

Abstract
The ubiquitin-proteasome and lysosome-autophagy pathways are the two major intracellular protein degradation systems that work cooperatively to maintain homeostasis. Proteasome inhibitors (PIs) have clinical activity in hematological tumors, and inhibitors of autophagy are also being evaluated as potential antitumor therapies. In the current study we found that chemical proteasome inhibitors and siRNA-mediated knockdown of the proteasome's enzymatic subunits promoted autophagosome formation, stimulated autophagic flux, and upregulated expression of the autophagy specific genes (ATGs) (ATG5 and ATG7) in some human prostate cancer cells and immortalized mouse embryonic fibroblasts (MEFs). Upregulation of ATG5 and ATG7 only occurred in cells displaying PIs-induced phosphorylation of the eukaryotic translation initiation factor 2alpha (eIF2α), an important component of the unfolded protein responses. Furthermore, PIs did not induce autophagy or upregulate ATG5 in MEFs expressing a phosphorylation-deficient mutant form of eIF2α. Combined inhibition of autophagy and the proteasome induced an accumulation of intracellular protein aggregates reminiscent of neuronal inclusion bodies and caused more cancer cell death than blocking either degradation pathway alone. Overall, our data demonstrate that proteasome inhibition activates autophagy via a phospho-eIF2α-dependent mechanism to eliminate protein aggregates and alleviate proteotoxic stress.
Keywords: Bortezomib, NPI-0052, autophagy, unfolded protein response, prostate cancer
Introduction
The 26S proteasome is a multicatalytic enzyme complex that degrades ubiquitinated proteins via an ATP-dependent mechanism. It plays a central role in cell cycle progression and cell death, and specific chemical inhibitors of the proteasome have recently emerged as effective antitumor therapies (Adams, 2004; Goldberg, 2007). Bortezomib (Velcade/PS-341, Millennium Pharmaceuticals, Inc.) is a peptide boronate inhibitor of the proteasome that received FDA approval for the treatment of multiple myeloma and mantle cell lymphoma. Its success has prompted the development of structurally distinct proteasome inhibitors (PIs), including NPI-0052 (salinosporamide A, Nereus Pharmaceuticals), which is an orally active irreversible PI that is distinct from bortezomib in terms of its effects on the proteasome's three active sites and its mechanisms of action (Chauhan et al., 2005; Miller et al., 2007). Their complex anti-tumor effects involve a number of mechanisms that include inhibition of the transcription factor nuclear factor kappaB (NFκB), stabilization of cell cycle regulators (p21WAF1 and p53) and pro-apoptotic proteins (Noxa, Bim and Bik) (McConkey and Zhu, 2008). PIs also induce endoplasmic reticulum (ER) stress and activate the unfolded protein response (UPR) since the proteasome plays an essential role in cellular protein quality control (Chauhan et al., 2008; McConkey and Zhu, 2008; Nawrocki et al., 2006). Though PIs have promising antitumor effects in hematological tumors, they have had more modest effects as single agent in solid tumors, and multiple myeloma patients who initially benefit from bortezomib-based therapy inevitably relapse (Anderson, 2004; McConkey and Zhu, 2008). Therefore, understanding the molecular mechanisms involved in the responses of cells to PIs should facilitate the development of PIs-based combination that will best exploit their unique antitumor activities.
Macroautophagy (hereafter referred to as “autophagy”) is a genetically programmed, evolutionarily conserved process that is involved in the degradation of long-lived proteins and protein aggregates and in the turnover of cytoplasmic organelles such as mitochondria and ER via lysosomes (Levine and Kroemer, 2008; Mizushima et al., 2008). Autophagy is regulated by a group of ATG genes that function to sense environmental stress, assemble double- or multi-membrane-bound autophagic vacuoles to sequester the cytoplasmic materials (termed autophagosome), and facilitate the transport of autophagosomes to lysosomes for degradation (Levine and Kroemer, 2008). Recent studies showed that autophagy is frequently activated in tumor cells exposed to chemotherapy or radiation and that it can confer therapeutic resistance by clearing damaged organelles and by providing nutrients when blood perfusion is limited (Ito et al., 2005; Kondo et al., 2005; Mizushima et al., 2008). Furthermore, inhibition of autophagy with chemical inhibitors or knockdown of key ATG expression induced cancer cell death and enhanced the effects of chemotherapy in some preclinical studies (Amaravadi et al., 2007; Apel et al., 2008; Carew et al., 2007; Kondo et al., 2005).
The ubiquitin-proteasome and autophagy-lysosome have been viewed as distinct degradation systems, but recent studies suggest that they are mechanistically linked. For example, ubiquitin-positive aggregates accumulate in Atg7-deficient hepatocytes and neurons (Komatsu et al., 2006; Komatsu et al., 2005), and autophagy is induced in response to proteasome inhibition under certain situations but the mechanisms have not been clarified (Ding et al., 2007; Fels et al., 2008; Pandey et al., 2007). We therefore wondered whether clinically relevant PIs stimulate autophagy in cancer cells and which mechanisms are involved in activating the process. We were also interested in determining how proteasome inhibitor-induced autophagy would influence cancer cell death, since autophagy has been implicated in both cancer cell death and survival. In a previous study we found that PIs induced PKR-like ER-resident kinase (PERK)/eIF2α arm of the UPR in some cancer cells but not in others (Zhu et al., 2009). Phosphorylation of eIF2α may regulate autophagy by mediating conversion of the ATG8/light chain 3 (LC3) from LC3-I (free form) to LC3-II [phosphatidylethanolamine(PE)-modified, membrane-bound form], which plays a key role in autophagosome maturation (Kouroku et al., 2007; Talloczy et al., 2002). Here we report that chemical PIs or knockdown of the proteasome's enzymatically active subunits induced increased expression of ATG genes and promoted autophagosome formation in human prostate cancer cells and MEFs via a phospho-eIF2α-dependent mechanism. Furthermore, combined inhibition of the proteasome and autophagy induced more cell death than were observed when either degradative pathway was blocked by itself. The current study provides a strong rationale for combining inhibitors of the proteasome and of autophagy in preclinical tumor models that possess an intact eIF2α arm of the UPR.
Materials and methods
Cell lines and culture
Human LNCaP-Pro5 prostate cancer cells were provided by Dr. Curtis Pettaway (Department of Urology, University of Texas M.D. Anderson Cancer Center, Houston, TX). Human PC3 prostate cancer cells were obtained from American Type Culture Collection (Rockville, MD) and PC3-R cells are spontaneously arising PIs-resistant variant of PC3. Prostate cancer cells were grown in RPMI-1640 (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum (Life Technologies), 1% vitamins (Life Technologies), sodium pyruvate (Bio Whittaker, Rockland, ME), L-glutamine (Bio Whittaker), penicillin/streptomycin solution (Bio Whittaker), and non-essential amino acids (Life Technologies) under conditions of 5% CO2 at 37°C in an incubator. eIF2α51SS wild-type and eIF2α51AA knock-in mutant mouse MEFs were kindly provided by Dr. Ron (New York University School of Medicine, New York, NY). ATF4 wild-type and ATF4 negative (ATF4−/−) mutant MEFs were generously provided by Dr. Wek (Indiana University School of Medicine, Indianapolis, IN). The MEFs were grown in dMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin solution, L-glutamine, non-essential amino acids, and 55μM β-mercaptoethanol.
Reagents, antibodies and plasmids
The proteasome inhibitor bortezomib was provided by Millenium Pharmaceuticals (Cambridge, MA) and NPI-0052 was provided by Nereus Pharmaceuticals (San Diego, CA). Propidium iodide, chloroquine (CQ), 3-methyladenine (3MA), thapsigargin (TG), and actinomycin D (ActD) were purchased from Sigma-Aldrich Co. (St. Louis, MO). The general caspase inhibitor zVAD-fmk (z-VAD) was purchased from R&D Systems, Inc. (Minneapolis, MN). Gemzar® (gemcitabine hydrochloride, Eli Lilly and Company, Indianapolis, IN) and SAHA (suberoylanilide hydroxamic acid) were obtained from the University of Texas MD Anderson Cancer Center. Antibodies were obtained from the following sources: ATF4, ATF3 and ubiquitin (Santa Cruz Biotechnology, Santa Cruz, CA); total and phosphorylated eIF2α(Ser52) (Invitrogen Biosource™, Carlsbad, CA); phosphorylated eIF2α(Ser51) (Cell Signaling Technology, Inc., Danvers, MA); proteasome subunits β1, β2 and β5 (BIOMOL International, L.P., Plymouth Meeting, PA); LC3, vinculin, green fluorescent protein(GFP) and actin (Sigma Chemical Co., St. Louis, MO); and Cy5-conjugated goat anti-mouse IgG and Sytox-green (Molecular Probes, Eugene, Oregon). Horseradish peroxidase-conjugated secondary antibodies were obtained from Amersham Pharmacia Biotech (Piscataway, NJ). GFP-tagged LC3 plasmids (GFP-LC3) were provided by Dr. Kondo (Department of Neurosurgery, University of Texas M.D. Anderson Cancer Center, Houston, TX).
Quantitative reverse transcription polymerase chain reaction (RT-PCR)
Total cellular mRNA was isolated using an RNeasy kit (QIAGEN Inc., Valencia, CA). One-step real-time RT-PCR was performed in triplicate using the AgPath-ID One-Step RT-PCR Kit (Ambion, Austin, TX) and the expression of each target gene was quantified using the StepOne™ Real-time PCR systems (Applied Biosystems, Foster City, CA). The following primer pairs were utilized for target gene amplification: hATG5, hATG7, cyclophilin A, mATG5, mATG7 and mTubulin5 (Applied Biosystems). The summary data were expressed as the average of ratios (relative expression to control)±SEM and the expression of the cyclophilin A (human prostate cancer cells) or tubulin5 (MEFs) served as internal control.
Transmission electron microscopy (TEM)
TEM of cells was performed in the High Resolution Electron Microscopy Facility in University of Texas M.D. Anderson Cancer Center as previously described (Nawrocki et al., 2006). Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques Corp., Danvers, MA).
Transient transfection assay
The GFP-LC3 plasmid was prepared using Qiagen plasmid DNA isolation kits (QIAGEN Inc., Valencia, CA). MEF cells were plated in 6-well plates and were transfected with GFP-LC3 plasmid using TransFast™ Transfection Reagent (Promega Co., Madison, WI). Fluorescence imaging was performed and digitally captured using an OLYMPUS 1×71 inverted fluorescence microscope.
Small interfering RNA (siRNA)–mediated gene silencing
LNCaP-Pro5 cells were transfected with siRNAs targeting human proteasome enzymatic active subunits (β1, 2, 5), ATG5 and ATG7, or siRNA non-specific control (Dharmacon RNA Technologies, Lafayette, CO) using the Oligofectamine reagent (Invitrogen Life Technologies, Carlsbad, CA). The efficiency of gene silencing was verified by quantitative RT-PCR.
Immunoblotting
Total cell lysates were prepared using a 1% Triton X-100 buffer as described previously (Zhu et al., 2009).
Quantification of DNA fragmentation and cell viability
After the indicated treatment, cells were collected and DNA fragmentation was measured by propidium iodide staining and FACS analysis. Cell viability was measured by PI-exclusion viability/FACS analysis. For the caspase inhibitor studies, cells were pre-incubated with 20μM zVAD-fmk for 1h before the indicated treatment.
Caspase-3/7 activity assay
Caspase-3/7 activities were measured using the Vybrant® FAM Caspase-3/7 Assay Kit (Invitrogen, Molecular Probe, Carlsbad, CA). LNCaP-Pro5 cells were exposed to various agents, harvested, pelleted by centrifugation, resuspended in media containing 1×FLICA solution and incubated for 1h at 37°C in the dark. Caspase-3/7 activities were measured using FACScan (Becton Dickinson, Mountain View, CA).
Confocal microscopy
LNCaP-Pro5 cells were plated in 4-well chamber slides and were treated with 100nM bortezomib, 100nM NPI-0052, 50μM chloroquine or a combination of PI and chloroquine for 12h. Ubiquitin staining was performed as previously described (Nawrocki et al., 2005). Slides were analyzed for ubiquitin-positive inclusion bodies using a Zeiss LSM510 confocal microscope (Oberkochem, Germany). The presence of ubiquitin-positive inclusion bodies was considered indicative of intracellular aggregates formation, and the aggregates were quantified in five different fields.
Data analysis
Experiments presented in the figures are representative of at least three independent repetitions. Statistical analyses were performed using GraphPad3.05 statistical software (GraphPad Software, San Diego, CA) using the Student's t test, or one-way ANOVA where appropriate (P<0.05 was considered statistically significant).
Results
Effects of the proteasome inhibition on autophagy
Since the ubiquitin-proteasome and autophagy-lysosome systems appear to be mechanistically linked, we suspected that autophagy might be activated to clear ubiquitinated/unfolded protein aggregates and promote cell survival when the proteasome is inhibited. Ultra-structural analyses by transmission electron microscopy (TEM) demonstrated that PIs induced autophagosome formation in about 30∼40% of LNCaP-Pro5 cells, as indicated by the presence of double or multiple membrane-bound vacuolar structures containing partially digested cytoplasmic contents (Figure 1A). PIs also induced structural alterations consistent with the formation of early-aggresomes, which are small patches of highly condensed electron-dense material in cytoplasm. Compared to typical aggresomes, they are smaller and are not necessarily concentrated around the perinuclear area. Moreover, some vacuoles and/or atypical autophagosomes accumulated near the early-aggresome-like structures, which may indicate that the early aggresomes transfer their cargo to the vacuoles as the mature autophagosomes form. We observed no obvious differences in the morphologies of the mitochondria and the ER in LNCaP-Pro5 cells exposed to PIs as compared to untreated controls. PIs induced ATG8/LC3 conversion from LC3-I to LC3-II, and the autophagy inhibitor chloroquine (a lysosomotropic agent that raises lysosomal pH, leading to inhibition of lysosome-autophagosome fusion and lysosomal protein degradation) enhanced the LC3-II accumulation, which further confirmed that PIs induced autophagy activation (Figure 7B).
Figure 1. Effects of proteasome inhibition on autophagy.
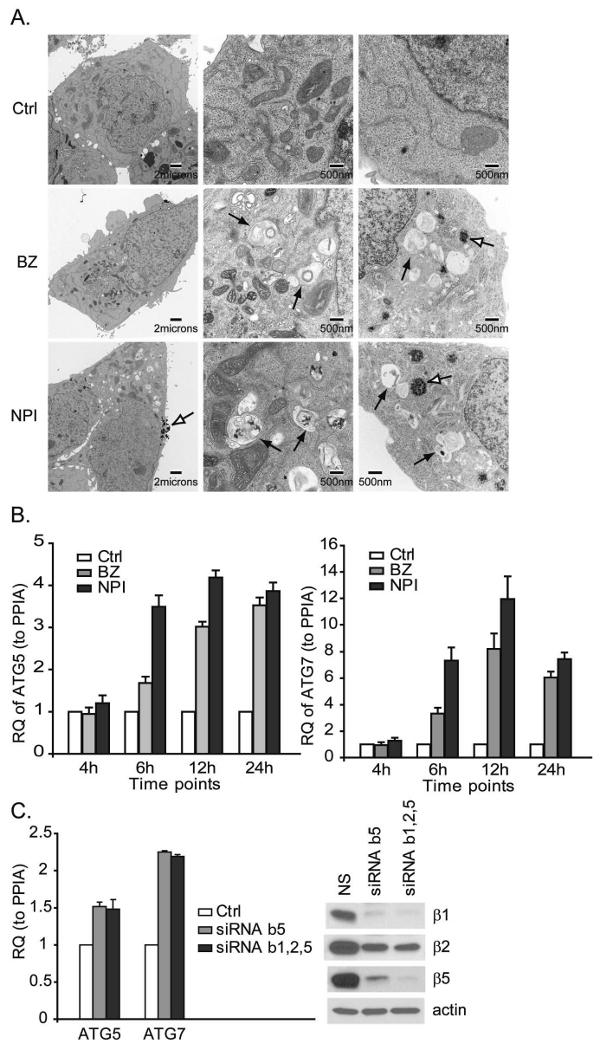
A. PIs induce autophagosome formation. LNCaP-Pro5 cells were treated with 100nM bortezomib (BZ) or NPI-0052 (NPI) for 24h. The black arrows identify autophagosome structures and the open arrows identify the early-aggresome like structures. B. PIs stimulate ATG gene expression. LNCaP-Pro5 cells were treated as above for indicated times and the expression of ATG5/7 was measured by one step-quantitative RT-PCR using the StepOne™ Real-time PCR systems. RQ, relative quantity; PPIA, cyclophilin A. C. Proteasome inhibition stimulates ATG gene expression. LNCaP-Pro5 cells were transfected with siRNA constructs specific for proteasome subunits β1, 2&5 or a non-targeted control siRNA for 72h. One step-quantitative RT-PCR for ATG5/7 was performed. Levels of the proteasome subunits were examined by immunoblotting and actin served as a loading control. Ctrl, Control; NS, Non-targeted control siRNA.
Figure 7. Effects of chloroquine and PIs on ubiquitin-positive aggregate formation.
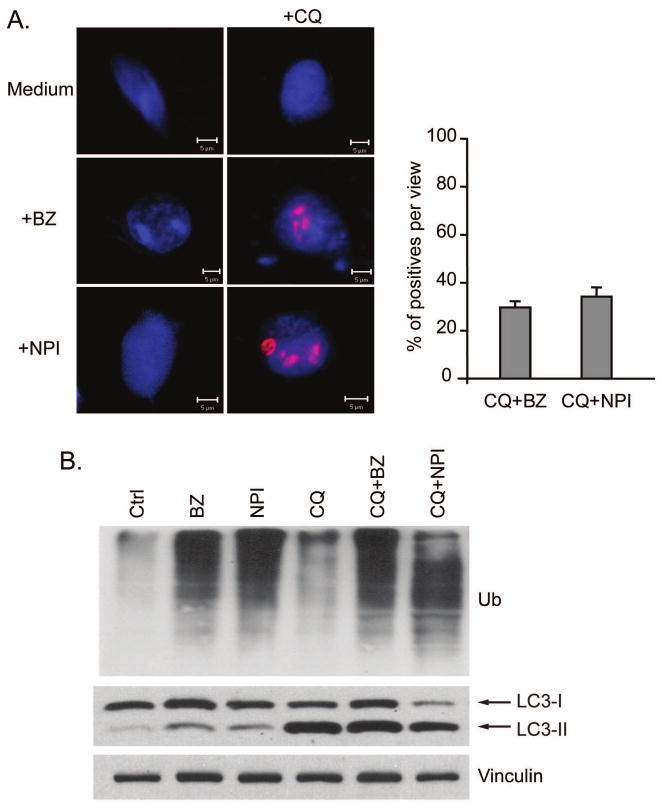
A. LNCaP-Pro5 cells were exposed to 100nM bortezomib (BZ), 100nM NPI-0052 (NPI), 50μM chloroquine (CQ) or a combination of chloroquine and PI for 12h. The nucleus was stained by sytox-green, shown as blue. Protein aggregates were stained using anti-ubiquitin, shown as red. The right panel is the quantification of cells that contain ubiquitin-positive aggregates per view. B. Effects of PIs and chloroquine on Atg8/LC3 expression. LNCaP-Pro5 cells were treated as above and the expression of ubiquitin and Atg8/LC3 was examined by immunoblot.
We next examined the effects of PIs on the expression of two critical autophagy pathway components (ATG5 and ATG7). ATG5 conjugates to ATG12 to form a complex that associates with the isolated membranes of the early autophagosome. ATG7 functions similar to an E1 ubiquitin-activating enzyme that is used in two ubiquitin-like modification systems in the formation of autophagosomes (Hara et al., 2006; Komatsu et al., 2006; Levine and Kroemer, 2008). Both PIs induced early induction of ATG5/7 mRNA levels and the effects were persistent in LNCaP-Pro5 cells (Figure 1B). Inhibition of the proteasome by specific silencing of the proteasome's β5 subunit (responsible for its “chymotryptic” activity) or triple silencing of all three proteasome active subunits (β1, 2&5, responsible for its “caspase-, trypsin-, and chymotrypsin-like activities respectively) also stimulated increases in ATG5/7 levels in LNCaP-Pro5 cells providing further evidence that the effects of PIs were “on target” (Figure 1C). Interestingly, single silencing of β5 and triple silencing of β1, 2&5 had similar effects on ATG5/7 gene induction and the proteasome β-subunits expression, suggesting that knockdown of β5 subunit alone might perturb overall proteasome assembly in the cells. Together these data demonstrated that autophagy is activated by PIs in LNCaP-Pro5 cells.
Role of eIF2α phosphorylation in PIs-mediated activation of autophagy
We recently found that PIs have heterogeneous effects on eIF2α phosphorylation in different cancer cells (Zhu et al., 2009). For example, PIs induced phosphorylation of eIF2α in LNCaP-Pro5 cells but not in PC3-R cells (Figure 2A). Nonetheless, PIs upregulated two downstream targets of phospho-eIF2α (ATF4 and ATF3) in both cell lines, possibly because PIs inhibit their proteasomal-dependent degradation (Wek et al., 2006). In contrast, PIs failed to induce ATG5/7 mRNA upregulation in PC3-R cells (Figure 2B). We also examined the effects of several other agents on ATG5/7 expression in LNCaP-Pro5 cells. Thapsigargin, which inhibits the sarcoplasmic/endoplasmic Ca2+-ATPase and induces ER stress and eIF2α phosphorylation, produced comparable increases in ATG5/7 expression. On the other hand, several other cytotoxic agents, including some that have been reported to regulate autophagy in previous studies (ceramide, SAHA and gemcitabine) (Iwata et al., 2005; Scarlatti et al., 2004; Shao et al., 2004), had milder effects on ATG gene induction (Figure 2C). Finally, actinomycin D (chemical inhibitor of transcription) blocked PI- or TG-induced upregulation of ATGs, which suggested that the effects of PIs/TG on ATG gene induction required transcription (Figure 2D).
Figure 2. Differential effects of PIs on activation of autophagy.
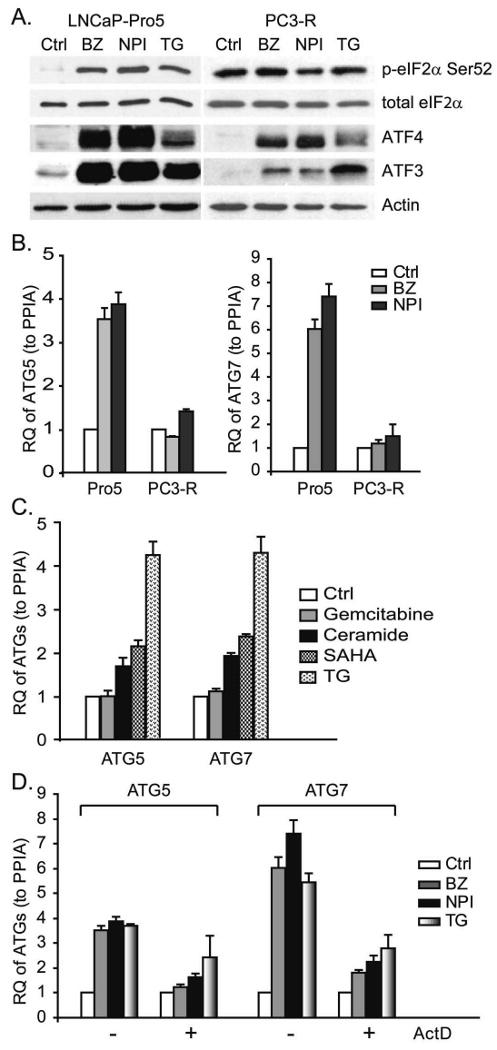
A. Differential effects of PIs on eIF2α phosphorylation. LNCaP-Pro5 and PC3-R cells were exposed to 100nM bortezomib (BZ), 100nM NPI-0052 (NPI) or 10μM thapsigargin (TG) for 4h, and phosphorylated eIF2α, total eIF2α, ATF4 and ATF3 levels were measured by immunoblotting. B. Differential effects of PIs on ATG5/7 expression. LNCaP-Pro5 and PC3-R cells were treated with 100nM bortezomib, 100nM NPI-0052 or 1μM thapsigargin for 24h and the expression of ATG5/7 was measured by one step-quantitative RT-PCR. C. Effects of other cytotoxic agents on ATG gene expression. LNCaP-Pro5 cells were exposed to 1μM thapsigargin, 30μM ceramide, 10μM SAHA or 10μM gemcitabine for 24h and the expression of ATG5/7 was measured by one step-quantitative RT-PCR. D. Effects of PIs on ATG expression after actinomycin D (ActD) treatment. LNCaP-Pro5 cells were pre-treated with 5μg/ml actinomycin D, and then were exposed to 100nM bortezomib, 100nM NPI-0052 or 1μM thapsigargin for 24h. The mRNA levels of ATG5/7 were measured by one step-quantitative RT-PCR. RQ, relative quantity; PPIA, cyclophilin A.
To more directly define the role of eIF2α phosphorylation in ATG gene induction, we examined the effects of PIs on autophagy in MEFs expressing wild-type (eIF2α51SS) or a phosphorylation-deficient knock-in mutant form of eIF2α (eIF2α51AA). Bortezomib, NPI-0052 and thapsigargin all promoted autophagosome formation as monitored by TEM and the clustering of punctate expression of GFP-LC3 in about 40% wild-type MEFs but failed to do so in the mutant MEFs (Figure 3A and 4A). The drugs also induced high expression of ATG5&7 (Figure 3B) and several other ATGs (supplemental figure 2) in the wild-type MEFs and had little to no effect on these genes in the mutant MEFs. Corresponding to the punctate expression, PIs and TG induced conversion of LC3-I to PE-modified LC3-II only in the wild-type MEFs and the LC3-II levels correlated with the number of autophagosomes (Figure 4B). Accumulation of LC3-II in the presence of lysosomal protease inhibitors is indicative of enhancement of autophagic flux (Mizushima and Yoshimori, 2007). The fact that chloroquine induced the accumulation of GFP-tagged and endogenous LC3-II provided further evidence that PIs and TG stimulated autophagic flux as chloroquine can efficiently block the lysosome mediated autophagic turnover (Figure 4C and Figure 7B). We were unable to observe punctate expression of GFP-LC3 in PI-exposed LNCaP-Pro5 cells. We speculate the difference may be related to the fact that the tumor cells might have heterogeneous abilities to modulate GFP-LC3 processing and the recruitment of LC3-II to autophagosomes (Geng et al., 2008). We also found that siRNA mediated knock down of the four known eIF2α kinases (PERK, GCN2, HRI and PKR) (Ron and Walter, 2007) partially suppressed the PI-mediated ATG gene induction in LNCaP-Pro5 cells (data not shown). Together, the results demonstrate that PIs induce ATG5/7 expression and activate autophagy via an eIF2α phosphorylation dependent mechanism.
Figure 3. Effects of PIs on autophagy in MEFs.
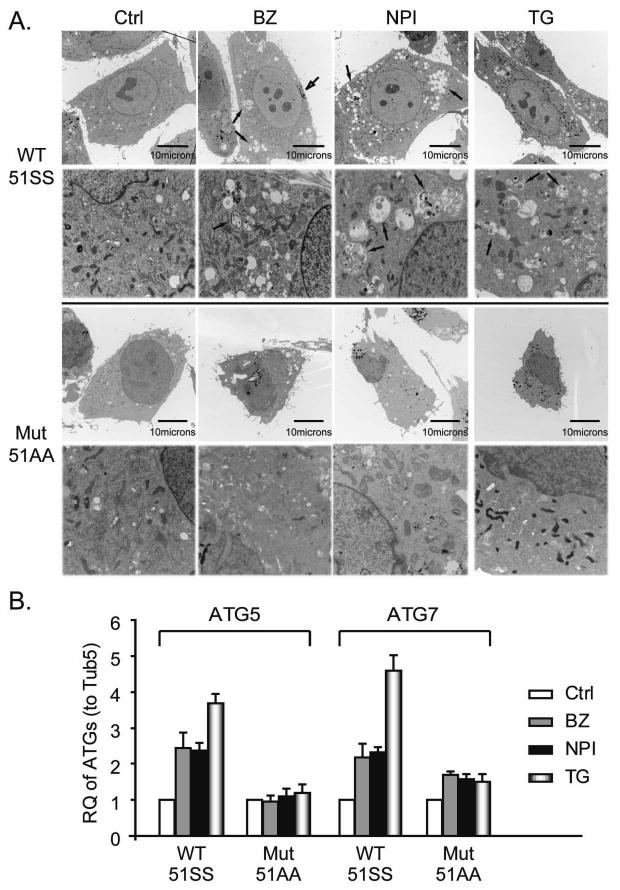
A. Effects of PIs on autophagosome formation in MEFs. MEFs were exposed to 100nM bortezomib (BZ), 100nM NPI-0052 (NPI) or 1μM thapsigargin (TG) for 24h. The arrows identify autophagosome structures. B. Effects of PIs and TG on ATG expression. MEFs were incubated with PIs or thapsigargin as described above and ATG5/7 mRNA levels were measured by one-step real-time RT-PCR. Tub5, tubulin5.
Figure 4. Effects of PIs on LC3 expression in MEFs.
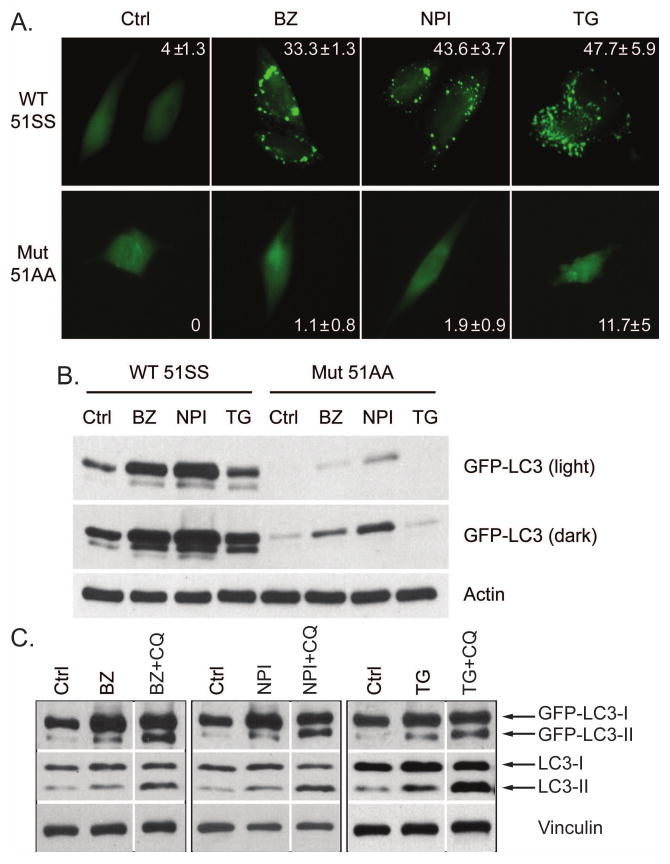
A. PIs and TG induce punctate expression of GFP-LC3. MEFs were transiently transfected with GFP-LC3 construct and then incubated with 100nM bortezomib (BZ), 100nM NPI-0052 (NPI) or 1μM thapsigargin (TG) for 12h. Punctate expression of GFP-LC3 was observed by fluorescence microscope and the percentage of cells displaying punctate GFP-LC3 was quantified (mean±S.E.). B. PIs and TG induce GFP-LC3 conversion. MEFs were incubated with PIs or thapsigargin as described above and the expression of GFP-LC3-I/II was examined by immunoblotting and actin severed as a loading control. C. Chloroquine promoted LC3-II accumulation. eIF2α51SS wild-type MEFs were transiently transfected with GFP-LC3 construct and were incubated with 100nM bortezomib (BZ), 100nM NPI-0052 (NPI) or 1μM thapsigargin (TG) with or without 50μM chloroquine(CQ) for 12h. The expression of GFP-LC3 and endogenous LC3 was examined by immunoblotting and vinculin served as a loading control.
Role of ATF4 in PI-mediated ATG gene induction
Our results suggest that phosphorylation of eIF2α stimulates ATG5 and ATG7 transcription because the transcription inhibitor actinomycin D blocked PIs-induced ATG5/7 accumulation. We next sought to identify the transcription factor(s) involved in the upregulation of the ATG transcripts. The transcription factor ATF4 is considered an essential downstream target of phospho-eIF2α within the PERK arm of the UPR (Ron and Walter, 2007) and the promoter regions of ATG5/7 contain ATF/CREB binding sites. Furthermore, a previous study demonstrated that nutrient deprivation or viral infection induced autophagy via a mechanism that was dependent on both eIF2α phosphorylation and ATF4 expression (Talloczy et al., 2002). We found that PIs- or TG-selectively induced the accumulation of ATF4 mRNA (data not shown) and protein (Figure 5A) in the wild-type but not in the mutant MEFs even though these chemicals promoted the accumulation of other labile proteins (i.e., ATF3 and p21) in the mutant MEFs. Nonetheless, several lines of evidence suggested to us that ATF4 is not required for PIs-/TG-mediated ATG gene induction regardless of the selective effects on ATF4 accumulation. First, PIs still induced strong increases of ATG5/7 mRNA in LNCaP-Pro5 cells in which ATF4 had been knocked down with siRNA (Figure 5B). Second, complete elimination of ATF4 expression (in ATF4−/− MEFs) also had no effect on PI- or TG-induced ATG5/7 accumulation (Figure 5C). Finally, PIs and TG induced comparable accumulation of ATF4 protein expression in both LNCaP-Pro5 and PC3-R cells, but they did not induce mRNA of ATG5/7 in the latter, presumably because PIs did not induce eIF2α phosphorylation in the PC3-R cells (Figure 2A). Therefore, the effects of PIs on ATG5/7 are phospho-eIF2α-dependent but ATF4-independent.
Figure 5. Lack of involvement of ATF4 in PI-induced ATGs' upregulation.
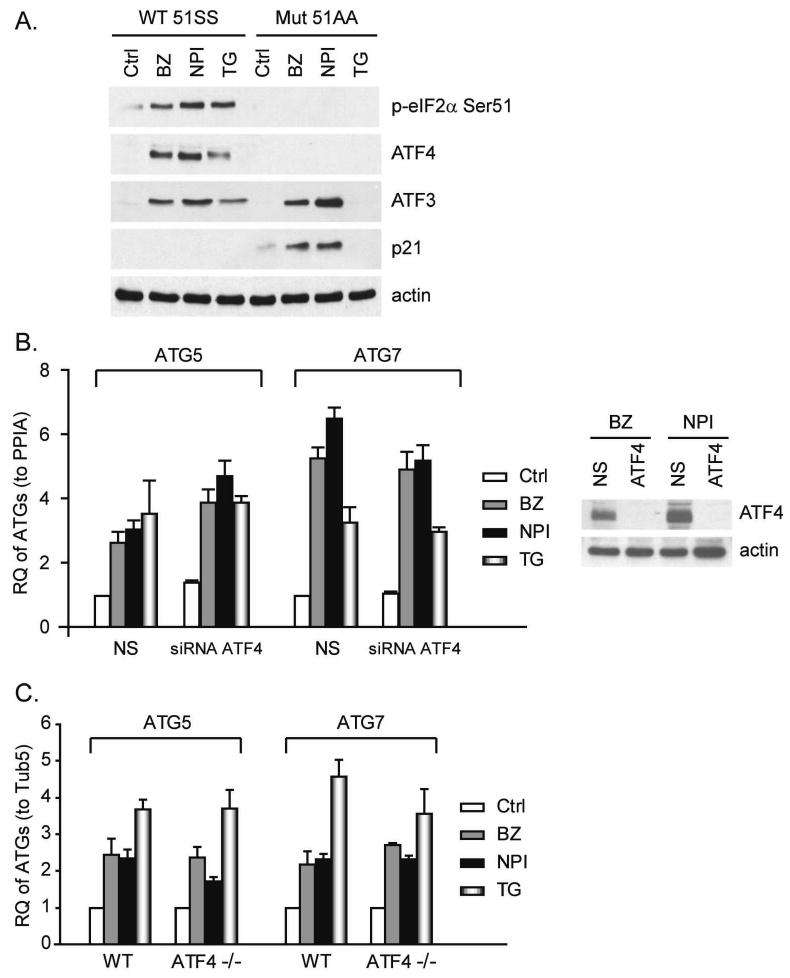
A. Effects of PIs of ATF4, ATF3 and p21WAF1 in MEFs. MEFs were exposed to 100nM bortezomib (BZ), 100nM NPI-0052 (NPI) or 10μM thapsigargin (TG) for 4h. Levels of phospho-eIF2α, ATF4, ATF3 and p21WAF1 in total lysates were measured by immunoblotting and actin served as loading control. B. Effects of knocking down ATF4 on ATG expression in LNCaP-Pro5 cells. LNCaP-Pro5 cells were transfected with siRNA constructs specific for ATF4 or a non-targeted control siRNA for 48h. Transfected cells were exposed to 100nM bortezomib, 100nM NPI-0052 or 1μM thapsigargin for 24h and the mRNA levels of ATG5/7 were measured by one-step real-time PCR. The expression of target genes in untreated group transfected with non-targeted control siRNA was arbitrarily set at 1 and data shown are mean±S.E., n=3. Silencing efficiency was examined by immunoblotting and actin served as loading control. C. Effects of PIs on ATGs in ATF4−/− MEFs. Wild type and ATF4−/− MEFs were incubated with PIs or thapsigargin as described above and the levels of ATG5/7 mRNAs were examined by one-step RT-PCR. Data shown are mean±S.E. RQ, relative quantity; PPIA, cyclophilin A; Tub5, tubulin5.
Effects of combined inhibition of the proteasome and autophagy on prostate cancer cell death
The results presented so far demonstrate that PIs activate autophagy, but how autophagy contributes to cell death is not clear. Therefore, we assessed the effects of combined inhibition of the proteasome and autophagy on apoptosis and necrosis in LNCaP-Pro5 cells. Two autophagy inhibitors (3-methyladenine and chloroquine) were used to block the autophagy activation; 3-methyladenine is a type III phosphatidylinositol 3-kinase (PI3K) inhibitor which could inhibit the autophagosome membrane formation (Amaravadi et al., 2007; Mizushima et al., 2008). Combining 3-methyladenine with PIs increased the levels of both types of cell death as determined by measuring apoptosis-associated DNA fragmentation or loss of plasma membrane integrity, whereas chloroquine primarily increased necrosis (Figure 6A). zVAD-fmk, a pan-caspase inhibitor, blocked caspases-3/7 activities and strongly reduced DNA fragmentation but did not affect plasma membrane permeabilization, indicating that caspases were not required for cell death (Figure 6B). The effects of 3-methyladenine on both types of cell death were stronger than those of chloroquine. Increased cell death was also observed when PIs and inhibitors of autophagy were combined in two other prostate cancer cell models (PC3-R and DU145) (data not shown).
Figure 6. Effects of combined inhibition of the proteasome and autophagy on cell death.
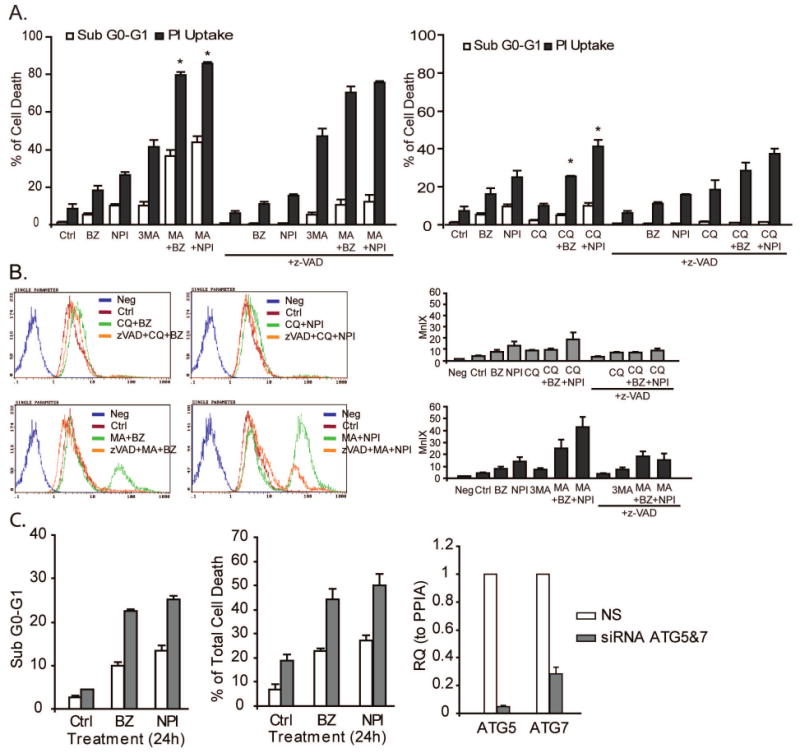
A. LNCaP-Pro5 cells were incubated with 100nM bortezomib (BZ), 100nM NPI-0052 (NPI), 5mM 3-methyladenine (3MA), 50μM chloroquine (CQ) or combinations for 24h. DNA fragmentation was measured by PI/FACS analysis and cell viability was measured by PI-exclusion cell viability/FACS analysis. *P<0.05, compared to single reagent treatment. B. LNCaP-Pro5 cells were treated as above and caspase-3/7 activities were measured using the Vybrant® FAM Caspase-3/7 Assay Kit and flow cytometry. Representative histograms are provided to show the activities of caspase-3/7. Note that peaks move to the left corresponding to lower MnIX values and weaker caspase-3/7 activities. Columns, mean of MnIX (n=3); bars, S.E. MnIX, mean fluorescence index. C. Effects of PIs on cell death after knocking down ATG5/7. Silencing efficiency was examined by one step-Quantitative RT-PCR for ATG5/7. RQ, relative quantity; PPIA, cyclophilin A.
To more directly examine the role of autophagy on cell death in response to proteasome inhibition, we compared the effects of PIs on cell death in cells transfected with siRNAs specific for ATG5/7 or an off-target control construct. In these experiments the silencing efficiency in untreated samples was approximately 90% (ATG5) and 80% (ATG7) as measured by quantitative RT-PCR. Consistent with the chemical inhibitor studies, knockdown of the ATGs increased PI-induced DNA fragmentation and necrosis (Figure 6C). Similarly, bortezomib and thapsigargin induced higher levels of cell death in the eIF2α phosphorylation deficient mutant MEFs than they did in the wild-type MEFs (data not shown and (Jiang and Wek, 2005)). Together, these data demonstrate that PIs-induced autophagy protects cells and combined inhibition of the proteasome and autophagy has greater cytotoxic effects than inhibition of either pathway alone.
Effects of combined inhibition of the proteasome and autophagy on the accumulation of intracellular protein aggregates
In a final series of experiments we assessed the effects of combined inhibition of the proteasome and autophagy on protein aggregate accumulation. We suspected that autophagy might function in a cytoprotective capacity to remove the protein aggregates that form when the proteasome degradation system is blocked. To test this hypothesis, we used immuno-fluorescent anti-ubiquitin staining and confocal microscopy to visualize intracellular protein aggregates in LNCaP-Pro5 cells exposed to PIs with or without chloroquine. Inclusion bodies were not visible in cells exposed to single agent bortezomib, NPI-0052, or chloroquine at any time point up to 24h (data not shown). However, exposure to combinations of bortezomib or NPI-0052 plus chloroquine led to clear inclusion body formation as early as 12h (Figure 7A). Chloroquine itself had no effects on ubiquitin accumulation which suggested that it could only alter the distribution pattern of ubiquitin and ubiquitinated protein (Figure 7B). These data strongly suggest that protein aggregates can be efficiently cleared by either system since visible protein aggregates did not appear when either pathway was blocked individually.
Discussion
The 26S proteasome and autophagy are considered the two major routes of protein degradation in eukaryotic cells and their mutual-exclusiveness and inter-dependence are of great interest (Kirkin et al., 2009). Here we report that proteasome inhibition activates autophagy via an eIF2α-dependent mechanism, which also causes global translational repression (Ron and Walter, 2007; Wek et al., 2006). Our results also show that phospho-eIF2α-dependent coupling of the proteasome inhibition to autophagy is important for limiting the accumulation of ubiquitin-containing protein aggregates that are reminiscent of the “inclusion bodies” found in neurodegenerative diseases (Komatsu et al., 2006; Levine and Kroemer, 2008; Mizushima et al., 2008; Rubinsztein, 2006). Importantly, these aggregates only became visible when both pathways were blocked, indicating that there must also be a signal that facilitates proteasome-mediated protein degradation when autophagy is blocked. Progressive accumulation of protein aggregates and defective organelles plays a critical role in neurodegenerative diseases by causing cellular toxicity and neuronal cell death (Hara et al., 2006; Komatsu et al., 2006; Rubinsztein, 2007). Therefore, the coupling between the proteasome and autophagy probably plays a central role in preventing proteotoxicity by maximizing efficient elimination of protein aggregates when either degradative system is overwhelmed.
We show that phospho-eIF2α-dependent coordination of protein degradation by the proteasome and autophagy is accompanied by increased transcription of ATGs (rather than mRNA stabilization). We excluded the involvement of ATF4 (Figure 5) and ATF5 (which contains two upstream open reading frame and phosphorylation of eIF2α could also selectively direct its translation in response to diverse stress conditions) (Zhou et al., 2008) (data not shown) in driving their expression and we are actively screening the potential transcription factors that are responsible for PIs-mediated ATG gene upregulation. We found that PIs and TG have broad effects on stress gene induction that are not restricted to ATG genes. PIs induced the accumulation of the mRNAs encoding the eIF2AKs, ATF5, ATF6, CHOP and NF-E2 related factor2 (Nrf2) in LNCaP-Pro5 cells (Supplemental table 1) and knocking down any of them did not block the PIs-/TG-induced ATG gene expression (data not shown). The global effects on gene induction suggest that complicated mechanisms are involved in PIs-mediated autophagy activation. Indeed, the current study suggests that phospho-eIF2α only partially account for the effects of PIs/TG on ATG gene induction in human prostate cancer cells. Recent studies also reported that the IRE1α-JNK pathway [Supplemental figure 1 and (Ding et al., 2007; Ogata et al., 2006)] and the Ca2+-PKCθ pathway (Sakaki et al., 2008) are required for autophagy activation. Precisely how the general stress response influences the outcome of PIs-based therapy will probably require a more global approach (e.g, RNAi library screening with high silencing efficiency in several different cell lines), but the idea of developing agents that selectively target this stress response to improve therapeutic efficacy in cancer or inhibit the progression of neurodegeneration is very attractive.
We found that the PIs and the inhibitors of autophagy studied here induced cell death via both apoptosis and necrosis, and necrosis predominated in cells exposed to both simultaneously (Figure 6). Given the complexities of their mechanisms of action and likely effects on cell homeostasis, the observation that combined inhibition of the proteasome and autophagy induces a complex mode of cell death is not unexpected. There is currently strong interest in evaluating the effects of triggering necrosis (rather than apoptosis) in cancer therapy because tumors accumulate molecular abnormalities that interrupt apoptosis-associated signaling pathways (Nelson and White, 2004). One potential concern with this approach is that apoptotic cells are cleared via well-organized mechanisms that do not trigger a secondary immune response, whereas necrotic cells are pro-inflammatory (Krysko et al., 2008; Nelson and White, 2004). Whether secondary inflammation promotes immune recognition of tumors to enhance therapeutic activity or causes excessive secondary toxicity will have to be determined in clinical trials.
Our findings have additional important implications for clinical translation. As discussed above, PIs do not have impressive single-agent activity in solid tumors, so there is interest in exploiting their unique mechanisms of action in the design of rational combinations. Chloroquine is already FDA-approved as an anti-malarial drug. Clinical trials evaluating its anti-cancer activity suggest that it may provide some benefit to patients with glioblastoma multiforme (Reyes et al., 2001; Savarino et al., 2006). On the other hand, laboratory studies have demonstrated that HDAC6 is required for the coupling between the proteasome inhibition and autophagy (Iwata et al., 2005; Pandey et al., 2007), and chemical HDAC inhibitors that block HDAC6 (including SAHA) are also being evaluated in clinical trials. Our results suggest that these combinations will be most active in cells that possess an intact PERK-eIF2α arm of the UPR, and we are testing this hypothesis in a Phase II clinical trial employing bortezomib and SAHA in patients. One potential concern is that proteotoxicity may be involved in drug efficacy in cancer cells and toxicity to normal tissues. One of bortezomib's toxic side effects is peripheral neuropathy, which could in theory be caused by inclusion body formation in peripheral neurons, and this toxicity could be exacerbated by combination therapy with inhibitors of autophagy. This possibility will be especially important to consider when employing PIs that cross the blood-brain barrier, as they may be capable of aggravating any neurodegenerative disorder that is present at baseline.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Department of Defense Prostate Cancer Research Program (PC050288). The TEM studies were supported by the MD Anderson Cancer Center Support Grant (CA16672) to the High Resolution Electron Microscopy Facility. The authors would also like to acknowledge Dr. Woonyoung Choi and Maosheng Huang's help with the array data analyses.
Footnotes
Supplementary information is available at Oncogene's website.
References
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–21. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KC. Bortezomib therapy for myeloma. Curr Hematol Rep. 2004;3:65. [PubMed] [Google Scholar]
- Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8:407–19. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111:1654–64. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, et al. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res. 2008;68:9323–30. doi: 10.1158/0008-5472.CAN-08-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J, Baba M, Nair U, Klionsky DJ. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J Cell Biol. 2008;182:129–40. doi: 10.1083/jcb.200711112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–7. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol. 2005;26:1401–10. [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280:14189–202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–69. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–9. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, Parthoens E, D'Herde K, Vandenabeele P. Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol. 2008;442:307–41. doi: 10.1016/S0076-6879(08)01416-X. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11:164–79. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Miller CP, Ban K, Dujka ME, McConkey DJ, Munsell M, Palladino M, et al. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood. 2007;110:267–77. doi: 10.1182/blood-2006-03-013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–9. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K, Jr, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–81. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- Nelson DA, White E. Exploiting different ways to die. Genes Dev. 2004;18:1223–6. doi: 10.1101/gad.1212404. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Reyes S, Rembao D, Sotelo J. The antimalarials quinacrine and chloroquine potentiate the transplacental carcinogenic effect of ethylnitrosourea on ependymal cells. Brain Tumor Pathol. 2001;18:83–7. doi: 10.1007/BF02479420. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron. 2007;54:854–6. doi: 10.1016/j.neuron.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem. 2008;283:15370–80. doi: 10.1074/jbc.M710209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino A, Lucia MB, Giordano F, Cauda R. Risks and benefits of chloroquine use in anticancer strategies. Lancet Oncol. 2006;7:792–3. doi: 10.1016/S1470-2045(06)70875-0. [DOI] [PubMed] [Google Scholar]
- Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–91. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008;283:7064–73. doi: 10.1074/jbc.M708530200. [DOI] [PubMed] [Google Scholar]
- Zhu K, Chan W, Heymach J, Wilkinson M, McConkey DJ. Control of HIF-1alpha expression by eIF2 alpha phosphorylation-mediated translational repression. Cancer Res. 2009;69:1836–43. doi: 10.1158/0008-5472.CAN-08-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.