

Abstract
The preferred ligand of AT2R-mediated natriuresis is Ang III. The major enzyme responsible for the metabolism of Ang III is aminopeptidase N (APN), which is selectively inhibited by compound PC-18. In this study, urine sodium excretion rates (UNaV), fractional excretion of sodium (FENa) and lithium (FELi), GFR, and mean arterial pressures (MAP) were studied in pre-hypertensive and hypertensive SHR, and compared to age-matched WKY. While renal interstitial (RI) infusion of AT1R blocker candesartan increased UNaV in WKY from a baseline of 0.05±0.01 to 0.17±0.04 μmol/min (P<0.01), identical infusions failed to increase UNaV in hypertensive SHR. Co-infusion of AT2R antagonist PD-123319 abolished the natriuretic responses to candesartan in WKY, indicating an AT2R-mediated effect. AT2R-mediated natriuresis was enabled in hypertensive SHR by inhibiting the metabolism of Ang III with PC-18 [0.05±0.01 to 0.11±0.03 μmol/min (P<0.05)]. The defects in sodium excretion were present prior to the onset of hypertension in SHR, since young WKY demonstrated double the UNaV of SHR (0.04±0.006 vs. 0.02±0.003 μmol/min, P<0.01) at baseline. The increased UNaV of young WKY was due to reduced renal proximal tubule sodium reabsorption, since increases in FENa were paralleled by increases in FELi. RI PC-18 infusion ameliorated defective AT2R-mediated natriuresis in young SHR by increasing FENa and FELi, without changing GFR. Thus, increased renal proximal tubule sodium retention is observed prior to the onset of hypertension in SHR, and inhibition of the metabolism of Ang III ameliorates this pathophysiologic defect in sodium excretion.
Keywords: Natriuresis, angiotensin receptors, hypertension, angiotensin III, aminopeptidase N, SHR
INTRODUCTION
Spontaneously hypertensive rats (SHR) develop hypertension at approximately 6 weeks of age, and are widely used as a model to study the development and maintenance of human genetic hypertension1. One of the proposed mechanisms of the initiation of hypertension in SHR involves a primary defect in renal sodium (Na+) excretion2-8. Over time, this defect necessitates an increase in renal perfusion pressure, an adaptation which becomes central to the development and maintenance of hypertension9, 10.
In normal rodents, both the intrarenal renin-angiotensin system and the renal dopaminergic system play important roles in renal proximal tubule Na+ handling. Basal Na+ excretion rates are generally determined by the activity of the intrarenal renin-angiotensin system, while the dopaminergic system regulates Na+ excretion in response to high salt intake.
While an acute sodium load or rise in blood pressure increase sodium excretion in normal rodents, the following three significant pharmacological manipulations also induce natriuresis: (1) blockade of intrarenal angiotensin type-1 receptors (AT1Rs), (2) stimulation of renal dopamine D1-like receptors and (3) activation of intrarenal angiotensin type-2 receptors (AT2Rs). Recent studies have shown that natriuresis resulting from both AT1R blockade and D1-like receptor stimulation are mediated, at least in part, by AT2R activation, since concomitant blockade of renal AT2Rs in these situations abolishes the natriuresis11, 12. Regarding direct renal AT2R-induced natriuresis, renal interstitial (RI) angiotensin III (Ang III), but not angiotensin II (Ang II), results in increased Na+ excretion when AT1Rs are blocked systemically11. This effect is also abolished by concomitant infusion of a selective AT2R antagonist, highlighting the important direct role of renal AT2R activation by Ang III in natriuresis11.
In the kidney, aminopeptidase A (APA), an enzyme normally expressed on the brush border of renal proximal tubule cells, is responsible for converting Ang II to Ang III. Ang III is subsequently degraded to angiotensin IV (Ang IV) by aminopeptidase N (APN). Inhibition of intrarenal APN results in augmented natriuretic responses to Ang III in the presence of systemic AT1R blockade13 and natriuresis engendered by inhibition of APN is abolished by concomitant inhibition of APA14. Taken together, these data suggest that renal AT2Rs mediate natriuresis engendered by D1-like receptor activation and AT1R blockade, and that Ang III, and not Ang II, is the preferred agonist of this response.
Thus far, studies regarding the etiology of increased Na+ reabsorption in young SHR have focused on alterations in renal dopaminergic and AT1R-mediated effects15-18. However, as mentioned previously, D1-like receptor mediated natriuresis and natriuresis due to AT1R blockade are dependent, at least in part, on renal AT2Rs11, 12. Thus, we hypothesize that rapid metabolism of Ang III, the preferred ligand of AT2R-mediated natriuresis, leads to abnormal sodium reabsorption previously demonstrated to occur in association with the development of hypertension in the SHR. The results indicate that renal AT1R blockade fails to induce natriuresis in hypertensive SHR unless the degradation of Ang III is inhibited, and that this defect is present prior to, rather than as a consequence of, established hypertension. Amelioration of AT2R-mediated natriuresis in pre-hypertensive SHR is achieved through inhibition of renal APN activity, and this effect is mediated by AT2Rs of the renal proximal tubule.
METHODS
Animal Preparation
The experiments, which were approved by the University of Virginia Animal Care and Use Committee, were conducted in 4- and12-week-old female WKY (Harlan) and SHR (Taconic), in accordance with the NIH Guide for Care and Use of Laboratory Animals. Rats were placed under general anesthesia with pentobarbital (50 mg/mL) given 5 mg/100 g of body weight intraperitoneally. A tracheostomy was performed, and arterial access was achieved by direct cannulation of the right carotid artery. Intravenous access was obtained via cannulation of the right internal jugular vein. Renal cortical interstitial infusion catheters were placed, as reported previously11-14. When more than one substance was simultaneously infused into the kidney, separate interstitial catheters were employed.
Rats were housed under controlled conditions (temperature: 21±1°C; humidity: 60±10%; and light: 8 to 20 h). Experiments were initiated at the same time each day to prevent any effect of diurnal variation in blood pressure. Mean arterial pressure (MAP) was measured by the direct intracarotid method with the use of a blodd pressure analyzer (Micromed Inc). MAP was recorded every 5 min and averaged for each of the control and experimental periods.
Pharmacological Agents
Candesartan, a specific, potent insurmountable inhibitor of AT1R (IC50 >1×10−5 mol/L and 2.9×10−8 mol/L for AT2Rs and AT1Rs, respectively), was used for AT1R blockade. PD-123319 (PD, Parke-Davis), a specific AT2R antagonist (IC50 2×10−8 mol/L and >1×10−4 mol/L for AT2Rs and AT1Rs, respectively), was used to block AT2Rs. A specific APN inhibitor, PC-18 (2-amino-4-methylsulfonyl-butane-thiol, Ki=8.0±1.7 nM)19, 20, was provided by Drs. Fournie-Zaluski and Roques, and infused interstitially to block the metabolism of Ang III to Ang IV. PC-18 has been shown to increase the half-life of endogenous Ang III in vivo19.
Measurement of GFR, FENa, and FELi
Urinary and plasma Na+ and Li+ concentrations were measured using a flame photometer (Instrumentation Laboratory-943). Glomerular filtration rate (GFR) was measured by inulin clearance utilizing a previously described method21. Tubular Na+ reabsorption was determined by calculating FENa and renal proximal tubule Na+ reabsorption was estimated using FELi as previously published22.
Effects of RI AT1R Blockade, RI AT1R Blockade + APN Inhibition, and RI AT1R Blockade + APN Inhibition + AT2R Blockade on UNaV, GFR, FENa, FELi, and MAP
4- and 12-week-old WKY and SHR were studied on normal Na+ intake (N=6 per group) 72 h following uninephrectomy. The remaining kidney was then infused for one hour with 5% dextrose in water (D5W), designated as the ‘Control Period’ in the Figures. Following the ‘Control Period’, the kidney was infused with one of the following: 1) D5W at 2.5 μL/ min, 2) candesartan (0.01 mg/kg/min), 3) candesartan + PD (10 μg/kg/min), 4) candesartan + PC-18 (25 μg/min), or 5) candesartan + PC-18 + PD directly into the RI space during three consecutive 1 h ‘Experimental Periods’. Inulin and lithium chloride in D5W were infused throughout the study via internal jugular catheter. UNaV, GFR, FENa, FELi and MAPs were calculated and/or recorded for each period.
Statistical Analysis
Comparisons among vehicle, AT1R blocker (candesartan), AT2R blocker (PD), and PC-18 (APN inhibitor) were estimated by ANOVA, including a repeated-measures term, by using the general linear models procedure of the Statistical Analysis System. Multiple comparisons of individual pairs of effect means were conducted by the use of least-square means pooled variance. Data are expressed as mean±1 SE. Statistical significance was identified at P<0.05.
RESULTS
Effects of RI Candesartan Infusion and Candesartan + PD Infusion on UNaV and MAP in 12-week-old WKY and SHR
As demonstrated in Figure 1A, in WKY, RI candesartan increased UNaV from a baseline of 0.05±0.01 to 0.16±0.03 μmol/min (P<0.0001) during experimental period 2 and to 0.17±0.04 μmol/min (P<0.01) during experimental period 3. PD co-infusion abolished the natriuretic responses to RI candesartan in WKY. In 12-week-old SHR, however, identical infusions of candesartan failed to increase UNaV (baseline: 0.04±0.01 to 0.03±0.01 μmol/min after 3 h of candesartan infusion; P=NS). As illustrated in Figure 1B, SHR had higher MAP values compared to WKY at baseline, but RI candesartan infusion did not significantly alter baseline MAP values in WKY or SHR. Similarly, co-infusion of PD with candesartan did not influence MAP in WKY.
FIGURE 1.
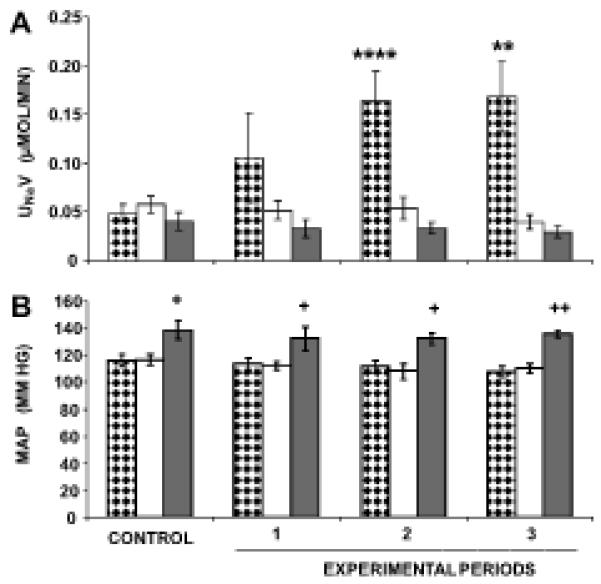
Direct renal interstitial (RI) infusion of candesartan, an AT1R antagonist, induces natriuresis that is blocked by the co-infusion of PD, a selective AT2R antagonist, in 12-week-old WKY, but not SHR. Mean arterial pressure (MAP) responses are significantly higher in SHR than WKY and remain unchanged in response to any of the experimental infusions. Panel A. During the “Control”, only 5% dextrose in water (D5W) is infused in each animal. During the “Experimental Periods”,  , (N=7) indicates urine sodium excretion rate (UNaV) in WKY in response to RI candesartan infusion,
, (N=7) indicates urine sodium excretion rate (UNaV) in WKY in response to RI candesartan infusion,  , (N=6) indicates UNaV in WKY in response to RI infusion of candesartan + PD, and
, (N=6) indicates UNaV in WKY in response to RI infusion of candesartan + PD, and  , (N=6) indicates UNaV in SHR in response to RI infusion of candesartan. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; **P<0.01 and ****P<0.0001 from respective control period, and +P<0.05 and ++P<0.01 between WKY and SHR.
, (N=6) indicates UNaV in SHR in response to RI infusion of candesartan. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; **P<0.01 and ****P<0.0001 from respective control period, and +P<0.05 and ++P<0.01 between WKY and SHR.
Effects of RI Candesartan ± APN Inhibition on UNaV and MAP in 12-Week-Old Hypertensive SHR
Figure 2A demonstrates that candesartan + PC-18 infusion increased UNaV from a baseline value of 0.05±0.01 to 0.10±0.02 μmol/min (P<0.05) during experimental period 1, to 0.12±0.02 μmol/min (P<0.01) during experimental period 2, and to 0.11±0.03 μmol/min (P<0.05) during experimental period 3. The addition of PD, an AT2R antagonist, abolished PC-18-engendered natriuresis in hypertensive SHR. Neither D5W- nor candesartan-infused kidneys demonstrated a significant change in UNaV across the duration of the experiment in these animals. MAP values remain unchanged in response to any of the pharmacological infusions in 12-week old SHR (Figure 2B).
FIGURE 2.
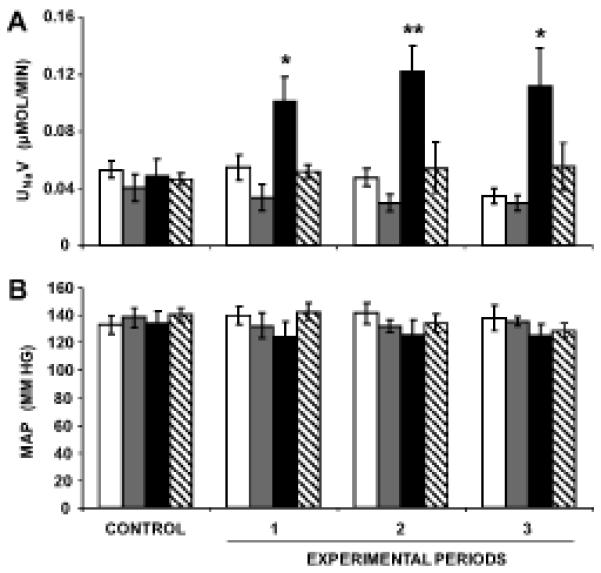
Direct renal interstitial (RI) infusion of candesartan, an AT1R antagonist, with and without PC-18, an inhibitor of aminopeptidase N (APN), induces natriuresis that is blocked by infusion of PD, an AT2R-antagonist, in 12-week-old SHR. Panel A.  , (N=6) indicates urine sodium (Na+) excretion rate (UNaV) in response to RI D5W infusion for the entire duration of the experiment.
, (N=6) indicates urine sodium (Na+) excretion rate (UNaV) in response to RI D5W infusion for the entire duration of the experiment.  , (N=6) indicates UNaV in response to RI infusion of candesartan, following a 1 h control period, when only D5W was infused.
, (N=6) indicates UNaV in response to RI infusion of candesartan, following a 1 h control period, when only D5W was infused.  , (N=6) indicates UNaV in response to RI co-infusion of candesartan + PC-18, following a 1 h control period, when only D5W was infused.
, (N=6) indicates UNaV in response to RI co-infusion of candesartan + PC-18, following a 1 h control period, when only D5W was infused.  , (N=7) indicates UNaV in response to RI co-infusion of candesartan + PC-18 + PD, following a 1 h control period, when only D5W was infused. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; *P<0.05 and **P<0.01 from respective control period.
, (N=7) indicates UNaV in response to RI co-infusion of candesartan + PC-18 + PD, following a 1 h control period, when only D5W was infused. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; *P<0.05 and **P<0.01 from respective control period.
Baseline Renal Function Studies on 4-Week-Old WKY and Pre-Hypertensive SHR
Figure 3A demonstrates reduced UNaV in 4-week-old pre-hypertensive SHR compared to WKY following 4 h of vehicle infusion with RI D5W (0.02±0.003 vs. 0.04±0.006 μmol/min, respectively). MAP values are not significantly different between 4-week-old WKY and SHR, with average values over 4 h of 120.3±4 and 120.0±4 mmHg, respectively (Figure 3B). Figure 4A demonstrates that 4- week-old WKY and SHR have similar GFRs following 4 h of vehicle infusion with RI D5W (0.51±0.04 and 0.45±0.08 mL/min/g of kidney weight, respectively). However, compared to 4-week-old SHR, age-matched WKY demonstrate significantly higher FENa (0.16±0.02 vs. 0.09±0.01%, P<0.05, Figure 4B) and FELi (11.7±0.9 vs. 7.7±0.9%, P<0.01, Figure 4C).
FIGURE 3.
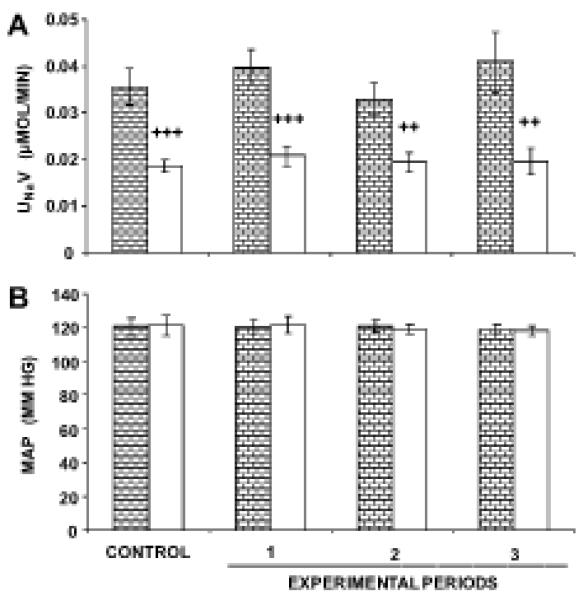
Baseline renal function studies on 4-week-old WKY and SHR. Panel A.  , (N=9) indicates urine sodium excretion rate (UNaV) in WKY in response to RI infusion of vehicle D5W.
, (N=9) indicates urine sodium excretion rate (UNaV) in WKY in response to RI infusion of vehicle D5W.  , (N=8) indicates UNaV in SHR in response to RI infusion of vehicle D5W. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; ++P<0.01 and +++P<0.001 from respective WKY period.
, (N=8) indicates UNaV in SHR in response to RI infusion of vehicle D5W. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; ++P<0.01 and +++P<0.001 from respective WKY period.
FIGURE 4.
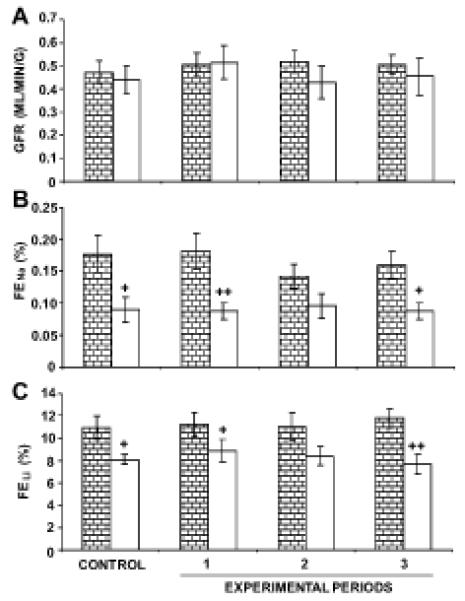
Baseline renal function studies on 4-week old WKY and SHR. Panel A.  , (N=9) indicates glomerular filtration rate (GFR) in WKY in response to RI infusion of vehicle D5W.
, (N=9) indicates glomerular filtration rate (GFR) in WKY in response to RI infusion of vehicle D5W.  , (N=8) indicates GFR in SHR in response to RI infusion of vehicle D5W. Panel B. Fractional excretion of sodium (FENa) responses to conditions in Panel A. Panel C. Fractional excretion of lithium (FELi) responses to conditions in Panel A. Data represent mean ± 1 SE; +P<0.05 and ++P<0.01 from WKY.
, (N=8) indicates GFR in SHR in response to RI infusion of vehicle D5W. Panel B. Fractional excretion of sodium (FENa) responses to conditions in Panel A. Panel C. Fractional excretion of lithium (FELi) responses to conditions in Panel A. Data represent mean ± 1 SE; +P<0.05 and ++P<0.01 from WKY.
Effects of RI Candesartan ± APN Inhibition on UNaV and MAP in 4-Week-Old WKY and Pre-Hypertensive SHR
Following RI candesartan infusion, 4-week-old WKY demonstrated an increase in UNaV from a baseline value of 0.05±0.01 to 0.15±0.02 μmol/min (P<0.05) following 3 h of candesartan infusion (Figure 5A). The increase in UNaV was abolished by co-infusion of PD. In 4-week-old SHR, RI candesartan infusion failed to increase UNaV. However, as demonstrated in Figure 5A, RI infusion of PC-18, an inhibitor of APN, enabled natriuretic responses to RI candesartan in 4-week-old SHR by increasing UNaV from a baseline value of 0.02±0.002 to 0.10±0.02 μmol/min (P<0.01). RI AT2R blockade with PD abolished PC-18 enabled natriuresis in 4-week-old SHR. MAP values remained unchanged in 4-week-old WKY or SHR in response to RI candesartan ± PC-18 ± PD (Figure 5B).
FIGURE 5.
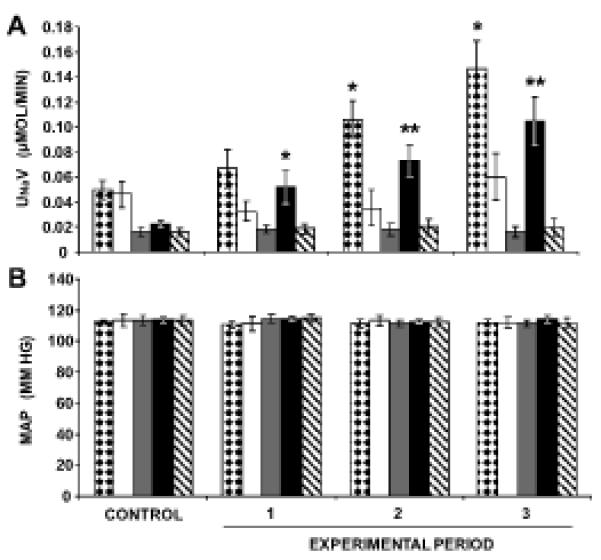
Direct renal interstitial (RI) infusion of candesartan, an AT1R antagonist induces natriuresis in 4-week-old WKY, but not SHR. The natriuresis is blocked by PD, an AT2R antagonist. RI co-infusion of candesartan + PC-18, an inhibitor of aminopeptidase N (APN), engenders natriuresis in 4-week-old SHR, and this is also blocked by PD. Panel A.  , (N=6) indicates urine sodium excretion rate (UNaV) in WKY in response to RI infusion of candesartan.
, (N=6) indicates urine sodium excretion rate (UNaV) in WKY in response to RI infusion of candesartan.  , (N=8) indicates UNaV in WKY in response to RI co-infusion of candesartan + PD.
, (N=8) indicates UNaV in WKY in response to RI co-infusion of candesartan + PD.  , (N=7) indicates UNaV in SHR in response to RI infusion of candesartan.
, (N=7) indicates UNaV in SHR in response to RI infusion of candesartan.  , (N=8) indicates UNaV in SHR in response to RI co-infusion of candesartan + PC-18.
, (N=8) indicates UNaV in SHR in response to RI co-infusion of candesartan + PC-18.  , (N=8) indicates UNaV in SHR in response to RI co-infusion of candesartan + PC-18 + PD. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; *P<0.05 and **P<0.01 from respective control period.
, (N=8) indicates UNaV in SHR in response to RI co-infusion of candesartan + PC-18 + PD. Panel B. Mean arterial pressure (MAP) responses to the conditions in Panel A. Data represent mean ± 1 SE; *P<0.05 and **P<0.01 from respective control period.
Renal Function Studies in 4-Week-Old WKY and Pre-Hypertensive SHR In Response To Natriuretic Stimuli
In 4-week old WKY, RI candesartan increased FENa (Figure 6B) and FELi (Figure 6C) from baseline values of 0.16±0.02% and 12.4±1.1%, to 0.31±0.03% (P<0.01) and 26.0±2.4% (P<0.001), respectively. RI AT1R blockade failed to induce changes in GFR (Figure 6A) in these animals. In 4-week-old SHR, RI candesartan infusion alone did not alter FENa, FELi, or GFR significantly. However, the addition of PC-18 to RI candesartan infusion increased the FENa (Figure 6B) and the FELi (Figure 6C) from 0.09±0.02% and 8.6±0.7% to 0.28±0.04% (P<0.001) and 29.3±4.2% (P<0.0001) after 3 h. GFR remained unaffected following the addition of PC-18 to candesartan in 4-week-old SHR (Figure 6A).
FIGURE 6.
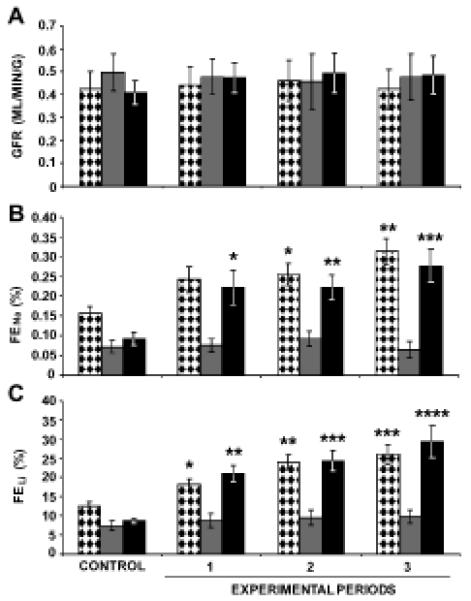
Renal function studies on 4-week-old WKY and SHR following the RI infusion of candesartan with and without PC-18. Panel A.  , (N=11) indicates glomerular filtration rate (GFR) in WKY in response to RI infusion of candesartan.
, (N=11) indicates glomerular filtration rate (GFR) in WKY in response to RI infusion of candesartan.  , (N=10) indicates GFR in SHR in response to RI infusion of candesartan.
, (N=10) indicates GFR in SHR in response to RI infusion of candesartan.  , (N=10) indicates GFR in SHR in response to RI co-infusion of candesartan + PC-18, an inhibitor of APN. Panel B. Fractional excretion of sodium (FENa) responses to conditions in Panel A. Panel C. Fractional excretion of lithium (FELi) responses to conditions in Panel A. Data represent mean ± 1 SE; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 from respective control period.
, (N=10) indicates GFR in SHR in response to RI co-infusion of candesartan + PC-18, an inhibitor of APN. Panel B. Fractional excretion of sodium (FENa) responses to conditions in Panel A. Panel C. Fractional excretion of lithium (FELi) responses to conditions in Panel A. Data represent mean ± 1 SE; *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 from respective control period.
DISCUSSION
One of the proposed mechanisms of the initiation of hypertension in SHR and humans involves a fundamental defect in the kidney's capacity to excrete Na+. Over time, a compensatory increase in renal perfusion pressure permits proper Na+ excretion, but also renders the animal hypertensive. Supporting this theory are the observations that transplantation of pre-hypertensive kidneys from SHR to WKY produces hypertension in WKY23, and that human subjects with genetic hypertension24 and SHR25, 26 excrete less Na+ and water than normotensive controls when renal perfusion pressure is lowered to normotensive levels. Chronic relationships between arterial pressure and urinary Na+ and water output are also shifted toward higher pressures in SHR compared to WKY, reflecting the kidneys adaptation to a higher perfusion pressure27.
In the present study, we hypothesized that AT2R-mediated natriuresis is dysfunctional in SHR due to rapid inactivation of the preferred ligand, Ang III. The major results provide insight into both the site and mechanisms of defective natriuresis in SHR, and are summarized as follows: (1) while selective intrarenal AT1R blockade induces significant AT2R-mediated natriuresis in 12-week-old WKY, identical infusions fail to do so in age-matched hypertensive SHR; (2) defective natriuresis is present in 4-week-old SHR prior to the onset of hypertension, and this occurs at the level of the renal proximal tubule; (3) inhibition of the activity of APN, the enzyme responsible for the degradation of Ang III, permits AT2R-mediated natriuresis in both 4- and 12-week old SHR; and (4) in 4-week old SHR, the natriuresis engendered by PC-18 occurs at the level of the renal proximal tubule.
Previous studies have shown that RI AT1R blockade with candesartan induces natriuresis that is mediated by renal AT2Rs in 12-week old Sprague-Dawley rats11. These results are not specific for the Sprague-Dawley strain, since the present study demonstrates similar AT2R-mediated natriuresis in response to RI candesartan infusion in WKY. The absence of MAP changes during RI AT1R blockade in WKY indicates that the observed natriuresis is due to direct intrarenal, and not systemic hemodynamic, factors. Low-dose candesartan has previously been reported to increase UNaV without affecting MAP values in WKY when administered systemically28, and the results of this study demonstrate that direct RI candesartan infusion at low doses, has the same effect.
In comparison, 12-week-old SHR fail to demonstrate an increase in UNaV following RI candesartan infusion. To investigate whether the lack of response was a consequence of established hypertension, both basal and stimulated natriuretic responses were assessed in young, 4-week-old pre-hypertensive SHR. Baseline UNaV was significantly reduced in young SHR compared to age-matched WKY, a finding which has been reported previously6, 26. However, in the present study, a defect in stimulated natriuresis, i.e., in response to AT1R blockade, was also observed in young SHR. Thus, not only is baseline Na+ excretion impaired before hypertension is established, but beneficial natriuretic responses mediated by renal AT2Rs are also compromised before hypertension develops in these animals.
The preferred ligand of AT2R-mediated natriuresis in normal rodents is Ang III, not Ang II11, 14. In the systemic circulation, Ang III is metabolized 2 to 4 times more rapidly than Ang II29, 30. APN is the major enzyme responsible for the metabolism of Ang III in the kidney31, and is expressed on brush border (apical) membranes of renal proximal tubule cells and enterocytes31. One of the first in vivo studies using PC-18 to inhibit the activity of APN was conducted in mice19, during which intracerebroventricular administration of PC-18 resulted in a 3.9-fold increase in the half-life of Ang III compared with control. The in vitro specificity of PC-18 toward APN, APA, and aminopeptidase B (APB), three zinc metalloproteases with significant identity between their amino acid sequences, was also tested19. The Ki values of this compound for APN were in the nanomolar range (Ki=8.0±1.7 nM), but PC-18 was 2150 and 125 times less active on APA and APB, respectively19. Thus, the infusion of PC-18 into the RI compartment in the present study allowed for examination of the effects mediated by Ang III within the intrarenal renin-angiotensin system.
In the present study, RI PC-18 infusion enabled natriuretic responses to AT1R blockade in both 4- and 12-week-old SHR. Thus, the decreased availability of intrarenal Ang III, whether due to increased degradation by APN, or decreased formation by APA, appears to be an important determinant of acute sodium excretion in SHR. Previous reports have suggested that SHR have increased renal proximal tubule cell APN protein expression compared to WKY, even though APN mRNA levels are similar between the two strains32. The increased expression of the major enzyme responsible for Ang III degradation may contribute to the lack of available Ang III for effective AT2R-mediated natriuresis in SHR, especially since natriuresis is ameliorated by APN inhibition. Furthermore, an inhibitory effect on APN during chronic AT1R blockade may provide an additional mechanism by which AT1R blockers improve hypertension in this strain. Chronic valsartan treatment has been reported to reduce renal APN activity in renovascular hypertension33, and chronic ARB administration may influence APN activity in SHR as well.
The nephron site at which AT2R-mediated natriuresis is stimulated in 4- and 12-week-old WKY and is defective in SHR, is the renal proximal tubule. Previous studies have validated the use of the FELi as a marker of renal proximal tubule Na+ transport in both WKY5 and SHR5, 34. In all of our studies, tubule events distal to the renal proximal tubule would have been detected by changes in FENa that were not accompanied by parallel changes in FELi. However, this was not the case in the baseline sodium excretion rates in young SHR, or the stimulated natriuresis in WKY or SHR.
Thus far, studies regarding the mechanisms of increased renal proximal tubule Na+ reabsorption in young SHR have focused on alterations in renal dopaminergic and AT1R-mediated effects. In the renal proximal tubule, increased activities of apical membrane sodium-hydrogen exchanger-3 (NHE-3) and basolateral membrane sodium-potassium ATPase (NKA) are associated with increased Na+ reabsorption. In young SHR, the ability of the dopamine D1-like receptor to inhibit NHE-3 or NKA is impaired due to an uncoupling of the D1-like receptor from its G-protein/effector complex 15-17. Furthermore, increased renal proximal tubule AT1R expression 18, elevated renal Ang II content 35, 36, and increased Ang II-AT1R mediated activation of NHE-3 37-39 have also been suggested as possible contributors to the excess Na+ retention of young SHR. However, as mentioned previously, D1-like receptor mediated natriuresis and natriuresis due to AT1R blockade are mediated, at least in part, by renal AT2Rs11, 12. Thus, the direct characterization of the natriuretic role of renal proximal tubule AT2Rs in this study, both in normal rodents and SHR, where excess sodium reabsorption actually contributes to the pathogenesis of the disease, permits a deeper understanding of the mechanisms underlying the initiation of hypertension in this model. The provision of APN as a potential therapeutic target for the amelioration of hypertension in SHR will be addressed in future studies.
PERSPECTIVES
In both SHR and hypertensive humans, increased Na+ reabsorption contributes to the eventual onset of genetic hypertension. To date, the only published studies examining the increased Na+ reabsorption of young pre-hypertensive SHR have focused on two defects: (1) elevated renal Ang II content causing increased Na+ retention via the AT1R and (2) functional hyposensitivity of renal proximal tubule cells to dopamine resulting in decreased Na+ excretion. The recently elucidated role of the renal AT2R and Ang III in the natriuretic responses of non-hypertensive rodents has become important to our understanding of the mechanisms which permit Na+ excretion in normal animals. The present study investigated the role of AT2Rs in natriuresis in young SHR, and identified a potentially compelling therapeutic target to overcome early defects in renal proximal tubule Na+ excretion in the initiation of hypertension.
Acknowledgments
Sources of Funding: This work was supported by National Institutes of Health grants K08-HL-093353 to SHP and R01-HL-081891 and R01-HL-087998 to RMC.
Footnotes
Disclosures: None
REFERENCES
- 1.Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension. Man and rat. Circ Res. 1981;48:309–319. doi: 10.1161/01.res.48.3.309. [DOI] [PubMed] [Google Scholar]
- 2.Christiansen RE, Roald AB, Tenstad O, Iversen BM. Renal hemodynamics during development of hypertension in young spontaneously hypertensive rats. Kidney Blood Press Res. 2002;25:322–328. doi: 10.1159/000066792. [DOI] [PubMed] [Google Scholar]
- 3.Nagaoka A, Kakihana M, Shibota M, Fujiwara K, Shimakawa K. Reduced sodium excretory ability in young spontaneously hypertensive rats. Jpn J Pharmacol. 1982;32:839–844. doi: 10.1254/jjp.32.839. [DOI] [PubMed] [Google Scholar]
- 4.Heckmann U, Zidek W, Schurek HJ. Sodium reabsorption in the isolated perfused kidney of normotensive and spontaneously hypertensive rats. J Hypertens Suppl. 1989;7:S172–173. doi: 10.1097/00004872-198900076-00082. [DOI] [PubMed] [Google Scholar]
- 5.Biollaz J, Waeber B, Diezi J, Burnier M, Brunner HR. Lithium infusion to study sodium handling in unanesthetized hypertensive rats. Hypertension. 1986;8:117–121. doi: 10.1161/01.hyp.8.2.117. [DOI] [PubMed] [Google Scholar]
- 6.Beierwaltes WH, Arendshorst WJ, Klemmer PJ. Electrolyte and water balance in young spontaneously hypertensive rats. Hypertension. 1982;4:908–915. doi: 10.1161/01.hyp.4.6.908. [DOI] [PubMed] [Google Scholar]
- 7.Firth JD, Raine AE, Ledingham JG. Sodium and lithium handling in the isolated hypertensive rat kidney. Clin Sci (Lond) 1989;76:335–341. doi: 10.1042/cs0760335. [DOI] [PubMed] [Google Scholar]
- 8.Mozaffari MS, Jirakulsomchok S, Shao ZH, Wyss JM. High-NaCl diets increase natriuretic and diuretic responses in salt-resistant but not salt-sensitive SHR. Am J Physiol. 1991;260:F890–897. doi: 10.1152/ajprenal.1991.260.6.F890. [DOI] [PubMed] [Google Scholar]
- 9.Guyton AC, Coleman TG, Young DB, Lohmeier TE, DeClue JW. Salt balance and long-term blood pressure control. Annu Rev Med. 1980;31:15–27. doi: 10.1146/annurev.me.31.020180.000311. [DOI] [PubMed] [Google Scholar]
- 10.Guyton AC, Coleman TG, Cowley AV, Jr., Scheel KW, Manning RD, Jr., Norman RA., Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1972;52:584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 11.Padia SH, Howell NL, Siragy HM, Carey RM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension. 2006;47:537–544. doi: 10.1161/01.HYP.0000196950.48596.21. [DOI] [PubMed] [Google Scholar]
- 12.Salomone LJ, Howell NL, McGrath HE, Kemp BA, Keller SR, Gildea JJ, Felder RA, Carey RM. Intrarenal dopamine D1-like receptor stimulation induces natriuresis via an angiotensin type-2 receptor mechanism. Hypertension. 2007;49:155–161. doi: 10.1161/01.HYP.0000251881.89610.ee. [DOI] [PubMed] [Google Scholar]
- 13.Padia SH, Kemp BA, Howell NL, Siragy HM, Fournie-Zaluski MC, Roques BP, Carey RM. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension. 2007;49:625–630. doi: 10.1161/01.HYP.0000254833.85106.4d. [DOI] [PubMed] [Google Scholar]
- 14.Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension. 2008;51:460–465. doi: 10.1161/HYPERTENSIONAHA.107.103242. [DOI] [PubMed] [Google Scholar]
- 15.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, Asico LD, Wang W, Zheng S, Yamaguchi I, Williams SM, Gainer J, Brown NJ, Hazen-Martin D, Wong LJ, Robillard JE, Carey RM, Eisner GM, Jose PA. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A. 2002;99:3872–3877. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, Li XX, Albrecht FE, Hopfer U, Carey RM, Jose PA. Dopamine(1) receptor, G(salpha), and Na(+)-H(+) exchanger interactions in the kidney in hypertension. Hypertension. 2000;36:395–399. doi: 10.1161/01.hyp.36.3.395. [DOI] [PubMed] [Google Scholar]
- 17.Sanada H, Jose PA, Hazen-Martin D, Yu PY, Xu J, Bruns DE, Phipps J, Carey RM, Felder RA. Dopamine-1 receptor coupling defect in renal proximal tubule cells in hypertension. Hypertension. 1999;33:1036–1042. doi: 10.1161/01.hyp.33.4.1036. [DOI] [PubMed] [Google Scholar]
- 18.Cheng HF, Wang JL, Vinson GP, Harris RC. Young SHR express increased type 1 angiotensin II receptors in renal proximal tubule. Am J Physiol. 1998;274:F10–17. doi: 10.1152/ajprenal.1998.274.1.F10. [DOI] [PubMed] [Google Scholar]
- 19.Reaux A, de Mota N, Zini S, Cadel S, Fournie-Zaluski MC, Roques BP, Corvol P, Llorens-Cortes C. PC18, a specific aminopeptidase N inhibitor, induces vasopressin release by increasing the half-life of brain angiotensin III. Neuroendocrinology. 1999;69:370–376. doi: 10.1159/000054439. [DOI] [PubMed] [Google Scholar]
- 20.Wright JW, Tamura-Myers E, Wilson WL, Roques BP, Llorens-Cortes C, Speth RC, Harding JW. Conversion of brain angiotensin II to angiotensin III is critical for pressor response in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284:R725–733. doi: 10.1152/ajpregu.00326.2002. [DOI] [PubMed] [Google Scholar]
- 21.Muchant DG, Thornhill BA, Belmonte DC, Felder RA, Baertschi A, Chevalier RL. Chronic sodium loading augments natriuretic response to acute volume expansion in the preweaned rat. Am J Physiol. 1995;269:R15–22. doi: 10.1152/ajpregu.1995.269.1.R15. [DOI] [PubMed] [Google Scholar]
- 22.Thomsen K, Shirley DG. The validity of lithium clearance as an index of sodium and water delivery from the proximal tubules. Nephron. 1997;77:125–138. doi: 10.1159/000190264. [DOI] [PubMed] [Google Scholar]
- 23.Kawabe K, Watanabe TX, Shiono K, Sokabe H. Influence on blood pressure of renal isografts between spontaneously hypertensive and normotensive rats, utilizing the F1 hybrids. Jpn Heart J. 1978;19:886–894. doi: 10.1536/ihj.19.886. [DOI] [PubMed] [Google Scholar]
- 24.Omvik P, Tarazi RC, Bravo EL. Regulation of sodium balance in hypertension. Hypertension. 1980;2:515–523. doi: 10.1161/01.hyp.2.4.515. [DOI] [PubMed] [Google Scholar]
- 25.Nagaoka A, Kakihana M, Suno M, Hamajo K. Renal hemodynamics and sodium excretion in stroke-prone spontaneously hypertensive rats. Am J Physiol. 1981;241:F244–249. doi: 10.1152/ajprenal.1981.241.3.F244. [DOI] [PubMed] [Google Scholar]
- 26.Arendshorst WJ, Beierwaltes WH. Renal tubular reabsorption in spontaneously hypertensive rats. Am J Physiol. 1979;237:F38–47. doi: 10.1152/ajprenal.1979.237.1.F38. [DOI] [PubMed] [Google Scholar]
- 27.Norman RA, Jr., Enobakhare JA, DeClue JW, Douglas BH, Guyton AC. Arterial pressure-urinary output relationship in hypertensive rats. Am J Physiol. 1978;234:R98–103. doi: 10.1152/ajpregu.1978.234.3.R98. [DOI] [PubMed] [Google Scholar]
- 28.Hiranyachattada S, Saetew S, Harris PJ. Acute effects of candesartan on rat renal haemodynamics and proximal tubular reabsorption. Clin Exp Pharmacol Physiol. 2005;32:714–720. doi: 10.1111/j.1440-1681.2005.04253.x. [DOI] [PubMed] [Google Scholar]
- 29.Wamberg C, Plovsing RR, Sandgaard NC, Bie P. Effects of different angiotensins during acute, double blockade of the renin system in conscious dogs. Am J Physiol Regul Integr Comp Physiol. 2003;285:R971–980. doi: 10.1152/ajpregu.00262.2003. [DOI] [PubMed] [Google Scholar]
- 30.Plovsing RR, Wamberg C, Sandgaard NC, Simonsen JA, Holstein-Rathlou NH, Hoilund-Carlsen PF, Bie P. Effects of truncated angiotensins in humans after double blockade of the renin system. Am J Physiol Regul Integr Comp Physiol. 2003;285:R981–991. doi: 10.1152/ajpregu.00263.2003. [DOI] [PubMed] [Google Scholar]
- 31.Ardaillou R, Chansel D. Synthesis and effects of active fragments of angiotensin II. Kidney Int. 1997;52:1458–1468. doi: 10.1038/ki.1997.476. [DOI] [PubMed] [Google Scholar]
- 32.Kemp BA, Keller SR, Padia SH. Renal Aminopeptidase N Expression Is Increased In SHR; Presented at: American Heart Association Council For High Blood Pressure Research Annual Scientific Meeting; Atlanta, GA. 2008. Abstract. [Google Scholar]
- 33.Prieto I, Hermoso F, Gasparo M, Vargas F, Alba F, Segarra AB, Banegas I, Ramirez M. Angiotensinase activities in the kidney of renovascular hypertensive rats. Peptides. 2003;24:755–760. doi: 10.1016/s0196-9781(03)00121-9. [DOI] [PubMed] [Google Scholar]
- 34.Holstein-Rathlou NH, Kanters JK, Leyssac PP. Exaggerated natriuresis and lithium clearance in spontaneously hypertensive rats. J Hypertens. 1988;6:889–895. doi: 10.1097/00004872-198811000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Matsushima Y, Kawamura M, Akabane S, Imanishi M, Kuramochi M, Ito K, Omae T. Increases in renal angiotensin II content and tubular angiotensin II receptors in prehypertensive spontaneously hypertensive rats. J Hypertens. 1988;6:791–796. [PubMed] [Google Scholar]
- 36.Correa FM, Viswanathan M, Ciuffo GM, Tsutsumi K, Saavedra JM. Kidney angiotensin II receptors and converting enzyme in neonatal and adult Wistar-Kyoto and spontaneously hypertensive rats. Peptides. 1995;16:19–24. doi: 10.1016/0196-9781(94)00150-5. [DOI] [PubMed] [Google Scholar]
- 37.Dagher G, Sauterey C. H+ pump and Na(+)-H+ exchange in isolated single proximal tubules of spontaneously hypertensive rats. J Hypertens. 1992;10:969–978. [PubMed] [Google Scholar]
- 38.Aldred KL, Harris PJ, Eitle E. Increased proximal tubule NHE-3 and H+-ATPase activities in spontaneously hypertensive rats. J Hypertens. 2000;18:623–628. doi: 10.1097/00004872-200018050-00016. [DOI] [PubMed] [Google Scholar]
- 39.LaPointe MS, Sodhi C, Sahai A, Batlle D. Na+/H+ exchange activity and NHE-3 expression in renal tubules from the spontaneously hypertensive rat. Kidney Int. 2002;62:157–165. doi: 10.1046/j.1523-1755.2002.00406.x. [DOI] [PubMed] [Google Scholar]