

Abstract
Importance to the field
Although most children with cancer are cured, there remain significant limitations of standard treatment, most notably chemotherapy resistance and non-specific toxicities. Novel immune-based therapies that target pediatric malignancies offer attractive adjuncts and/or alternatives to commonly employed cytotoxic regimens of chemotherapy or radiotherapy. Elucidation of the principles of tumor biology and the development of novel laboratory technologies over the last decade have led to substantial progress in bringing immunotherapies to the bedside.
Areas covered in the review
Current immunotherapeutic clinical trials in pediatric oncology and the science behind their development are reviewed.
What the reader will gain
Most of the immune-based therapies studied to date have been well tolerated, and some have shown promise in the setting of refractory or high-risk malignancies, demonstrating that immunotherapy has the potential to overcome resistance to conventional chemotherapy.
Take home message
Some immune-based therapies, such as ch14.18 and MTP-PE, have already been proven effective in phase III randomized trials. Further studies are needed to optimize and integrate these therapies into standard regimens, and to test them in randomized trials for patients with childhood cancer.
Keywords: Cancer, childhood, chimeric antigen receptors, cytokines, immunotherapy, monoclonal antibodies, natural killer cells, pediatric, toll-like receptors, vaccines
1. Introduction
The majority of children with cancer are cured, but the limits of currently employed cytotoxic regimens appear to have been reached for many pediatric malignancies1. Toxicity continues to be substantial despite advances in supportive care, and while survival rates have improved over the last 40 years for most childhood cancers, cures for many with high risk and metastatic disease are not achievable despite aggressive surgical, chemotherapeutic and radiotherapy combination therapies. In pediatrics, many malignancies can be treated into a state of minimal residual disease (MRD), but in high-risk disease, relapse inevitably occurs and is often refractory to further therapy. There is a great need to develop alternative anticancer therapies that are less toxic and more effective.
Immune-based therapies represent an approach that might be integrated into current multimodal regimens or that might have efficacy when used alone. Dramatic progress in scientific discovery and technology has led to rapid development and translation of such therapies for the clinic. Importantly, resistance to conventional therapies does not appear to confer resistance to immune-based therapies2-4.
A current paradigm in cancer immunology is that tumor-associated antigens can induce immune reactivity, but that tumors have mechanisms to block and/or circumvent immune responses5, 6. Therapies that exploit known tumor antigens, or target the mechanisms of immune evasion, have the potential to amplify host antitumor immune responses to the point of completely eradicating established or microscopic disease. Pediatric oncology is especially attractive because of the ability for conventional therapies to establish a period of MRD, a setting in which immune-based therapies may be more likely to be effective7-9.
2. Allogeneic Hematopoietic Stem Cell Transplantation
One of the clearest demonstrations that the immune system is capable of curing patients with resistant cancer is the powerful graft-versus-leukemia (GVL) effect mediated by donor T cells after allogeneic hematopoietic stem cell transplant (HSCT). It has been well established that the benefits of HSCT arise in part from donor T cells targeting allogeneic antigens presented in the context of major histocompatibility complex (MHC), as well as minor histocompatibility antigens, expressed on leukemia cells10, 11. This effect is highly dependent on the type of leukemia12, 13, timing and dose of T cells14, 15, presence of graft-versus-host disease (GVHD)16, disease burden17, 18 and rate of tumor progression19.
2.1 Graft-versus-leukemia effect – T cells
Allogeneic HSCT laid a critical foundation for understanding the biology of T cell-based therapies. The presence of GVHD, which is mediated by donor T cells, is associated with a GVL effect, demonstrated by improved leukemia-free survival of patients with mild GVHD over those without any GVHD20, 21. Additionally, patients without GVHD have lower leukemia relapse rates than recipients of HSCT from identical twin donors, evidence that an allogeneic GVL effect can occur in the absence of clinically evident GVHD11. Lastly, recent data suggests that recurrent leukemias following haploidentical HSCT mutate mismatched MHC alleles from the donor as an escape mechanism from the GVL effect22.
Importantly, the GVL effect is not equipotent across leukemias, with myeloid leukemias being more susceptible than lymphoid leukemias13. T cell depletion of the stem cell graft has been associated with increased risk of relapse in myeloid malignancies14, 23, 24, but the risk of relapse is not increased with lymphoid malignancies, like acute lymphoblastic leukemia (ALL)25, 26. Even within myeloid leukemias, chronic myelogenous leukemia is more susceptible to GVL mediated by donor lymphocyte infusion than acute myelogenous leukemia (AML)19, 24, although reports of pediatric AML responses exist27. Thus, the T cell-mediated GVL effect occurs with a wide range of efficacy across and within leukemia sub-types. Notably, leukemic eradication occurs much more slowly than that observed after chemotherapy or radiotherapy28. Quantitative effects are also important since the timing and dose of T cell administration, tumor burden, and rate of leukemic growth substantially impact the efficacy of the GVL effect. Results are best with low disease burden, for example in the setting of MRD13, 17.
Given the risks of GVHD and associated immunosuppression, the “holy grail” of HSCT is to discover and implement strategies that induce GVL in the absence of GVHD. Cancer vaccines are one approach that might be utilized to direct the T cell repertoire preferentially toward tumor-associated antigens rather than to normal host tissue antigens. There are numerous antigens that have been explored in that regard, for example, the Wilms tumor-1 (WT1) protein, a transcription factor expressed in various malignancies.29 Three clinical trials are in progress in pediatrics that target WT1 post-allogeneic HSCT in an attempt to augment the GVL effect (Table 1). Peptide vaccination targeting the myeloid antigen proteinase-3 is undergoing Phase I/II study in adults with myeloid leukemias and myelodysplastic syndrome30, and may become a viable approach for pediatric AML in the future. Future studies will likely use adoptive immunotherapy of antigen-specific T cells following allogeneic HSCT in lieu of unselected lymphocytes as currently administered. Not only would this strategy increase the numbers of T cells available to target the malignancy, but it could also diminish the risk for GVHD if the targeted antigen showed limited tissue distribution.
Table 1. Current Pediatric Clinical Trials: T Cell Therapies for WT1+ Tumors.
| Clinical trial number | Title | Primary Site / Sponsor |
|---|---|---|
| NCT00608166 | Phase I/II Vaccine Therapy and Donor Lymphocyte Infusions in Treating Patients With Progressive or Relapsed Hematologic Malignancies After Donor HSCT | National Cancer Institute |
| NCT00620633 | Phase I Dose Escalation Trial of WT1-Sensitized T Cells for Residual or Relapsed Leukemia After Allogeneic HSCT | Memorial Sloan-Kettering Cancer Center |
| NCT00052520 | Phase I/II Study Of Adoptive Immunotherapy With CD8+ WT1-Specific CTL Clones for Patients With Advanced MDS, CML, AML or ALL After Allogeneic HSCT | Fred Hutchinson Cancer Research Center |
2.2 Graft-versus-leukemia effect - Natural killer cells
In the last decade, natural killer (NK) cells have drawn considerable interest for their potential to mediate a GVL effect. NK cells are the first lymphoid cells to appear after HSCT, they produce growth factors that can enhance engraftment and immune reconstitution, they do not seem to initiate GVHD, and they have the capacity to kill virally-infected cells31. NK cells utilize critical inhibitory and activating receptors32 and it is well established that NK cells are usually inactivated by self-MHC class I through their killer immunoglobulin-like receptors (KIRs). In addition, NK cells possess activating receptors that signal NK cells to directly kill tumor cells in the absence of inflammatory signals, and NK cells are critical mediators of antibody-dependent cellular cytotoxicity (ADCC).
Initial preclinical studies demonstrated that blockade of inhibitory receptors on NK cells could enhance their cytotoxicity after autologous HSCT33. Blockade was not needed with haploidentical NK cells when the appropriate MHC class I molecules were not present to inactivate KIRs on donor NK cells. In clinical trials, enhanced survival was observed in adults with AML after haploidentical NK KIR-mismatched HSCT34, and this seemed to be dependent on a T cell-depleted platform35, possibly due to the absence of immunosuppressive agents and/or the absence of regulatory T cells (Tregs) that might inhibit NK cell function36.
Currently, clinical trials in pediatrics include studies of haploidentical HSCT for hematologic malignancies and solid tumors designed to provide a source of KIR-mismatched NK cells. In addition, trials that incorporate infusions of purified allogeneic NK cells in a non-transplant setting for a variety of hematologic malignancies are also being conducted (Table 2). The optimal approaches and settings to utilize adoptive NK cell therapy have not yet been defined. For example, it is not known how long adoptively transferred NK cells can persist in vivo, and given they do not form immunological memory, multiple infusions might be required to have a lasting effect. It is also not clear whether NK cells will better utilized as single agents, or in combination with antibodies and/or cytokines to achieve maximal benefit.
Table 2. Current Pediatric Clinical Trials: NK Cell Therapies.
| Clinical trial number | Title | Primary Site / Sponsor |
|---|---|---|
| NCT00187096 | Pilot Study Of Haplo-Identical NK Cell Transplantation For AML | St. Jude Children's Research Hospital |
| NCT00640796 | Phase I Expanded, Activated Haploidentical NK Cell Infusions for Non-B Lineage Hematologic Malignancies | St. Jude Children's Research Hospital |
| NCT00697671 | Phase I Haploidentical NK Cell Infusions for Poor Prognosis Non-AML Hematologic Malignancies | St. Jude Children's Research Hospital |
| NCT00145626 | Phase II HLA-Nonidentical Stem Cell and NK Cell Transplantation for Children Less the Two Years of Age With Hematologic Malignancies | St. Jude Children's Research Hospital |
| NCT00703820 | Phase II Randomized Trial of Clofarabine + Cytarabine Versus Conventional Induction Therapy with a Phase II Study Of NK Cell Transplantation In Newly Diagnosed AML | St. Jude Children's Research Hospital |
| NCT00274846 | Pilot study of Allogeneic NK Cells in Patients With Relapsed AML | University of Minnesota |
| NCT00660166 | Phase I HLA Class I Haplotype Mismatched NK Cell Infusions After Autologous HSCT for Hematological Malignancies | Tufts Medical Center |
| NCT00582816 | Phase I/II Reduced Intensity Haploidentical Transplantation With NK Cell Infusion for Pediatric Acute Leukemia and High Risk Solid Tumors | University of Wisconsin, Madison |
| NCT00698009 | Phase II Haploidentical NK Cells in Relapsed Neuroblastoma Post Autologous HSCT | M.D. Anderson Cancer Center |
3. Tumor vaccines
Similar to vaccines used to prevent infection, cancer vaccines seek to generate sustained responses to a specific antigen through the formation of immunologic memory. However in oncology, the targeted antigens are often weakly immunogenic tumor-associated antigens, or overexpressed self-antigens. Self-antigens are problematic because the T cells with high affinity for self-antigens are deleted in the thymus early in life and peripherally anergized throughout life, a mechanism that limits the risk of autoimmunity. Nevertheless, it appears that substantial numbers of T cells with reactivity toward self-antigens are present in the lymphoid system of normal humans, and the goal of tumor vaccines is to activate and expand these populations. While vaccines targeting self-antigens expressed by tumors can have antitumor effects, the potency of such appears to be less than when foreign antigens are targeted.
There are several challenges that make uniform adoption of vaccines difficult, especially in pediatrics. First, vaccines can be derived from a variety of sources. Peptides can be administered with adjuvant, whole protein may be administered directly or with adjuvants, and DNA vaccines encoded by viral vectors can be used37. In addition, some groups have genetically modified autologous or allogeneic tumors themselves to generate a vaccine. Dendritic cells (DCs) loaded with tumor lysate, peptides or apoptotic bodies are commonly used in pediatrics since they avoid the need to limit accrual of patients to those with unique MHC alleles, as is necessary when peptide based vaccines are used. Importantly, it remains unknown which, if any, of the approaches currently under study are superior as a tumor vaccine38. Second, optimal antigens have not been defined for most pediatric tumors. Third, standard therapies administered to nearly all pediatric cancer patients are highly immunosuppressive and immune recovery following such may be quite prolonged (e.g., 6-12 months)39, 40, limiting the number of effector cells that might respond to the vaccine. Thus, effective immunotherapies for children with cancer will require approaches that can both optimize immune reconstitution and potently immunize. Fourth, as with tumor immunotherapies in general, optimal techniques for tumor vaccination have not been defined. Further, the kinetics of treatment-induced immune responses may result in slow tumor kill, which is less likely to be effective in the setting of highly proliferative, drug-resistant cancers of childhood. Thus current efforts are based on identifying appropriate antigens, augmenting the potency of the vaccine itself, administering tumor vaccines in the setting of MRD and combining tumor vaccines with other immune based therapies.
Sizable numbers of cancer vaccine trials have been conducted in adults over the last 15 years, with only a small fraction of patients with established tumors demonstrated tumor shrinkage after vaccine therapy alone. Whether clinical benefit can be achieved independent of gross tumor shrinkage, such as in the prevention of recurrence, remains unknown. Only a limited number of randomized studies of tumor vaccination have been conducted. Encouraging results of Phase III studies of tumor vaccination in B cell lymphoma41 and prostate cancer42 raise the prospect that Food and Drug Administration (FDA)-approved tumor vaccines may soon become available.
In pediatrics, early non-randomized phase vaccine trials have been conducted or are underway in a wide range of malignancies (Table 3). Clinical responses have been observed in Epstein Barr virus (EBV)-associated lymphomas after manipulating the tumor to overexpress the subdominant antigen LMP243, and a complete response (CR) was reported in a patient with metastatic fibrosarcoma treated with a vaccine comprised of tumor lysate-pulsed DCs44, 45. Interleukin (IL)-2 transduced into autologous neuroblastoma cells resulted in a 20% objective tumor response and 30% stable disease rate in 10 patients46. No systemic toxicity was reported in that study. Follow-up studies using autologous and allogeneic neuroblastoma cells attempted to improve outcome by cotransfecting IL-2 with the chemokine lymphotactin. Two CRs and one partial response (PR), a 14% total response rate, were observed in the allogeneic vaccine trial47, while only one stable disease (13%) was noted with the autologous vaccine48. Thus, there has been evidence for clinical benefit in a small percentage of patients with pediatric cancer treated with a variety of different tumor vaccines. However much further work is needed to determine whether and how tumor vaccines can mediate reproducible rates of clinical benefit, and many challenges remain.
Table 3. Current Pediatric Clinical Trials: Tumor Vaccines.
| Clinical trial number | Title | Primary Site / Sponsor |
|---|---|---|
| NCT00526240 | A Pilot Study of Tumor Vaccination in Patients With Neuroblastoma and Pediatric Sarcomas and Altered T-Cell Homeostasis | National Cancer Institute |
| NCT00058799 | Phase I CD40 Ligand & IL-2 gene modified leukemia vaccine | Baylor College of Medicine |
| NCT00048386 | Phase I/II IL-2 gene modified autologous neuroblastoma vaccine | Baylor College of Medicine |
| NCT00703222 | Phase I/II post autologous HSCT | Baylor College of Medicine |
| NCT00002748 | Phase I IL-2 gene modified autologous or partially matched allogeneic neuroblastoma vaccine | St. Jude Children's Research Hospital |
| NCT00186862 | Phase I Lymphotactin and IL-2 gene modified neuroblastoma vaccine post chemotherapy | St. Jude Children's Research Hospital |
| NCT00258687 | Phase I Vaccination with GM-CSF gene modified, autologous, lethally irradiated clear cell sarcoma, alveolar soft part sarcoma, renal cell carcinoma or melanoma vaccine | Dana-Farber Cancer Institute |
| NCT00069940 | Phase I Vaccination with telomerase peptide + GM-CSF for brain tumors, GIST and sarcomas | Dana-Farber Cancer Institute |
| NCT00085397 | Randomized Phase II Immunization against melanoma comparing autologous dendritic cells pulsed with gp100 peptide to autologous dendritic cells fused with autologous tumor cells | Dana-Farber Cancer Institute |
| NCT00576641 | Phase l Tumor-associated antigen pulsed dendritic cell vaccine for brain stem glioma and glioblastoma | Cedars-Sinai Medical Center |
| NCT00576537 | Phase ll Tumor lysate-pulsed dendritic cell vaccine for brain tumors | Cedars-Sinai Medical Center |
| NCT00101309 | Phase I EBV-transformed fusion tumor cell vaccine for relapsed Ewing's sarcoma and neuroblastoma | Milton S. Hershey Medical Center |
| NCT00107185 | Phase I Dose escalation study of autologous tumor lysate-pulsed dendritic cell vaccine for malignant gliomas | Jonsson Comprehensive Cancer Center |
| NCT00405327 | Phase II Tumor lysate-pulsed dendritic cell vaccine for high-risk solid tumor patients following autologous HSCT | University of Michigan Cancer Center |
As with many immune-based therapies, tumor vaccines may be more effective in patients with MRD7-9, but early phase clinical trials are typically conducted in the setting of rapidly progressive, chemotherapy-refractory tumors, making assessments of both vaccine activity and safety difficult49. Because of this, recent studies have begun to incorporate vaccines into standard cytoreductive therapy regimens, wherein the immunotherapy is administered in the setting of MRD. This paradigm of “consolidative immunotherapy” is particularly pertinent to pediatric oncology, where even very high-risk tumors are usually responsive to front line therapies, and patients with high-risk disease can often be reduced to a state of MRD. In this regard, a study of adoptive T-cell transfer with peptide-pulsed DC vaccines targeting the specific translocation breakpoints in patients with metastatic and recurrent Ewing sarcoma and alveolar rhabdomyosarcoma following completion of dose-intensive chemotherapy was recently conducted50. Here, potential benefits could occur due to the tumor vaccine itself and/or to the effects of autologous lymphocyte infusions on immune reconstitution following lymphodepleting chemotherapy, as seen in animal models51. Using an intent-to-treat analysis of all patients entered onto the study, irrespective of whether immunotherapy was eventually administered or not, an overall 5 year survival of 31% was seen. For patients who actually received immunotherapy, the 5 year overall survival was 43%, which is favorable compared to historical controls, but no doubt includes some selection bias, since only patients whose tumors responded to frontline therapy were able to receive immunotherapy. At the same time, patients were not required to be in remission prior to receiving immunotherapy and this relatively favorable survival leaves open the possibility that incorporating immunotherapy into this regimen may have benefited some patients with high-risk sarcomas. Importantly, biologic responses measured to the vaccine itself were inconsistent, however all immunized patients demonstrated the capacity to generate T cell responses to influenza vaccination within 3 months following chemotherapy, indicating that vaccine induced T cell responses can be observed early after cytotoxic chemotherapy. A subsequent study targeting patients with metastatic and recurrent pediatric sarcomas is underway with a modified DC vaccine, which incorporates approaches to deplete regulatory or suppressive CD4+ T cells, and also incorporates recombinant human interleukin-7 (rhIL-7) to enhance immune reconstitution (Table 3). It is important to note that single arm studies of this type cannot definitively demonstrate benefit, but they can be used to optimize vaccine strategies and to study the impact of conventional cytotoxic therapy on vaccine responsiveness. Ultimately, any firm conclusions regarding the value of any immunotherapy requires demonstration of benefit in a multi-institutional randomized Phase III trial.
4. Adoptive Immunotherapy and Chimeric Antigen Receptors
One inherent factor limiting the efficacy of T cell based vaccines is the inability to rapidly generate large numbers of antigen specific cells in vivo. Especially when targeting sizable tumor burdens, it is clear that large numbers of effector T cells are necessary. Adoptive immunotherapy, ex vivo approaches to expand antigen specific T cells, allows the generation of very large numbers of antigen specific T cells and can also enhance the function of cells by removing them from the immunosuppressive tumor environment. Although it remains labor intensive, many new approaches are now available to enhance the capacity to generate T cells and NK cells for adoptive immunotherapy. Among these are artificial antigen presenting cells (APCs) that express MHC, costimulatory molecules and/or cytokines have been developed to promote expansion of effector cells47, 48, 52. Artificial APCs can be employed as an “off the shelf” reagent in that a putative tumor antigen can be inserted into the MHC. The selective expansion of tumor-reactive cells and the potential for generating memory cells ex vivo, represents one strategy to attempt to overcome the common immune system impairments sustained from dose-intensive chemotherapy and radiation.
Genetic engineering can endow cytotoxic T cells and NK cells with specific antigenic specificities, and such antigen specific cell populations can be expanded ex vivo and then administered as adoptive immunotherapy. Both genetically engineered T cell receptors which recognize antigen in an MHC restricted manner, as well as genetically engineered receptors that incorporate the binding sites of moAbs (chimeric antigen receptors or CARs) have been used. The CARs are transduced into a patient's T cells or NK cells, which have the potential to bind their target through a single-chain Fv fragment fused to the signaling chain of the T-cell receptor (Figure 1). CAR-transduced cells have been shown to traffic to tumor sites53, 54. Clinical trials using a CAR against GD2 have been completed in children with neuroblastoma, with no toxicity noted9, 55. EBV-specific cytotoxic lymphocytes engineered to recognize GD2 were active against neuroblastoma, with 50% objective responses including one sustained CR56. Clinical trials with genetically engineered TCRs and CARs in pediatric patients are ongoing (Table 5) and additional studies are in development for children with ALL, lymphoma, medulloblastoma, and glioblastoma multiforme9.
Figure 1. Components of a chimeric antigen receptor.
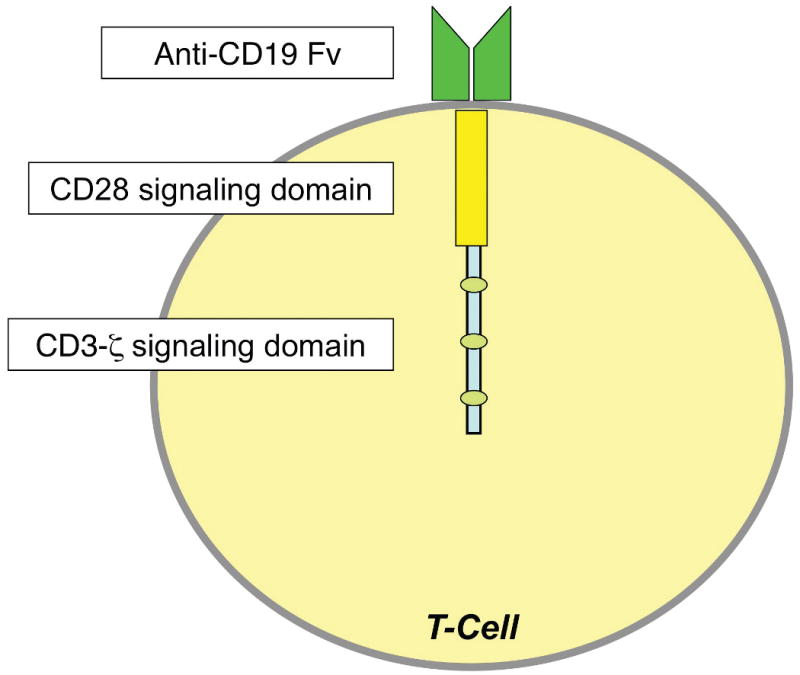
The binding portion of the CAR consists of the Fv fragment from an antibody against a target antigen, in this case CD19. Typically a T cell receptor signaling domain is attached to the Fv fragment, in this case using a CD28/CD3ζ tail.
Table 5. Current Pediatric Clinical Trials: Chimeric Antigen Receptors.
| Clinical trial number | Title | Primary Site / Sponsor |
|---|---|---|
| NCT00586391 | Phase I CD19 Chimeric Receptor Expressing T Lymphocytes In B-Cell Non Hodgkin's Lymphoma And CLL | Baylor College of Medicine |
| NCT00608270 | Phase I Study of CD19 Chimeric Receptor Expressing T Lymphocytes in B-Cell Non Hodgkin's Lymphoma and CLL | Baylor College of Medicine |
| NCT00840853 | Phase I/II Study of the Administration of Multi-Virus-Specific CTLs Expressing CD19 Chimeric Receptors for Prophylaxis of Therapy of Relapse of ALL Post-HSCT | Baylor College of Medicine |
| NCT00924326 | Phase I/II Study of B Cell Malignancies Using T Cells Expressing an Anti-CD19 Chimeric Receptor: Assessment of the Impact of Lymphocyte Depletion Prior to T Cell Transfer | National Cancer Institute |
5. Monoclonal Antibodies
There has been significant progress in the clinical development of monoclonal antibodies (moAbs) as cancer therapies in adults, with promising results now emerging from pediatric studies (Table 4). Importantly, antibodies can recognize tumor antigens, are not limited to processed peptides, and do not require peptide presentation by MHC molecules like T cells do. For effective targeting, the targeted antigen should be relatively tumor-specific and highly expressed.
Table 4. Current Pediatric Clinical Trials: Monoclonal Antibody-Based Trials.
| Clinical trial number | Title | Primary Site / Sponsor |
|---|---|---|
| NCT00831844 | A Phase II Study of IMC-A12 (Anti-IGF-I Receptor Monoclonal Antibody, IND #100947, NSC #742460) in Children With Relapsed/Refractory Solid Tumors | Children's Oncology Group |
| NCT00354107 | A Phase I/II Pilot Study of Ifosfamide, Carboplatin and Etoposide Therapy (ICE) and SGN-30 (NSC# 731636, IND#) in Children With CD30+ Recurrent Anaplastic Large Cell Lymphoma | Children's Oncology Group |
| NCT00098839 | A Feasibility Pilot and Phase 2 Study Of Chemoimmunotherapy With Epratuzumab For Children With Relapsed CD22-Positive Acute Lymphoblastic Leukemia | Children's Oncology Group |
| NCT00666588 | A Phase II Pilot Study of Bortezomib Combined With Reinduction Chemotherapy in Children and Young Adults With Recurrent, Refractory or Secondary Acute Myeloid Leukemia | Children's Oncology Group |
| NCT00372593 | A Phase III Randomized Trial of Gemtuzumab Ozogamicin (Mylotarg) Combined With Conventional Chemotherapy for De Novo Acute Myeloid Leukemia (AML) in Children, Adolescents, and Young Adults | Children's Oncology Group |
| NCT00659425 | A Phase 1, Multicenter, Dose Escalation Study of CAT-8015 in Children, Adolescents and Young Adults With Refractory CD22+ Acute Lymphoblastic Leukemia (ALL) or Non-Hodgkin's Lymphoma (NHL) | National Cancer Institute |
| NCT00428272 | A Phase I Trial of Monoclonal Antibody HGS-ETR2 (Lexatumumab) With or Without Interferon Gamma in Patients With Refractory Pediatric Solid Tumors | National Cancer Institute |
| NCT00556881 | A Phase I Study of Ipilimumab (Anti-CTLA-4) in Children, Adolescents and Young Adults With Treatment Refractory Cancer | National Cancer Institute |
| NCT00020384 | Short-Course EPOCH-Rituximab For Untreated CD-20+ HIV-Associated Lymphomas | National Cancer Institute |
| NCT00445965 | Phase II Study of Intrathecal I-3F8 in Patients With GD2-Expressing Central Nervous System and Leptomeningeal Neoplasms | Memorial Sloan Kettering Cancer Center |
| NCT00089245 | Phase I Study Of Inthrathecal Radioimmunotherapy Using I-8H9 For Central Nervous System/Leptomeningeal Neoplasms | Memorial Sloan Kettering Cancer Center |
| NCT00492167 | Phase I Study of Oral Yeast β-Glucan and Intravenous Anti-GD2 Monoclonal Antibody 3F8 Among Patients With Metastatic Neuroblastoma | Memorial Sloan Kettering Cancer Center |
| NCT00450307 | 3F8 Antibody Dose Escalation Plus Granulocyte-Macrophage Colony-Stimulating Factor in High-Risk Neuroblastoma: A Phase I Trial | Memorial Sloan Kettering Cancer Center |
| NCT00058370 | A Trial Of Radioimmunotherapy, Reduced-Dose External Beam Craniospinal Radiation Therapy With IMRT Boost, And Chemotherapy For Patients With Standard-Risk Medulloblastoma | Memorial Sloan Kettering Cancer Center |
| NCT00072358 | Phase II Study of Anti-GD2 3F8 Antibody and GM-CSF for High-Risk Neuroblastoma | Memorial Sloan Kettering Cancer Center |
| NCT00450827 | Combination of Targeted I -3F8-Mediated Radioimmunotherapy and Bevacizumab in Patients With Relapsed or Refractory Neuroblastoma: A Phase I Study | Memorial Sloan Kettering Cancer Center |
| NCT00877110 | Phase I Study of Anti-GD2 3F8 Antibody and Allogeneic Natural Killer Cells for High-Risk Neuroblastoma | Memorial Sloan Kettering Cancer Center |
| NCT00720174 | A Phase I/II Study of Doxorubicin and A12 in Advanced Soft Tissue Sarcoma | University of Chicago |
| NCT00643240 | Phase I Open Label, Single Arm Escalation Trial to Evaluate the Biodistribution and Safety of BU-12 in Patients With Advanced Leukemia | Masonic Cancer Center |
| NCT00600054 | Phase 2 Study of Safety and Efficacy of Nimotuzumab in Pediatric Patients With Recurrent Diffuse Intrinsic Pontine Glioma | YM Biosciences |
| NCT00642941 | A Phase II Trial of R1507, a Recombinant Human Monoclonal Antibody to the Insulin-Like Growth Factor-1 Receptor for the Treatment of Patients With Recurrent or Refractory Ewing's Sarcoma, Osteosarcoma, Synovial Sarcoma, Rhabdomyosarcoma and Other Sarcomas. | Hoffmann-La Roche |
| NCT00761644 | A Phase I Trial of Doxil, Bevacizumab and Temsirolimus | M.D. Anderson Cancer Center |
| NCT00667342 | A Study of Bevacizumab, a Humanized Monoclonal Antibody Against Vascular Endothelial Growth Factor (VEGF), in Combination With Chemotherapy for Treatment of Osteosarcoma | St. Jude's Children's Research Hospital |
| NCT00381797 | Phase II Study of Bevacizumab Plus Irinotecan in Children With Recurrent, Progressive, or Refractory Malignant Gliomas, Diffuse/Intrinsic Brain Stem Gliomas, Medulloblastomas, Ependymomas and Low Grade Gliomas | Pediatric Brain Tumor Consortium |
| NCT00866047 | A Phase 2 Study of SGN-35 in Treatment of Patients With Relapsed or Refractory Systemic Anaplastic Large Cell Lymphoma (ALCL) | Seattle Genetics, Inc. |
| NCT00649584 | A Phase I Dose Escalation Study of SGN-35 Alone and in Combination With Gemcitabine in Patients With Relapsed/Refractory CD30-Positive Hematologic Malignancies | Seattle Genetics, Inc. |
| NCT00658658 | A Phase 1 Study to Evaluate the Safety and Pharmacokinetics of Panitumumab in Children With Solid Tumors | Amgen |
| NCT00563680 | A Phase 2 Study of AMG 479 in Relapsed or Refractory Ewing's Family Tumor and Desmoplastic Small Round Cell Tumors | Amgen |
| NCT00589706 | A Phase II Study o the Adjuvant Use of Anti-Epidermal Growth Factor Receptor-425 (Anti-EGFR-425) Monoclonal Antibody Radiolabeled With I-125 for High Grade Gliomas | Drexel University College of Medicine |
| NCT00668148 | A Five-Tier, Phase 2 Open-Label Study of IMC-A12 Administered as a Single Agent Every 2 Weeks in Patients With Previously- Treated, Advanced or Metastatic Soft Tissue and Ewing's Sarcoma/PNET | ImClone Systems |
| NCT00039130 | Phase II Study Of Rituximab And Short Duration, High Intensity Chemotherapy With G-CSF Support In Previously Untreated Patients With Burkitt Lymphoma/Leukemia | Cancer and Leukemia Group B |
MoAbs have the potential to kill tumors through immune mediated or non-immune mediated effector pathways. Immune mediated pathways of cell death that follow binding of the moAb to its target can occur through a variety of mechanisms (Figure 2). The specific mechanism involved, or whether killing occurs at all, varies with different cancers and target antigens. Antibody-dependent cellular cytotoxicity (ADCC) appears to be the primary mechanism of killing for moAbs targeting the GD2 disialoganglioside in neuroblastoma. GD2 is expressed at high densities on nearly all neuroblastoma cells, is not shed from the cell surface, and is restricted to neuroectodermal tissues, thus representing a potentially good target for moAb therapy. Well-studied candidate antibodies include ch14.18, hu14.18 and 3F857-59. These moAbs have toxicities that are manageable in an outpatient setting, have demonstrated responses in patients with refractory neuroblastoma, and seem to be more effective in a MRD setting than in bulky disease9, 58, 60.
Figure 2. Potential mechanisms of tumor cell lysis by monoclonal antibodies.
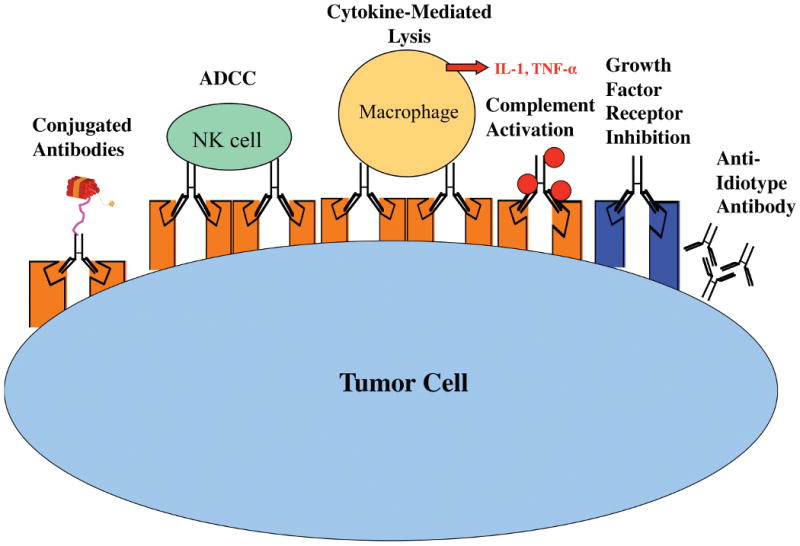
MoAbs can (1) be conjugated to toxins or radionuclides, (2) recruit effector cells to participate in ADCC or (3) release inflammatory cytokines that contribute to tumor killing, (4) activate complement, (5) interfere with growth factor signaling, or (6) bind anti-idiotype antibodies.
To enhance recruitment of effector cells for ADCC, ch14.18, hu14.18 and 3F8 have also been co-administered with, or conjugated to, cytokines and growth factors such as IL-2 and granulocyte-macrophage colony-stimulating factor (GM-CSF)58, 59, 61. This approach led to the development of “immunocytokines,” which use moAbs to both target the tumor and to transport factors designed to enhance the immune response within the tumor microenvironment. After a Phase I trial demonstrated tolerability59, a Phase II trial was conducted with a hu14.18-IL2 fusion protein in which a 21% CR rate was observed in children with neuroblastoma and MRD62.
Early trials with 3F8 demonstrated that moAbs can treat MRD after autologous BMT for stage IV neuroblastoma63, leading investigators to study anti-GD2 therapy in the post transplant setting. A recent randomized Phase III trial with ch14.18 plus GM-CSF and IL-2 versus standard therapy following autologous BMT for high risk neuroblastoma was stopped prematurely because of enhanced event-free and overall survival in the treatment arm64. These results were the first to clearly demonstrate a survival benefit in a randomized phase III trial of neuroblastoma through the addition of moAb-based therapy and pave the way for incorporation of moAbs into frontline regimens for patients with high risk neuroblastoma. Interestingly, this regimen had essentially no activity in patients with recurrent bulk neuroblastoma, consistent with the paradigm that immune-based therapies which are ineffective against bulky tumor may provide substantial survival benefit when administered in the setting of MRD. Ongoing studies are also underway to treat GD2+ brain tumors by conjugating the moAb 3F8 to a radioisotope65.
Trastuzumab, a moAb directed against the human epidermal growth factor receptor 2 (HER2), is being explored as a therapy for osteosarcoma. HER2 expression correlates with survival in osteosarcoma, although since HER2 is not overexpressed in this tumor, efficacy may be limited66, 67. Phase II trials of the anti-CD20 moAb rituximab for relapsed Hodgkin lymphoma have demonstrated high overall response rates (86-100%), with approximately equal numbers of CRs and PRs68, 69.
New moAbs that target the host immune system can also have antitumor effects through the augmentation of endogenous immune responses. For example, CTLA4 is a receptor on the surface of T cells that diminishes autoimmune reactivity. Treatment with blocking anti-CTLA4 moAbs inhibits this suppressive signal and leads to widespread augmentation of T cell mediated immune reactivity. This agent has shown reproducible antitumor effects in melanoma, prostate cancer and other tumors in adults70, and a pediatric Phase I trial is in progress (NCT00556881, National Cancer Institute). Anti-4-1BB moAbs also have the potential to mediate antitumor effects through interaction with activating receptors on T cells71.
Certain moAbs can induce direct cytotoxicity to cancer cells upon binding independent of immune effects, likely by interrupting signaling pathways critical for tumor survival. For example, moAbs to vascular endothelial growth factor (i.e. bevacizumab) and insulin-like growth factor-1 receptor (IGF-1R) have shown activity against pediatric solid tumors, with sometimes dramatic responses72-74. Antibodies can also initiate apoptosis, such as the moAb against tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor, which has been shown to lead to stable disease in sarcomas75.
To date, MoAbs against malignant lymphoma in adults have proven to be most effective when used in combination with chemotherapy regimens76. The cytotoxic potency of moAbs can be dramatically increased by conjugation with toxins, radioisotopes, or other cytotoxic agents. MoAbs against CD20 conjugated to radionuclides for children with Hodgkin lymphoma must include stem cell rescue because of the associated bone marrow toxicity77. MoAbs against CD25 and CD30 conjugated to ricin have induced clinical responses in Hodgkin lymphoma78. Recombinant immunotoxins that incorporate anti-CD22 Fv and modified Pseudomonas exotoxin are undergoing study, and these have shown clinical activity in pediatric ALL79 (NCT00659425, National Cancer Institute). The FDA has approved anti-CD33 conjugated to calicheamicin (gemtuzumab ozogamicin) for the treatment of AML in adults. Approximately 30% of children with relapsed and refractory AML respond to gemtuzumab ozogamicin as a single agent. Currently, this agent is being tested both alone and in combination with chemotherapy in children with AML80, 81. Phase III trials of gemtuzumab ozogamicin in combination with chemotherapy in pediatric AML are being conducted by the Children's Oncology Group (COG)80 (NCT00372593), while the Nordic Society of Paediatric Haematology and Oncology is studying its use prior to HSCT (NCT00476541). This agent has also been used to treat occasional cases of ALL with CD33 expression and there have been anecdotal reports of CRs being achieved82.
A variety of mechanisms of resistance to moAb-based therapies are well described. Patients may develop antibodies to foreign protein epitopes, resulting in binding and neutralization of therapeutic activity. Tumors can shed antigens from their surface, which may serve as a decoy, competing for moAb binding. Malignant cells may also down-regulate antigens, reducing moAb binding to the cell surface target76. Lastly tumors can downregulate expression of MHC class I.
6. Activation of Innate Immunity
Activation of innate immunity has the potential to induce antitumor effector cells and to upregulate antigen presentation and co-stimulatory molecules, thus boosting T-cell responses. Stimulation of toll-like receptors (TLRs), which recognize highly conserved structural and molecular patterns on pathogens and inflammatory mediators released during cell death, called damage-associated molecular patterns (DAMPs), are critical to initiating activation of APCs. Many investigations of TLR activation to augment innate immunity are underway or in development. The cell-wall skeleton of Bacillus Calmette-Guérin (BCG), a TLR2 and TLR4 agonist used in the treatment of bladder cancer83, and imiquimod, a TLR7 agonist used for basal cell skin cancer, represent two examples84. Clinical trials are ongoing in several other adult malignancies. Preliminary results suggest that TLR agonists may not be sufficient as single agents to induce regression of established bulky tumors85. A pediatric Phase I trial of BCG with the anti-idiotypic moAb A1G4 in high-risk patients with GD2+ tumors such as neuroblastoma is in progress (NCT00003023, Memorial Sloan-Kettering Cancer Center).
Muramyl tripeptide phosphatidylethanolamine (L-MTP-PE) is an immunogenic analog of muramyl dipeptide from the cell wall of mycobacteria that is encapsulated in multilamellar liposomes. It binds to TLR4, activating monocytes and macrophages, and promotes antitumor activity86. MTP-PE was studied in a Phase III trial in patients with nonmetastatic osteosarcoma and improved overall survival rates occurred in patients receiving MTP-PE (p=0.03), with a trend toward improved event free survival (p=0.08). 87
Although chemotherapy and radiation have been traditionally viewed as immunosuppressive due to cytotoxic elimination of immune cells, they may paradoxically activate the immune system via endogenous inflammatory mediators released in response to cell death88. It has been demonstrated that tumor cells can release or expose DAMPs in response to chemotherapy-mediated cytotoxicity. The DAMPs then bind to TLRs and cause activation of APCs. Moreover, the release of endogenous self-TLR agonists, collectively termed alarmins, such as high-mobility group box protein 1 (HMGB1), may also stimulate innate immunity89. HMGB1 is a nuclear protein that binds the receptor for advanced glycation end products (RAGE), TLR2, and/or TLR490. In rhabdomyosarcoma preclinical models, HMGB1 binds to RAGE to simulate myogenesis, and as a means of survival, tumors may reduce RAGE expression91. Thus, since alarmins are a byproduct of tumor cell death capable of inducing an immune response, the receptors for alarmins may be purposefully underexpressed by tumors to avoid detection by the immune system. Whether receptors for alarmins are a potential novel target for immunotherapy remains to be seen88.
Natural killer-T (NKT) cells and gamma-delta T cells represent more primitive innate immune effectors that may have a role in anti-tumor immunity. NKT cells have been demonstrated to induce immune activation and antitumor effects by producing gamma interferon (IFNγ). Type I NKT cells recognizes the glycolipid α-galactosylceramide (αGalCer) in association with a nonclassical MHC class I molecule called CD1d. Clinical trials were performed in adults with lung cancer using either αGalCer-pulsed DCs or infusion of autologous NKT cells in patients with lung cancer. Neither treatment yielded responses, but both therapies were well tolerated in Phase I trials92, 93. The addition of type I NKT cells may improve vaccine responses94. Gamma-delta T cells are found primarily in the gut mucosa, and are believed to target lipid antigens. A Phase I trial of autologous gamma-delta T cells in adults with renal cell carcinoma demonstrated a 60% stable disease rate, and was well tolerated95. Presently, no trials in pediatric cancers have been opened with either NKT cells or gamma-delta T cells.
It remains to be determined how best to target innate immune activation in the treatment of cancer. Clearly TLR agonists have therapeutic potential in the setting of bladder and skin cancers. Benefits in other tumors may require stimulation of multiple TLRs and the targeting of pathways used by tumors to escape immune recognition. Importantly, whole cell infusions may contain combinations of both immune-activating and inhibitory NKT populations, the latter of which could diminish anti-tumor responses. While there is great hope that gamma-delta T cells will have efficacy, caution must be exercised in extrapolating observations from mouse models where these cell subsets form a larger component of the immune system than humans.
7. Cytokines and Growth Factors
As noted above, studies of combination immune–based therapies are being conducted in attempt to amplify the magnitude of anti-cancer immune responses. Cytokines and growth factors that expand and activate T cells are under investigation in that regard. IL-2 is a gamma-c cytokine produced by T helper 1 cells that causes proliferation of B and T cells, as well as NK cells. IL-2 is FDA-approved for renal cell carcinoma and malignant melanoma; however, for pediatric tumors, several trials of IL-2 have been performed with no antitumor effects observed96-98. Moreover, recent studies have clearly demonstrated that in addition to activating T and NK cells, IL-2 also substantially expands and activates CD4+CD25+ Tregs, a potential mediator of tumor-induced immune suppression99. IL-7, a member of the gamma-c cytokine family, is a key regulator of lymphocyte homeostasis100. IL-7 has been studied in two clinical trials in adults with refractory malignancies, and has led to increases in CD4+ and CD8+ T cells with no overt toxicity noted101, 102. It remains unknown whether IL-7 will be active as direct anti-tumor therapy, although it is likely to be an effective adjuvant103. It is anticipated that IL-7 will facilitate more effective integration of T cell-based therapies following cytotoxic chemotherapy, since it dramatically increases T cell recovery in that setting. Lastly IL-21, another member of the gamma-c cytokine family, is produced by activated T helper 2 cells and is synergistic with other cytokines such as IL-2. IL-21 has been studied in three clinical trials in adults with metastatic melanoma and renal cell carcinoma, with 1 CR and 1-4 PRs noted in each trial104.
Cytokines that work on a more broad range of immune cells, such as interferons, are also under investigation. Alpha interferons (IFN-α) are produced by lymphocytes, macrophages and plasmacytoid DCs, primarily in response to viral infections. They stimulate macrophages and NK cells to elicit an anti-viral response, although subtypes have shown activity against a variety of malignancies. IFN-α2a is FDA-approved for the adjuvant therapy of adults with stage III melanoma, hairy cell leukemia, AIDS-related Kaposi's sarcoma, and CML. An ongoing clinical trial examines the use of IFN-α2a in children with melanoma105. Phase I trials are also exploring the role of pegylated IFN-α2a for plexiform neurofibromas (NCT00678951, National Cancer Institute) and brain tumors in children (NCT00041145, National Cancer Institute). Interferon-α2b is approved in adults for the treatment of hairy cell leukemia, malignant melanoma, and AIDS-related Kaposi's sarcoma. IFN-α2b is undergoing study in combination with GM-CSF for ALL, AML, blast phase CML and myelodysplastic syndrome (NCT00548847, Emory University). How IFN-α mediates antitumor effects remains unclear, but it likely activates innate immunity.
IFN-γ is produced by NK cells, NKT cells, DCs and CD4+ and CD8+ T cells in response to viral and intracellular bacterial infections, as well as during anti-tumor responses. It acts mainly on macrophages, DCs, NK cells and T cells. IFN-γ is FDA-approved for the treatment of children with osteopetrosis and chronic granulomatous disease. It has shown activity against Ewing sarcoma when combined with a TRAIL agonist in preclinical models106 and is currently being studied in children with solid tumors or lymphomas in combination with the TRAIL receptor agonist moAb, lexatumumab (NCT00428272, National Cancer Institute).
Tumor necrosis factor-alpha (TNF-α) is a cytokine that is an acute phase reactant secreted mainly by macrophages at the onset of an inflammatory response. It acts predominantly on neutrophils and macrophages, but also has functions in the liver and brain. Regional therapy with TNF-α has been performed in patients with sarcoma, and some antitumor responses were observed in Ewing sarcoma and Wilms tumor, although this approach is limited by the development of systemic toxicity107.
Growth factors are also being explored as a means to enhance antigen presentation by tumor cells directly, or through the recruitment of APCs to the site of the tumor. Recombinant GM-CSF (sargramostim) is a myeloid growth factor that stimulates hematopoietic stem cells to make granulocytes and monocytes. When given before and during induction chemotherapy, GM-CSF may make leukemic blast cells more susceptible to the cytotoxic effects of chemotherapy108, 109. It causes upregulation of costimulatory molecule expression on leukemia blasts in vitro and, in combination with IFN-α, can induce antitumor immune responses in relapsed AML and ALL after allogeneic HSCT110. Tumor cells engineered to secrete GM-CSF are particularly effective as antitumor vaccines, and the addition of GM-CSF to standard vaccines may increase their activity by recruiting DCs to the site of vaccination111. Inhaled GM-CSF is undergoing study in children with pulmonary metastases from osteosarcoma (NCT00673179, M.D. Anderson Cancer Center; NCT0066365, COG). One child with Ewing sarcoma demonstrated a CR112, 113 and 48% of patients had disease stabilization or partial regression for a mean duration of 10 months. This included 8 of 13 (62%) with sarcoma113.
8. Modulation of Inhibitory Cell Subsets
While the majority of therapies are aimed at optimizing the efficacy of immune effector cells, future trials will likely incorporate strategies that also reduce cell subsets that inhibit immune responses. For example, initial trials with IL-2 in pediatrics produced no clinical responses114, 115. It is possible that IL-2 primarily activated Tregs, which express the high affinity IL-2 receptor CD25 and can inhibit immune responses through secretion of transforming growth factor-β and IL-10116. Depletion of Tregs has improved immunotherapy in preclinical models117. As described above, a current consolidative immunotherapy trial underway incorporates ex vivo Treg depletion of autologous lymphocyte infusions in attempt to enhance vaccine induced immune responses for patients high-risk pediatric sarcomas and neuroblastoma (NCT00526240, National Cancer Institute). Another example is with type II NKT cells, which bind CD1d but lack the classic T cell receptor that defines NKT cells, and with rare exception, do not recognize αGalCer118. This subset is generally thought to suppress anti-tumor activity119. Trials have not yet attempted to target this subset.
Tumor-associated macrophages (TAMs) appear to consist primarily of so-called M2 macrophages, that localize into hypoxic regions of tumors and secrete various immunosuppressive cytokines, and facilitate angiogenesis and invasion120. In many animal models, macrophage depletion results in diminished tumor cell survival, increased tumor rejection, or both121. Pediatric tumors show a predominance of macrophage infiltration, suggesting that M2 macrophages may be important in childhood cancer122. To date, no specific strategies to deplete TAMs in the context of immunotherapy trials have been advanced. Lastly, the role of myeloid-derived suppressor cells (MDSCs) as an important component of the immunosuppressive microenvironment of tumors has been demonstrated in experimental models. Indeed, tumor growth results in expansion of MDSCs, increasing nitric oxide in tumors, which inhibits antigen-specific responses123. Their role in pediatric cancers remains unclear.
Thus, there are a variety of inhibitory cell subsets that might need to be targeted in parallel with activation of anti-tumor effectors. It remains to be seen if this will enhance presently employed immunotherapeutic strategies, although this seems likely to have clinical relevance given that such inhibitory pathways have been demonstrated to be active in a variety of malignancies.
9. Expert opinion
Treatment of childhood cancer has experienced great strides during the last 50 years. However, the benefits afforded by cytotoxic therapies appear to have plateaued, and the late effects of current standard regimens are substantial. The challenge to the field of pediatric oncology is to develop biologic based approaches that enhance the benefits of standard therapies, lessen toxicity, and extend the gains in survival to those high-risk groups that have not benefited from chemotherapy. Immunotherapies are biologic based approaches that have shown promise in preclinical models and clinical trials in this regard. The development of immunotherapeutic modalities for cancer is advancing rapidly from pre-clinical studies into clinical trials. In some settings, the benefit of immune based therapy in pediatric oncology has already been demonstrated. For instance, graft-versus-leukemia effects clearly contribute to the benefit of allogeneic HSCT for leukemia. Randomized studies have demonstrated improved event free survival when ch14.18 plus GM-CSF and IL-2 are incorporated into standard therapy for patients with high-risk neuroblastoma and have demonstrated improved overall survival when MTP-PE is incorporated into standard therapy for osteosarcoma. Perhaps most importantly, emerging science suggests that this may only represent the tip of the iceberg with regard to potential benefits of immunotherapy in pediatric oncology.
However, navigating the challenging terrain between scientific discovery, preclinical development, early clinical trials and large scale Phase III testing is arduous and slow. Especially in pediatric oncology, where essentially all of the cancers are rare, collaboration across groups and building consensus regarding optimal approaches to move forward in clinical trials is critical to move from early phase trials to definitive Phase III studies in the most efficient and expedient manner. Continued improvements in technology open new possibilities for therapies with enhanced potency, but many current approaches for administering immunotherapies are already technically challenging to produce and administer. Thus, a balance needs to be struck between the practical ability to apply new therapies and the benefits that are afforded by new technologies. For instance, while individualized tumor vaccines may have a strong biologic basis, ultimately such therapies will be available to more patients if off-the-shelf reagents can be generated that are equally effective.
Finally, it is understandable that current approaches seek to fully optimize the efficacy of specific therapies, whether moAbs, tumors vaccines, adoptive immunotherapy, etc. However, we must keep in mind that the immune system represents more than a collection of parts, and it is designed to be interactive with cross-talk across the elements. Thus, our long term vision should seek to combine immunotherapies in ways that synergize with one another, in order to fully exploit the natural complexity and interactive nature of immunity.
Acknowledgments
The authors acknowledge Dr. Kristin Baird, Dr. Terry Fry, and Dr. Melinda Merchant, colleagues in the Immunology Section of the Pediatric Oncology Branch, National Cancer Institute.
Research Support: This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI), Center for Cancer Research.
Footnotes
Conflict of Interest
The authors declare no potential conflicts of interest.
Publisher's Disclaimer: The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Wayne AS, Reaman GH, Helman LJ. Progress in the curative treatment of childhood hematologic malignancies. J Natl Cancer Inst. 2008 Sep 17;100(18):1271–3. doi: 10.1093/jnci/djn306. [DOI] [PubMed] [Google Scholar]
- 2.Cullen KV, Davey RA, Davey MW. Drug resistance does not correlate with resistance to Fas-mediated apoptosis. Leukemia Research. 2001;25(1):69–75. doi: 10.1016/s0145-2126(00)00085-0. [DOI] [PubMed] [Google Scholar]
- 3.Giavazzi RR, Bucana CCD, Hart IIR. Correlation of tumor growth inhibitory activity of macrophages exposed to adriamycin and adriamycin sensitivity of the target tumor cells. Journal of the National Cancer Institute. 1984;73(2):447–55. doi: 10.1093/jnci/73.2.447. [DOI] [PubMed] [Google Scholar]
- 4.Kontny HU, Lehrnbecher TM, Chanock SJ, Mackall CL. Simultaneous Expression of Fas and Nonfunctional Fas Ligand in Ewing's Sarcoma. Cancer Res. 1998 December 15;58(24):5842–9. 1998. [PubMed] [Google Scholar]
- 5.Mapara MY, Sykes M. Tolerance and Cancer: Mechanisms of Tumor Evasion and Strategies for Breaking Tolerance. J Clin Oncol. 2004 March 15;22(6):1136–51. doi: 10.1200/JCO.2004.10.041. 2004. [DOI] [PubMed] [Google Scholar]
- 6.Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annual Review of Immunology. 2004;22(1):329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brichard VG, Lejeune D. Cancer immunotherapy targeting tumour-specific antigens: towards a new therapy for minimal residual disease. Expert Opinion on Biological Therapy. 2008;8(7):951–68. doi: 10.1517/14712598.8.7.951. [DOI] [PubMed] [Google Scholar]
- 9.Capitini CM, Cooper LJN, Egeler RM, Handgretinger R, Locatelli F, Sondel PM, et al. Highlights of the First International “Immunotherapy in Pediatric Oncology: Progress and Challenges” Meeting. Journal of pediatric hematology/oncology. 2009;31(4):227–44. doi: 10.1097/MPH.0b013e31819a5d8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rassam S, Katz F, Chessells JGM. Successful allogeneic bone marrow transplantation in juvenile CML: conditioning or graft-versus-leukaemia effect? Bone Marrow Transplant. 1993;11(3):247–50. [PubMed] [Google Scholar]
- 11.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990 February 1;75(3):555–62. 1990. [PubMed] [Google Scholar]
- 12.Passweg JJR, Tiberghien PP, Cahn JJY, Vowels MMR, Camitta BBM, Gale RRP, et al. Graft-versus-leukemia effects in T lineage and B lineage acute lymphoblastic leukemia. Bone marrow transplantation. 1998;21(2):153–8. doi: 10.1038/sj.bmt.1701064. [DOI] [PubMed] [Google Scholar]
- 13.Loren AW, Porter DL. Donor leukocyte infusions for the treatment of relapsed acute leukemia after allogeneic stem cell transplantation. Bone Marrow Transplant. 2008;41(5):483–93. doi: 10.1038/sj.bmt.1705898. [DOI] [PubMed] [Google Scholar]
- 14.Maraninchi DD, Gluckman EE, Blaise DD, Guyotat DD, Rio BB, Pico JJL, et al. Impact of T-cell depletion on outcome of allogeneic bone-marrow transplantation for standard-risk leukaemias. The lancet. 1987;2(8552):175–8. doi: 10.1016/s0140-6736(87)90763-x. [DOI] [PubMed] [Google Scholar]
- 15.Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Blood. 1995 September;86(5):2041–50. see comments. 1995. [PubMed] [Google Scholar]
- 16.Sullivan KM, Weiden PL, Storb R, Witherspoon RP, Fefer A, Fisher L, et al. Influence of acute and chronic graft-versus-host disease on relapse and survival after bone marrow transplantation from HLA-identical siblings as treatment of acute and chronic leukemia[published erratum appears in Blood 1989 Aug 15;74(3):1180] Blood. 1989 May 1;73(6):1720–8. 1989. [PubMed] [Google Scholar]
- 17.Bader P, Kreyenberg H, Henze GHR, Eckert C, Reising M, Willasch A, et al. Prognostic Value of Minimal Residual Disease Quantification Before Allogeneic Stem-Cell Transplantation in Relapsed Childhood Acute Lymphoblastic Leukemia: The ALL-REZ BFM Study Group. J Clin Oncol. 2009 January 20;27(3):377–84. doi: 10.1200/JCO.2008.17.6065. 2009. [DOI] [PubMed] [Google Scholar]
- 18.Krejci O, van der Velden VHJ, Bader P, Kreyenberg H, Goulden N, Hancock J, et al. Level of minimal residual disease prior to haematopoietic stem cell transplantation predicts prognosis in paediatric patients with acute lymphoblastic leukaemia: a report of the Pre-BMT MRD Study Group. Bone Marrow Transplant. 2003;32(8):849–51. doi: 10.1038/sj.bmt.1704241. [DOI] [PubMed] [Google Scholar]
- 19.Collins RH, Shpilberg O, Drobyski WR, Porter DL, Giralt S, Champlin R, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. Journal of clinical oncology. 1997;15(2):433–44. doi: 10.1200/JCO.1997.15.2.433. [DOI] [PubMed] [Google Scholar]
- 20.Gratwohl A, Hermans J, Apperley J, Arcese W, Bacigalupo A, Bandini G, et al. Acute graft-versus-host disease: grade and outcome in patients with chronic myelogenous leukemia. Working Party Chronic Leukemia of the European Group for Blood and Marrow Transplantation. Blood. 1995 July 15;86(2):813–8. 1995. [PubMed] [Google Scholar]
- 21.Zecca M, Prete A, Rondelli R, Lanino E, Balduzzi A, Messina C, et al. Chronic graft-versus-host disease in children: incidence, risk factors, and impact on outcome. Blood. 2002 July 30;100(4):1192–200. doi: 10.1182/blood-2001-11-0059. 2002. [DOI] [PubMed] [Google Scholar]
- 22.Vago L, Perna S, Zanussi M, Mazzi B, Barlassina C, Stanghellini MTL, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. New England Journal of Medicine, The. 2009;361(5):478–88. doi: 10.1056/NEJMoa0811036. [DOI] [PubMed] [Google Scholar]
- 23.Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood. 2001 December 1;98(12):3192–204. doi: 10.1182/blood.v98.12.3192. 2001. [DOI] [PubMed] [Google Scholar]
- 24.Sierra J, Perez WS, Rozman C, Carreras E, Klein JP, Rizzo JD, et al. Bone marrow transplantation from HLA-identical siblings as treatment for myelodysplasia. Blood. 2002 August 28;100(6):1997–2004. 2002. [PubMed] [Google Scholar]
- 25.Aversa F. Haploidentical haematopoietic stem cell transplantation for acute leukaemia in adults: experience in Europe and the United States. Bone Marrow Transplant. 2008;41(5):473–81. doi: 10.1038/sj.bmt.1705966. [DOI] [PubMed] [Google Scholar]
- 26.Green A, Clarke E, Hunt L, Canterbury A, Lankester A, Hale G, et al. Children With Acute Lymphoblastic Leukemia Who Receive T-Cell-Depleted HLA Mismatched Marrow Allografts From Unrelated Donors Have an Increased Incidence of Primary Graft Failure but a Similar Overall Transplant Outcome. Blood. 1999 October 1;94(7):2236–46. 1999. [PubMed] [Google Scholar]
- 27.Bonnanomi S, Connor P, Webb D, Ancliff P, Amrolia P, Rao K, et al. Successful outcome of allo-SCT in high-risk pediatric AML using chemotherapy-only conditioning and post transplant immunotherapy. Bone Marrow Transplant. 2008;42(4):253–7. doi: 10.1038/bmt.2008.160. [DOI] [PubMed] [Google Scholar]
- 28.Cross NC, Hughes TP, Feng L, O'Shea P, Bungey J, Marks DI, et al. Minimal residual disease after allogeneic bone marrow transplantation for chronic myeloid leukaemia in first chronic phase: correlations with acute graft-versus-host disease and relapse. British journal of haematology. 1993;84(1):67–74. doi: 10.1111/j.1365-2141.1993.tb03026.x. [DOI] [PubMed] [Google Scholar]
- 29.Rezvani K, Yong ASM, Savani BN, Mielke S, Keyvanfar K, Gostick E, et al. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood. 2007 September 15;110(6):1924–32. doi: 10.1182/blood-2007-03-076844. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heslop HE, Stevenson FK, Molldrem JJ. Immunotherapy of Hematologic Malignancy. Hematology. 2003 January 1;2003(1):331–49. doi: 10.1182/asheducation-2003.1.331. 2003. [DOI] [PubMed] [Google Scholar]
- 31.Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9(5):486–94. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 32.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh CY, Blazar BR, George T, Welniak LA, Capitini CM, Raziuddin A, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood. 2001;97(10):3132–7. doi: 10.1182/blood.v97.10.3132. [DOI] [PubMed] [Google Scholar]
- 34.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 35.Davies SM, Ruggieri L, DeFor T, Wagner JE, Weisdorf DJ, Miller JS, et al. Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants. Blood. 2002 November 15;100(10):3825–7. doi: 10.1182/blood-2002-04-1197. 2002. [DOI] [PubMed] [Google Scholar]
- 36.Ruggeri LL, Mancusi AA, Burchielli EE, Aversa FF, Martelli MFMF, Velardi AA. Natural killer cell alloreactivity in allogeneic hematopoietic transplantation. Current opinion in oncology. 2007;19(2):142–7. doi: 10.1097/CCO.0b013e3280148a1a. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg SSA, White DDE. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy. Journal of immunotherapy with emphasis on tumor immunology. 1996;19(1):81–4. [PubMed] [Google Scholar]
- 39.Alanko S, Salmi TT, Pelliniemi TT. Recovery of blood T-cell subsets after chemotherapy for childhood acute lymphoblastic leukemia. Pediatric hematology and oncology. 1994;11(3):281–92. doi: 10.3109/08880019409141671. [DOI] [PubMed] [Google Scholar]
- 40.Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. The New England journal of medicine. 1995;332(3):143–9. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 41.Schuster SJ, Neelapu SS, Gause BL, Muggia FM, Gockerman JP, Sotomayor EM, et al. Idiotype vaccine therapy (BiovaxID) in follicular lymphoma in first complete remission: Phase III clinical trial results. J Clin Oncol. 2009 June 20;27(18S):2. Meeting Abstracts. 2009. [Google Scholar]
- 42.Celestia SH, Paul FS, Eric JS, Patrick AB, John N, Lianng Y, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;9999(9999) doi: 10.1002/cncr.24429. NA. [DOI] [PubMed] [Google Scholar]
- 43.Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood. 2007 October 15;110(8):2838–45. doi: 10.1182/blood-2007-05-091280. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, et al. Vaccination Of Pediatric Solid Tumor Patients with Tumor Lysate-pulsed Dendritic Cells Can Expand Specific T Cells and Mediate Tumor Regression. Cancer Res. 2001 December 1;61(23):8513–9. 2001. [PubMed] [Google Scholar]
- 45.Geiger Jd, Hutchinson RR, Hohenkirk LL, McKenna EE, Chang AA, Mulé JJ. Treatment of solid tumours in children with tumour-lysate-pulsed dendritic cells. The lancet. 2000;356(9236):1163–5. doi: 10.1016/S0140-6736(00)02762-8. [DOI] [PubMed] [Google Scholar]
- 46.Bowman L, Grossmann M, Rill D, Brown M, Zhong Wy, Alexander B, et al. IL-2 Adenovector-Transduced Autologous Tumor Cells Induce Antitumor Immune Responses in Patients With Neuroblastoma. Blood. 1998 September 15;92(6):1941–9. 1998. [PubMed] [Google Scholar]
- 47.Rousseau RF, Haight AE, Hirschmann-Jax C, Yvon ES, Rill DR, Mei Z, et al. Local and systemic effects of an allogeneic tumor cell vaccine combining transgenic human lymphotactin with interleukin-2 in patients with advanced or refractory neuroblastoma. Blood. 2003 March 1;101(5):1718–26. doi: 10.1182/blood-2002-08-2493. 2003. [DOI] [PubMed] [Google Scholar]
- 48.Russell HV, Strother D, Mei Z, Rill D, Popek E, Biagi E, et al. Phase I trial of vaccination with autologous neuroblastoma tumor cells genetically modified to secrete IL-2 and lymphotactin. Journal of immunotherapy. 2007;30(2):227–33. doi: 10.1097/01.cji.0000211335.14385.57. [DOI] [PubMed] [Google Scholar]
- 49.Simon RM, Steinberg SM, Hamilton M, Hildesheim A, Khleif S, Kwak LW, et al. Clinical Trial Designs for the Early Clinical Development of Therapeutic Cancer Vaccines. J Clin Oncol. 2001 March 15;19(6):1848–54. doi: 10.1200/JCO.2001.19.6.1848. 2001. [DOI] [PubMed] [Google Scholar]
- 50.Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A Pilot Study of Consolidative Immunotherapy in Patients with High-Risk Pediatric Sarcomas. Clin Cancer Res. 2008 August 1;14(15):4850–8. doi: 10.1158/1078-0432.CCR-07-4065. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merchant M, Melchionda F, Sinha M, Khanna C, Helman L, Mackall C. Immune reconstitution prevents metastatic recurrence of murine osteosarcoma. Cancer immunology, immunotherapy. 2007;56(7):1037–46. doi: 10.1007/s00262-006-0257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of Highly Cytotoxic Human Natural Killer Cells for Cancer Cell Therapy. Cancer Res. 2009 May 1;69(9):4010–7. doi: 10.1158/0008-5472.CAN-08-3712. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kershaw MH, Teng MWL, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5(12):928–40. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 54.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular therapy. 2007;15(4):825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 56.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. 1998 June 1;16(6):2169–80. doi: 10.1200/JCO.1998.16.6.2169. 1998. [DOI] [PubMed] [Google Scholar]
- 58.Kushner BH, Kramer K, Cheung NKV. Phase II Trial of the Anti-GD2 Monoclonal Antibody 3F8 and Granulocyte-Macrophage Colony-Stimulating Factor for Neuroblastoma. J Clin Oncol. 2001 November 15;19(22):4189–94. doi: 10.1200/JCO.2001.19.22.4189. 2001. [DOI] [PubMed] [Google Scholar]
- 59.Osenga KL, Hank JA, Albertini MR, Gan J, Sternberg AG, Eickhoff J, et al. A Phase I Clinical Trial of the hu14.18-IL2 (EMD 273063) as a Treatment for Children with Refractory or Recurrent Neuroblastoma and Melanoma: a Study of the Children's Oncology Group. Clin Cancer Res. 2006 March 15;12(6):1750–9. doi: 10.1158/1078-0432.CCR-05-2000. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Modak S, Cheung NK. Disialoganglioside directed immunotherapy of neuroblastoma. Cancer investigation. 2007;25(1):67–77. doi: 10.1080/07357900601130763. [DOI] [PubMed] [Google Scholar]
- 61.Ozkaynak MF, Sondel PM, Krailo MD, Gan J, Javorsky B, Reisfeld RA, et al. Phase I Study of Chimeric Human/Murine Anti-Ganglioside GD2 Monoclonal Antibody (ch14.18) With Granulocyte-Macrophage Colony-Stimulating Factor in Children With Neuroblastoma Immediately After Hematopoietic Stem-Cell Transplantation: A Children's Cancer Group Study. J Clin Oncol. 2000 December 15;18(24):4077–85. doi: 10.1200/JCO.2000.18.24.4077. 2000. [DOI] [PubMed] [Google Scholar]
- 62.Schusterman S, London W, Gillies S, Hank J, Voss S, Seeger R, et al. Anti-neuroblastoma activity of hu14.18-IL2 against minimal residual disease in a Children's Oncology Group (COG) phase II study. J Clin Oncol. 2008;26(May 20 suppl) doi: 10.1200/JCO.2009.27.8861. abstr 3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheung NK, Kushner BH, Cheung IY, Kramer K, Canete A, Gerald W, et al. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. J Clin Oncol. 1998 September 1;16(9):3053–60. doi: 10.1200/JCO.1998.16.9.3053. 1998. [DOI] [PubMed] [Google Scholar]
- 64.Yu A, Gilman A, Ozkaynak M, London W, Kreissman S, Chen H, et al. A phase III randomized trial of the chimeric anti-GD2 antibody ch14.18 with GM-CSF and IL2 as immunotherapy following dose intensive chemotherapy for high-risk neuroblastoma: Children's Oncology Group (COG) study ANBL0032. J Clin Oncol. 2009;27(15s) abstr 10067z. [Google Scholar]
- 65.Kramer K, Humm JL, Souweidane MM, Zanzonico PB, Dunkel IJ, Gerald WL, et al. Phase I Study of Targeted Radioimmunotherapy for Leptomeningeal Cancers Using Intra-Ommaya 131-I-3F8. J Clin Oncol. 2007 December 1;25(34):5465–70. doi: 10.1200/JCO.2007.11.1807. 2007. [DOI] [PubMed] [Google Scholar]
- 66.Gorlick R, Huvos AG, Heller G, Aledo A, Beardsley GP, Healey JH, et al. Expression of HER2/erbB-2 Correlates With Survival in Osteosarcoma. J Clin Oncol. 1999 September 1;17(9):2781. doi: 10.1200/JCO.1999.17.9.2781. 1999. [DOI] [PubMed] [Google Scholar]
- 67.Scotlandi K, Manara MC, Hattinger CM, Benini S, Perdichizzi S, Pasello M, et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing's sarcoma. European journal of cancer. 2005;41(9):1349–61. doi: 10.1016/j.ejca.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 68.Rehwald U, Schulz H, Reiser M, Sieber M, Staak JO, Morschhauser F, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood. 2003 January 15;101(2):420–4. doi: 10.1182/blood.V101.2.420. 2003. [DOI] [PubMed] [Google Scholar]
- 69.Ekstrand BC, Lucas JB, Horwitz SM, Fan Z, Breslin S, Hoppe RT, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood. 2003 June 1;101(11):4285–9. doi: 10.1182/blood-2002-08-2644. 2003. [DOI] [PubMed] [Google Scholar]
- 70.Movva S, Verschraegen C. The monoclonal antibody to cytotoxic T lymphocyte antigen 4, ipilimumab (MDX-010), a novel treatment strategy in cancer management. Expert Opinion on Biological Therapy. 2009;9(2):231–41. doi: 10.1517/14712590802643347. [DOI] [PubMed] [Google Scholar]
- 71.David HL. The promise of 4-1BB (CD137)-mediated immunomodulation and the immunotherapy of cancer. Immunological Reviews. 2008;222(1):277–86. doi: 10.1111/j.1600-065X.2008.00621.x. [DOI] [PubMed] [Google Scholar]
- 72.Benesch MM, Windelberg MM, Sauseng WW, Witt VV, Fleischhack GG, Lackner HH, et al. Compassionate use of bevacizumab (Avastin) in children and young adults with refractory or recurrent solid tumors. Annals of Oncology. 2008;19(4):807–13. doi: 10.1093/annonc/mdm510. [DOI] [PubMed] [Google Scholar]
- 73.Bender JLG, Adamson PC, Reid JM, Xu L, Baruchel S, Shaked Y, et al. Phase I Trial and Pharmacokinetic Study of Bevacizumab in Pediatric Patients With Refractory Solid Tumors: A Children's Oncology Group Study. J Clin Oncol. 2008 January 20;26(3):399–405. doi: 10.1200/JCO.2007.11.9230. 2008. [DOI] [PubMed] [Google Scholar]
- 74.Kolb EAEA, Gorlick RR, Houghton PJPJ, Morton CLCL, Lock RR, Carol HH, et al. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the pediatric preclinical testing program. Pediatric blood & cancer. 2008;50(6):1190–7. doi: 10.1002/pbc.21450. [DOI] [PubMed] [Google Scholar]
- 75.Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, et al. Phase 1 and Pharmacokinetic Study of Lexatumumab in Patients with Advanced Cancers. Clin Cancer Res. 2007 October 15;13(20):6187–94. doi: 10.1158/1078-0432.CCR-07-0950. 2007. [DOI] [PubMed] [Google Scholar]
- 76.Wilson WWH. Chemotherapy sensitization by rituximab: experimental and clinical evidence. Seminars in oncology. 2000;27(6 Suppl 12):30–6. [PubMed] [Google Scholar]
- 77.Cooney-Qualter E, Krailo M, Angiolillo A, Fawwaz RA, Wiseman G, Harrison L, et al. A Phase I Study of 90Yttrium-Ibritumomab-Tiuxetan in Children and Adolescents with Relapsed/Refractory CD20-Positive Non Hodgkin's Lymphoma: A Children's Oncology Group Study. Clin Cancer Res. 2007 September 15;13(18):5652s–60. doi: 10.1158/1078-0432.CCR-07-1060. 2007. [DOI] [PubMed] [Google Scholar]
- 78.Schnell R, Borchmann P, Staak JO, Schindler J, Ghetie V, Vitetta ES, et al. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin's lymphoma. Ann Oncol. 2003 May 1;14(5):729–36. doi: 10.1093/annonc/mdg209. 2003. [DOI] [PubMed] [Google Scholar]
- 79.Wayne A, Kreitman R, Pastan I. Monoclonal antibodies and immunotoxins as new therapeutic agents for childhood acute lymphoblastic leukemia. Alexandria: 2007. [Google Scholar]
- 80.Aplenc R, Alonzo TA, Gerbing RB, Lange BJ, Hurwitz CA, Wells RJ, et al. Safety and efficacy of gemtuzumab ozogamicin in combination with chemotherapy for pediatric acute myeloid leukemia: a report from the Children's Oncology Group. Journal of clinical oncology. 2008;26(14):2390–3295. doi: 10.1200/JCO.2007.13.0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arceci RJ, Sande J, Lange B, Shannon K, Franklin J, Hutchinson R, et al. Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33+ acute myeloid leukemia. Blood. 2005 August 15;106(4):1183–8. doi: 10.1182/blood-2004-10-3821. 2005. [DOI] [PubMed] [Google Scholar]
- 82.Zwaan C, Reinhardt D, Jurgens H, Huismans DR, Hahlen K, Smith OP, et al. Gemtuzumab ozogamicin in pediatric CD33-positive acute lymphoblastic leukemia: first clinical experiences and relation with cellular sensitivity to single agent calicheamicin. Leukemia. 2003;17(2):468–70. doi: 10.1038/sj.leu.2402749. [DOI] [PubMed] [Google Scholar]
- 83.Brandau S, Suttmann H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: A success story with room for improvement. Biomedicine & Pharmacotherapy. 2007;61(6):299–305. doi: 10.1016/j.biopha.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 84.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13(5):552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 85.Dudek AZ, Yunis C, Harrison LI, Kumar S, Hawkinson R, Cooley S, et al. First in Human Phase I Trial of 852A, a Novel Systemic Toll-like Receptor 7 Agonist, to Activate Innate Immune Responses in Patients with Advanced Cancer. Clin Cancer Res. 2007 December 1;13(23):7119–25. doi: 10.1158/1078-0432.CCR-07-1443. 2007. [DOI] [PubMed] [Google Scholar]
- 86.Mori K, Ando K, Heymann D. Liposomal muramyl tripeptide phosphatidyl ethanolamine: a safe and effective agent against osteosarcoma pulmonary metastases. Expert Review of Anticancer Therapy. 2008;8(2):151–9. doi: 10.1586/14737140.8.2.151. [DOI] [PubMed] [Google Scholar]
- 87.Meyers PA, Schwartz CL, Krailo MD, Healey JH, Bernstein ML, Betcher D, et al. Osteosarcoma: The Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival--A Report From the Children's Oncology Group. J Clin Oncol. 2008 February 1;26(4):633–8. doi: 10.1200/JCO.2008.14.0095. 2008. [DOI] [PubMed] [Google Scholar]
- 88.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 89.Roses RE, Xu M, Koski GK, Czerniecki BJ. Radiation therapy and Toll-like receptor signaling: implications for the treatment of cancer. Oncogene. 2008;27(2):200–7. doi: 10.1038/sj.onc.1210909. [DOI] [PubMed] [Google Scholar]
- 90.Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2007;15(1):3–12. doi: 10.1038/sj.cdd.4402269. [DOI] [PubMed] [Google Scholar]
- 91.Riuzzi F, Sorci G, Donato R. RAGE Expression in Rhabdomyosarcoma Cells Results in Myogenic Differentiation and Reduced Proliferation, Migration, Invasiveness, and Tumor Growth. Am J Pathol. 2007 September 1;171(3):947–61. doi: 10.2353/ajpath.2007.070049. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A Phase I Study of In vitro Expanded Natural Killer T Cells in Patients with Advanced and Recurrent Non-Small Cell Lung Cancer. Clin Cancer Res. 2006 October 15;12(20):6079–86. doi: 10.1158/1078-0432.CCR-06-0114. 2006. [DOI] [PubMed] [Google Scholar]
- 93.Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A Phase I Study of {alpha}-Galactosylceramide (KRN7000)-Pulsed Dendritic Cells in Patients with Advanced and Recurrent Non-Small Cell Lung Cancer. Clin Cancer Res. 2005 March 1;11(5):1910–7. doi: 10.1158/1078-0432.CCR-04-1453. 2005. [DOI] [PubMed] [Google Scholar]
- 94.Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9(1):28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 95.Bennouna J, Bompas E, Neidhardt EM, Rolland F, Philip I, Galéa C, et al. Phase-I study of Innacell gammadelta, an autologous cell-therapy product highly enriched in gamma9delta2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer immunology, immunotherapy. 2008;57(11):1599–609. doi: 10.1007/s00262-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roper M, Smith MA, Sondel PM, Gillespie A, Reaman GH, Hammond GD, et al. A phase I study of interleukin-2 in children with cancer. Journal of pediatric hematology/oncology. 1992;14(4):305–11. [PubMed] [Google Scholar]
- 97.Bauer M, Reaman GH, Hank JA, Cairo MS, Anderson P, Blazar BR, et al. A phase II trial of human recombinant interleukin-2 administered as a 4-day continuous infusion for children with refractory neuroblastoma, non-Hodgkin's lymphoma, sarcoma, renal cell carcinoma, and malignant melanoma. A Childrens Cancer Group study Cancer. 1995;75(12):2959–65. doi: 10.1002/1097-0142(19950615)75:12<2959::aid-cncr2820751225>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 98.Kalwak K, Ussowicz M, Gorczyńska E, Turkiewicz D, Toporski J, Dobaczewski G, et al. Immunologic effects of intermediate-dose IL-2 i.v. after autologous hematopoietic cell transplantation in pediatric solid tumors. Journal of interferon & cytokine research. 2003;23(4):173–81. doi: 10.1089/107999003765027375. [DOI] [PubMed] [Google Scholar]
- 99.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+ regulatory T cells. Nature medicine. 2005;11(11):1238–43. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 100.Fry TJ, Mackall CL. Interleukin-7: from bench to clinic. Blood. 2002 May 13;99(11):3892–904. doi: 10.1182/blood.v99.11.3892. 2002. [DOI] [PubMed] [Google Scholar]
- 101.Rosenberg SA, Sportès C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. Journal of immunotherapy. 2006;29(3):313–9. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J Exp Med. 2008 July 7;205(7):1701–14. doi: 10.1084/jem.20071681. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y, Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. The journal of clinical investigation. 2005;115(5):1177–87. doi: 10.1172/JCI23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andorsky DJ, Timmerman JM. Interleukin-21: biology and application to cancer therapy. Expert Opinion on Biological Therapy. 2008;8(9):1295–307. doi: 10.1517/14712598.8.9.1295. [DOI] [PubMed] [Google Scholar]
- 105.Navid F, Furman WL, Fleming M, Rao BN, Kovach S, Billups CA, et al. The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer. 2005;103(4):780–7. doi: 10.1002/cncr.20860. [DOI] [PubMed] [Google Scholar]
- 106.Merchant MS, Yang X, Melchionda F, Romero M, Klein R, Thiele CJ, et al. Interferon {gamma} Enhances the Effectiveness of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Receptor Agonists in a Xenograft Model of Ewing's Sarcoma. Cancer Res. 2004 November 15;64(22):8349–56. doi: 10.1158/0008-5472.CAN-04-1705. 2004. [DOI] [PubMed] [Google Scholar]
- 107.Seibel NL, Dinndorf PA, Bauer M, Sondel PM, Hammond GD, Reaman GH. Phase I study of tumor necrosis factor-alpha and actinomycin D in pediatric patients with cancer: a Children's Cancer Group study. Journal of immunotherapy with emphasis on tumor immunology. 1994;16(2):125–31. doi: 10.1097/00002371-199408000-00006. [DOI] [PubMed] [Google Scholar]
- 108.Baek JH, Sohn SK, Kim DH, Kim JG, Yang DH, Kim YK, et al. Pilot Remission Induction Therapy with Idarubicin, plus an Intensified Dose of Ara-C and Priming with Granulocyte Colony-Stimulating Factor for Acute Myeloid Leukemia. Acta Haematologica. 2007;117(2):109–14. doi: 10.1159/000097386. [DOI] [PubMed] [Google Scholar]
- 109.Thomas X, Raffoux E, Botton Sd, Pautas C, Arnaud P, de Revel T, et al. Effect of priming with granulocyte-macrophage colony-stimulating factor in younger adults with newly diagnosed acute myeloid leukemia: a trial by the Acute Leukemia French Association (ALFA) Group. Leukemia. 2007;21(3):453–61. doi: 10.1038/sj.leu.2404521. [DOI] [PubMed] [Google Scholar]
- 110.Arellano ML, Langston A, Winton E, Flowers CR, Waller EK. Treatment of relapsed acute leukemia after allogeneic transplantation: a single center experience. Biology of blood and marrow transplantation. 2007;13(1):116–23. doi: 10.1016/j.bbmt.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 111.Zhang WG, Liu SH, Cao XM, Cheng YX, Ma XR, Yang Y, et al. A phase-I clinical trial of active immunotherapy for acute leukemia using inactivated autologous leukemia cells mixed with IL-2, GM-CSF, and IL-6. Leukemia Research. 2005;29(1):3–9. doi: 10.1016/j.leukres.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 112.Anderson PM. Aerosol granulocyte macrophage-colony stimulating factor: A low toxicity, lung-specific biological therapy in patients with lung metastases. Clinical cancer research. 1999;5(9):2316–23. [PubMed] [Google Scholar]
- 113.Rao RD. Aerosolized granulocyte macrophage colony-stimulating factor (GM-CSF) therapy in metastatic cancer. American journal of clinical oncology: cancer clinical trials. 2003;26(5):493–8. doi: 10.1097/01.coc.0000037664.04141.D0. [DOI] [PubMed] [Google Scholar]
- 114.Negrier S, Michon J, Floret D, Bouffet E, Gentet JC, Philip I, et al. Interleukin-2 and lymphokine-activated killer cells in 15 children with advanced metastatic neuroblastoma [published erratum appears in J Clin Oncol 1992 Jun;10(6):1026] J Clin Oncol. 1991 August 1;9(8):1363–70. doi: 10.1200/JCO.1991.9.8.1363. 1991. [DOI] [PubMed] [Google Scholar]
- 115.Casper JT, Camitta BM, McOlash L, Kirchner P, Piaskowski V, Truitt RL. Immunological evaluation of pediatric cancer patients receiving recombinant interleukin-2 in a phase I trial. Journal of immunotherapy. 1992;11(4):274–85. doi: 10.1097/00002371-199205000-00006. [DOI] [PubMed] [Google Scholar]
- 116.Finn O. Cancer immunology. New England Journal of Medicine, The. 2008;358(25):2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 117.Johnson BD, Jing W, Orentas RJ. CD25+ regulatory T cell inhibition enhances vaccine-induced immunity to neuroblastoma. Journal of immunotherapy. 2007;30(2):203–14. doi: 10.1097/01.cji.0000211336.91513.dd. [DOI] [PubMed] [Google Scholar]
- 118.Berzofsky JA, Terabe M. NKT Cells in Tumor Immunity: Opposing Subsets Define a New Immunoregulatory Axis. J Immunol. 2008 March 15;180(6):3627–35. doi: 10.4049/jimmunol.180.6.3627. 2008. [DOI] [PubMed] [Google Scholar]
- 119.Kumar V, Halder R, Arrenberg P. Cross-regulation between distinct natural killer T cell subsets influences immune response to self and foreign antigens. Journal of cellular physiology. 2009;218(2):246–50. doi: 10.1002/jcp.21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer and Metastasis Reviews. 2006;25(3):315–22. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 121.Mayumi O. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Science. 2008;99(8):1501–6. doi: 10.1111/j.1349-7006.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vakkila J, Jaffe R, Michelow M, Lotze MT. Pediatric Cancers Are Infiltrated Predominantly by Macrophages and Contain a Paucity of Dendritic Cells: a Major Nosologic Difference with Adult Tumors. Clin Cancer Res. 2006 April 1;12(7):2049–54. doi: 10.1158/1078-0432.CCR-05-1824. 2006. [DOI] [PubMed] [Google Scholar]
- 123.Ostrand-Rosenberg S, Sinha P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol. 2009 April 15;182(8):4499–506. doi: 10.4049/jimmunol.0802740. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]