreversibly terminated if its level fell below an autoactivation threshold (Fig. 2A). In a biological context, the irreversible termination could represent inactivation of a regulatory pathway or cell death caused by transient loss of an essential factor. Although positive autoregulation drove the system to maximum expression, random inactivation events resulted in occasional transient declines in product level (Fig. 2B). If the threshold level necessary to maintain expression was established well below the average expression level (Pth = 0.05 uP, or 10% of the product level produced in the two-gene model), then expression persisted indefinitely in the two-gene model (Fig. 2B, upper trace and histogram). If one gene was inactivated, the mean product level decreased by half but was still almost 5-fold above the threshold. Occasional prolonged lapses of expression, however, resulted in product levels below the threshold (Fig. 2B, lower trace and histogram), a terminal event in our model system. As expected from a stochastic system, a histogram of survival times for the one-gene model were distributed exponentially (Fig. 2C). Of survival times, ≈63% were less than the average survival time of ≈20 Tp whereas a small percentage survived as long as 100 Tp. Because the probability of survival depends on the relative values of the expression kinetics, the product half-life, and the threshold value, the average survival time could range from minutes to decades. In this regard, inactivating a single gene in a diploid organism could result in the gradual accumulation over the lifetime of the organism of low probability random events affecting cell survival or function.
Figure 2.

Maintenance of autoregulated gene expression is sensitive to copy number in a stochastic manner. (A) The model of Fig. 1 was modified to simulate simultaneous binding (with affinity Kp) of two P molecules to a receptor that activates gene expression, and the expression deactivation rate was decreased 10-fold (Td = 10Tp/4) to simulate a relatively stable gene with infrequent expression lapses. The expression rate, Jp, was decreased to 12.2 to maintain an average product level of ≈0.5 uP. Setting Kp = 0.2 of the full-scale value of P = 1.0 established an all-or-none activation/deactivation threshold at a value Pth = 0.05. (B) When two genes were active (upper trace and histogram), P levels were maintained well above the threshold, and the genes were expressed indefinitely. However, when one gene was inactivated, random lapses of expression allowed P levels to fall below the threshold level (lower trace continued as a second line, and lower histogram). (C) Histogram of survival times for 100 trials of a one-gene model starting with active expression and an initial P of 0.25 (the steady-state level of P for persistent expression). As expected for a stochastic process, survival times were distributed exponentially where ≈63% of trials lasted less than the average survival time of ≈23 Tp.
Instability Enhances Signal Discrimination.
To achieve relatively stable product levels, genes could use either stable expression kinetics (kd = 0) or unstable expression kinetics that are fast relative to the product half-life (illustrated in Fig. 1 B and D, respectively). Because unstable gene expression increases the susceptibility to transient declines in product level, there might be selective pressure for highly stable gene expression. To explore the relative properties of stable and unstable stochastic gene expression systems, we modeled the generation of a threshold level of product in response to varying amounts of inducer in a fixed time period (10 Tp). In this model, the activation rate constant was proportional to the binding of an inducing stimulus, S, with a Michaelis–Menten constant of Ks such that k′a = ka S/Ks/(1 + S/Ks) (Fig. 3A). At saturating levels of stimulus (S/Ks ≫ 1), the system behaved as in Fig. 1 whereas at lower values of S, the activation of transcription was delayed and reactivation was less frequent.
Figure 3.

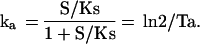 |
In the case of stable gene expression (kd = 0), the curve simply reflected the probability of initiating gene expression at a given concentration of stimulus (Fig. 3B) because initiated expression inevitably led to the accumulation of product above the threshold level. For stable genes, the fast kinetic model (Ta = Tp/40) was approximately one log unit more sensitive to the concentration of S than the slow kinetic model (Ta = Tp/4) because of the increased probability of gene activation in the trial period, but both had similar slopes. When a Hill function was fitted to each curve, the Hill coefficients (the curves’ slopes at the half-maximal value on a log-log plot) were 2.0 and 2.4 for the slow-stable and fast-stable kinetics, respectively.
In contrast, when unstable gene expression parameters were used, the slow-unstable model (Ta = Td = Tp/4) had a similar sensitivity to the stimulus as the fast-unstable model (Ta = Td = Tp/40), but the fast-unstable model generated a substantially steeper curve. The Hill coefficients were 2.4 and 7.0 for the slow-unstable and fast-unstable models, respectively. In the unstable gene models, the increased activation kinetics of the fast model did not result in an increased sensitivity to stimulus because the deactivation rate also was increased. In contrast to the stable system, where a single initiation event leads to threshold levels of product accumulation, the unstable system repeatedly deactivates expression and needs a series of initiating events to attain expression periods sufficient for the product to reach threshold. Therefore, one potential advantage of unstable gene expression is the opportunity to inactivate expression repeatedly and to resample the stimulus intensity before a commitment event.
Haploinsufficiency and Signal Discrimination.
In the context of achieving a threshold level in a defined period of time, the fast-unstable system exhibited a steeper slope than the other simulations. In a biological context, this would reflect a more predictable response for a cell at any point in a gradient of inducing signal. This enhanced signal discrimination, however, was strongly influenced by gene copy number (Fig. 3C). (In contrast to Fig. 3B, the simulations in Fig. 3C were plotted on a linear S/Ks coordinate, and the threshold level was set at 0.05 uP, or 10% of the average product level in the diploid system at maximum stimulus.) Without dosage compensation (solid lines in Fig. 3C), the haploid system demonstrated a significantly decreased slope compared with the diploid system whereas four genes yielded a steeper slope than the diploid system.
Although the threshold level for these simulations (0.05 uP) was below the average steady state product level produced by a single gene (0.25 uP), some of the decrease in the haploid slope was caused by the increased amount of time required for a single gene to produce the threshold amount of product. When the rate of gene expression was doubled in the haploid system to maintain the same production rate as two active genes (dosage compensation), the haploid system slope was still less steep than the diploid. The failure of dosage compensation in the haploid system to achieve signal discrimination comparable to the diploid system reflects the enhanced predictability achieved by integrating independent stochastic events.
Together, these curves illustrate that the stability of gene expression and gene copy number are both important variables in achieving predictable outcomes in stochastic systems. In this regard, developmental diseases caused by a heterozygous null mutation may be attributable to the kinetics of gene expression at submaximal stimulus, rather than solely to decreased steady-state product levels at maximal stimulus.
DISCUSSION
Several experimental systems have supported the stochastic nature of gene initiation in response to an inductive signal. When cells containing a transgene of the mouse mammary tumor virus regulatory region driving the β-galactosidase reporter were exposed to increasing amounts of dexamethasone, the population produced correspondingly increasing amounts of β-galactosidase activity (2). The increase in activity reflected an increasing number of cells expressing β-galactosidase rather than a graded increase in each expressing cell, indicating that the concentration of dexamethasone was predominantly effecting the probability of transgene expression. Similarly, a β-galactosidase reporter driven by binding sites for the inducible transcription factor NF-AT (nuclear factor of activated T cells) that was integrated into a T cell line showed that intermediate ranges of inducer elicited a bimodal response in the cell population, with higher levels of inducer increasing the percentage of β-galactosidase expressing cells (3). The initiation of hepatocyte gene expression in response to glucocorticoids also has been shown to be stochastic. Immunofluorescent detection of carbamoylphosphate synthase, phosphoenolpyruvate carboxykinase, and arginase in cultured hepatocytes exposed to glucocorticoid demonstrated a randomly initiated expression of each of the three genes (4). Although these studies examined the response of genes to graded levels of inducers, other studies demonstrated that enhancer elements increased the probability that a gene would be expressed (6, 7) but did not modulate the level of expression from an active template. Together, these results are consistent with the interpretation that inducers of gene expression predominantly act by increasing the probability that a gene will be expressed.
Although experimental evidence supports the stochastic character of transcription initiation, very little is known about the in vivo stability of an established transcription complex. Theoretical considerations of reaction kinetics and protein stability necessitate a finite off-rate for any transcription complex, but direct measurement of in vivo transcription deactivation rates remains technically difficult. A study of pigment expression during melanocyte differentiation showed a random initiation of pigment expression in an isogenic population and also demonstrated that, at early time points of pigment expression, some cells reverted to the nonpigmented state, indicating a stochastic deactivation rate for pigment expression (14). In this type of analysis, however, the long half-life of the gene products, both RNA and protein, might mask a higher incidence of transitions between active and inactive expression states. In situ hybridization to nascent RNA transcripts by using intron probes has been used to more directly assess the stability of gene transcription. In situ hybridization to carbamoylphosphate synthase intronic RNA in glucocorticoid treated hepatocytes indicated that expression was both random and intermittent, supporting a stochastic transition between active and inactive states (5). At the globin locus, during a period of developmental transition, transcription of the δ, γ, and β globin genes appeared to be intermittent, with estimated half-lives of gene expression that ranged from as short as 4 min for the ɛ gene to 45–80 min for the β gene (15).
Recently, the stochastic nature of gene activation and product formation in cellular regulatory networks was used to model phenotypic variation in isogenic populations (8). In this case, random variance between successive regulatory events produced probabilistic outcomes that can partition isogenic cell populations into distinct phenotypes. Although probabilistic processing can randomly generate different responses to an identical signal, an intrinsic random response presents a problem for highly predictable discrimination between similar signals. In this regard, the importance of expression deactivation rates for gradient discrimination in stochastic signaling systems has been analyzed in the elegant work of Ko (9, 10). Although Ko demonstrated that increasing the deactivation rate of a transcription unit resulted in decreased interunit variance in expression levels when the probability of activation was <1, he concluded that other mechanisms would need to be used for a relatively stable expression model to discriminate a gradient. Our modeling illustrates that relatively unstable gene expression systems (i.e., fast-unstable kinetics) can compensate partly for the signal degradation secondary to stochastic initiation because the system requires a series initiation events to maintain continued expression. A consequence of decreased expression stability, however, was a dependence on gene copy number to achieve and/or maintain a minimal threshold level of product. Therefore, systems that exploit expression instability for signal discrimination in diploid organisms would be susceptible to degraded signal discrimination in the haploid state. This was true even if the amount of product produced from the remaining gene(s) was increased to achieve the same average steady-state level as before gene reduction (i.e., dosage compensation).
These findings predict that a subset of genes that generate a response to a signaling gradient in diploid organisms should have unstable expression kinetics during the critical period of signal detection and that some haploinsufficiency syndromes might be associated with mutations in this subset of genes. Although almost nothing is known about expression instability during development, numerous genes have been identified that cause haploinsufficiency syndromes (16). The high representation of transcription factors and signaling molecules associated with haploinsufficiency syndromes suggests that signal transduction during embryonic development is particularly sensitive to gene copy number. For example, heterozygous inactivating mutations of the transcription factor PAX6 are associated with an aniridia syndrome (17); mutations of GLI-3 are associated with Greig cephalopolysyndactyly syndrome (18); mutations of ZNF-141 are associated with Wolf–Hirschhorn syndrome (19); mutations of TUPLE-1 are associated with CATCH22 (cardiac defect, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia, and 22q11 deletions) (20, 21); mutations of OSF2/CBFA1 are associated with cleidocranial dysplasia (22); mutations of SOX9 are associated with campomelic dysplasia (23); and mutations of both SOX10 and MITF are associated with neural crest syndromes, Waardenburg–Hirschsprung disease and Waardenburg syndrome type 2A, respectively (24, 25). Heterozygous deletion of the signal transduction protein LIS-1, a member of the G-protein family, has been shown to cause Miller–Dieker lissencephaly syndrome (26); mutations of the RET tyrosine kinase has been associated with Hirschsprung’s disease (27, 28); and mutations of the notch ligand Jagged1 cause Alagille syndrome characterized by liver, heart, skeleton, eye, face, and kidney developmental abnormalities (29, 30). Our modeling suggests that some of these genes might show unstable expression kinetics during critical developmental periods and that loss of one allele might result in stochastic delays of gene initiation or interruptions of gene expression.
Loss of one allele, either through germ-line or somatic mutation, is generally viewed as a “first hit” that is biologically silent until the second allele is inactivated by mutation. In contrast, stochastic models of gene expression that incorporate random gene deactivation, even at very low rates, predict that intermittent deactivation of the remaining allele would transiently mimic the homozygous null state. In addition, genes that respond to inductive stimuli would have an increased average response time. Therefore, single allele mutations could degrade the fidelity of regulatory processes and have profound consequences on critical aspects of cell biology—such as cell cycle regulation, DNA synthesis and repair, or apoptosis. Furthermore, gene redundancy could decrease the variance in stochastic systems and could increase fidelity.
A common interpretation of haploinsufficiency is that the sensitivity to copy number represents a requirement for >50% of the normal diploid product level. In stochastic models of gene expression, copy number influences the probability of gene expression, with the variance of product level determined by the relative rates of expression kinetics and of product degradation. In this regard, decreasing the copy number might lead to a haploinsufficiency phenotype by decreasing the probability of a critical event. Just as we suggest that developmental haploinsufficiency disease might result from stochastic gene expression, it is possible that some dominantly inherited late onset degenerative diseases result from single allele null mutations that confer a finite lifetime risk for the gene product to fall below a critical threshold because of occasionally prolonged off-times of the remaining wild-type allele. Even if the average gene off-period is short relative to the product half-life, over the life-span of an organism, the accumulation of low probability events could result in a late-onset disease phenotype. As a further speculation, we suggest that non-zero expression deactivation rates might have evolved such that regions of the haploid genome can be sampled early in development to eliminate embryos with deleterious mutations in a subset of critical genes. This would diminish the representation of serious recessive mutations in the population and significantly save reproductive energy.
Acknowledgments
We thank our many colleagues that critically commented on this work, particularly Mark Groudine, Dan Gottschling, Ed Giniger, and members of the Weintraub Group. S.J.T. was supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR45203).
References
- 1.Beaudet A L. In: Harrison’s Principles of Internal Medicine. Fauci A S, Braunwald E, Isselbacher K J, Wilson J D, Martin J B, Kasper D L, Hauser S L, Longo D L, editors. New York: McGraw–Hill; 1998. pp. 365–395. [Google Scholar]
- 2.Ko M S H, Nakauchi H, Takahashi N. EMBO J. 1990;9:2835–2842. doi: 10.1002/j.1460-2075.1990.tb07472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiering S, Northrop J P, Nolan G P, Mattila P S, Crabtree G R, Herzenberg L A. Genes Dev. 1990;4:1823–1834. doi: 10.1101/gad.4.10.1823. [DOI] [PubMed] [Google Scholar]
- 4.van Roon M A, Aten J A, van Oven C H, Charles R, Lamers W H. Dev Biol. 1989;136:508–516. doi: 10.1016/0012-1606(89)90276-5. [DOI] [PubMed] [Google Scholar]
- 5.Dingemanse M A, de Boer P A, Moorman A F, Charles R, Lamers W H. Differentiation. 1994;56:153–162. doi: 10.1046/j.1432-0436.1994.5630153.x. [DOI] [PubMed] [Google Scholar]
- 6.Weintraub H. Proc Natl Acad Sci USA. 1988;85:5819–5823. doi: 10.1073/pnas.85.16.5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walters M C, Fiering S, Eidemiller J, Magis W, Groudine M, Martin D I K. Proc Natl Acad Sci USA. 1995;92:7125–7129. doi: 10.1073/pnas.92.15.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAdams H H, Arkin A. Proc Natl Acad Sci USA. 1997;94:814–819. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko M S H. J Theor Biol. 1991;153:181–194. doi: 10.1016/s0022-5193(05)80421-7. [DOI] [PubMed] [Google Scholar]
- 10.Ko M S H. BioEssays. 1992;14:341–346. doi: 10.1002/bies.950140510. [DOI] [PubMed] [Google Scholar]
- 11.Ferrell J E, Jr, Machleder E M. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 12.Goss P J E, Peccoud J. Proc Natl Acad Sci USA. 1998;95:6750–6755. doi: 10.1073/pnas.95.12.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colquhoun D, Hawkes A G. In: Single Channel Recording. Sakmann B, Neher E, editors. New York: Plenum; 1983. [Google Scholar]
- 14.Bennett D C. Cell. 1983;34:445–453. doi: 10.1016/0092-8674(83)90378-1. [DOI] [PubMed] [Google Scholar]
- 15.Wijgerde M, Grosveld F, Fraser P. Nature (London) 1995;377:209–213. doi: 10.1038/377209a0. [DOI] [PubMed] [Google Scholar]
- 16.Fisher E, Scambler P. Nat Genet. 1994;7:5–7. doi: 10.1038/ng0594-5. [DOI] [PubMed] [Google Scholar]
- 17.Ton C C, Hirvonen H, Miwa H, Weil M M, Monaghan P, Jordan T, van Heyningen V, Hastie N D, Meijers-Heijboer H, Dreschsler M, et al. Cell. 1991;67:1059–1074. doi: 10.1016/0092-8674(91)90284-6. [DOI] [PubMed] [Google Scholar]
- 18.Vortkamp A, Gessler M, Grzeschik K H. Nature (London) 1991;352:539–540. doi: 10.1038/352539a0. [DOI] [PubMed] [Google Scholar]
- 19.Tommerup N, Aagaard L, Lund C L, Boel E, Baxendale S, Bates G P, Lehrach H, Vissing H. Hum Mol Genet. 1993;2:1571–1575. doi: 10.1093/hmg/2.10.1571. [DOI] [PubMed] [Google Scholar]
- 20.Halford S, Wadey R, Roberts S, Daw S C, Whiting J A, O’Donnell H, Dunham I, Bentley D, Lindsay E, Baldini A, et al. Hum Mol Genet. 1993;2:2099–2107. doi: 10.1093/hmg/2.12.2099. [DOI] [PubMed] [Google Scholar]
- 21.Wilson D I, Burn J, Scrambler P, Goodship J. J Med Genet. 1993;30:852–856. doi: 10.1136/jmg.30.10.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Nat Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 23.Cameron F J, Hageman R M, Cooke-Yarborough C, Kwok C, Goodwin L L, Sillence D O, Sinclair A H. Hum Mol Genet. 1996;5:1625–1630. doi: 10.1093/hmg/5.10.1625. [DOI] [PubMed] [Google Scholar]
- 24.Pingault V, Bondurand N, Kuhlbrodt K, Goerich D E, Prehu M O, Puliti A, Herbarth B, Hermans-Borgmeyer I, Legius E, Matthijs G, et al. Nat Genet. 1998;18:171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 25.Nobukuni Y, Watanabe A, Takeda K, Skarka H, Tachibana M. Am J Hum Genet. 1996;59:76–83. [PMC free article] [PubMed] [Google Scholar]
- 26.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns W B, Caskey C T, Ledbetter D H. Nature (London) 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 27.Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kaariainen H, et al. Nature (London) 1994;367:377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 28.Edery P, Lyonnet S, Mulligan L M, Pelet A, Dow E, Abel L, Holder S, Nihoul-Fekete C, Ponder B A, Munnich A. Nature (London) 1994;367:378–380. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Krantz I D, Deng Y, Genin A, Banta A B, Collins C C, Qi M, Trask B J, Kuo W L, Cochran J, et al. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 30.Oda T, Elkahloun A G, Pike B L, Okajima K, Krantz I D, Genin A, Piccoli D A, Meltzer P S, Spinner N B, Collins F S, et al. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 31.Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M. EMBO J. 1997;16:1381–1390. doi: 10.1093/emboj/16.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Press W H, Vetterling W T, Teukolsky S A, Flannery B P. Numerical Recipes in C. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]