

Abstract
Purpose
Solid tumors contain microenvironmental regions of hypoxia that present a barrier to traditional radiotherapy and chemotherapy, and this work describes a novel approach to circumvent hypoxia. We propose to overcome hypoxia by augmenting the effectiveness of drugs that are designed to specifically kill hypoxic tumor cells.
Experimental Design
We have constructed RKO colorectal tumor cells that express a small RNA hairpin that specifically knocks down the hypoxia-inducible factor 1a (HIF1a) transcription factor. We have used these cells in vitro to determine the effect of HIF1 on cellular sensitivity to the hypoxic cytotoxin PR-104, and its role in cellular oxygen consumption in response to the pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA). We have further used these cells in vivo in xenografted tumors to determine the role of HIF1 in regulating tumor hypoxia in response to DCA using 18F-fluoroazomycin arabinoside positron emission tomography, and its role in regulating tumor sensitivity to the combination of DCA and PR-104.
Results
HIF1 does not affect cellular sensitivity to PR-104 in vitro. DCA transiently increases cellular oxygen consumption in vitro and increases the extent of tumor hypoxia in vivo as measured with 18F-fluoroazomycin arabinoside positron emission tomography. Furthermore, we show that DCA-dependent alterations in hypoxia increase the antitumor activity of the next-generation hypoxic cytotoxin PR-104.
Conclusions
DCA interferes with the HIF-dependent “adaptive response,” which limits mitochondrial oxygen consumption. This approach transiently increases tumor hypoxia and represents an important method to improve antitumor efficacy of hypoxia-targeted agents, without increasing toxicity to oxygenated normal tissue.
Hypoxia is a common feature of solid tumors. It results from an inadequate supply of oxygen to meet the demands of tumor cells (1). The malformed vasculature of solid tumors produces chronic spatial heterogeneity in oxygen concentrations termed diffusion-limited hypoxia. This is further complicated by temporal instability in blood flow which leads to intermittent periods of perfusion-dependent hypoxia. Clinically, hypoxia is associated with poor patient outcome and metastasis in a variety of tumor types (1, 2). Although there have been many clinical trials aimed at improving tumor oxygenation, none have shown significant enough effects to warrant their adoption into standard clinical practice. An alternative approach to the problem is to exploit tumor hypoxia by developing therapies that target this unique condition, such as the hypoxic cytotoxins (3, 4).
The first hypoxic cytotoxin to enter clinical trials was tirapazamine (5). In advanced trials, tirapazamine has been shown to be most effective in patients with hypoxic tumors as assessed by hypoxia-sensitive positron emission tomography (PET) imaging (6). One second-generation hypoxic cytotoxin is PR-104, a dinitrobenzimide mustard that undergoes bioreduction under severe hypoxia to produce reactive nitrogen mustard metabolites that damage DNA (7). Although the prodrug PR-104A has some toxicity, the activation of PR-104A to its more toxic form is confined to lower oxygen concentrations compared with tirapazamine (7). The activated cytotoxic metabolites are thought to diffuse from hypoxic cells, creating a bystander effect, and resulting in significant single-agent activity in model tumors (7).
Because PR-104 relies on severe hypoxia for its enhanced cytotoxicity, it is predicted that increasing tumor hypoxia during drug exposure will provide a therapeutic benefit. Our group (8, 9), along with others (10), has shown that under hypoxic conditions, the transcription factor hypoxia-inducible factor 1 (HIF1) causes an increase in pyruvate dehydrogenase kinase (PDK), which leads to a reduction in pyruvate flux through the mitochondria, decreasing oxygen consumption. Interfering with this adaptive response transiently increases tumor oxygen consumption and thereby increases tumor hypoxia (11). These data are consistent with mathematical models of tumor oxygenation predicting that even modest changes in oxygen consumption will have a profound effect on tumor oxygenation (12, 13). We have therefore tested whether manipulating tumor oxygen consumption using the PDK inhibitor dichloroacetate (DCA; ref. 14) is able to increase the uptake of hypoxia-dependent radiotracers, and increase the effectiveness of PR-104 in model tumors.
Materials and Methods
Cell lines and tumor xenografts
RKO and RKOshHIF1α cell lines have been previously described (9). The pre-prodrug PR-104 was a gift from the Proacta Corporation (Auckland, New Zealand). The PR-104A prodrug was a gift from William Denny (Auckland, New Zealand). DCA was purchased from Acros. Tumor xenografts were established by injecting 5 × 106 cells s.c. into the flanks or shoulders of 6- to 8-week-old nude mice. Caliper measurements of two perpendicular diameters were used to monitor tumor growth [volume = (d1)(d2)2 (0.52)]. All animal protocols were approved by the Stanford Administrative Panel on Laboratory Animal Care. For clonogenic survival, cells were plated in 6 cm dishes, and 6 h later, the dishes were placed in a normoxic incubator or a hypoxic chamber at <0.02% O2 (Sheldon Corp., Cornelius, OR). Pro-drug PR-104A was added in pre-equilibrated medium for the indicated times and then removed. Dishes were re-fed with standard medium and placed in a normoxic incubator for colony formation.
Oxygen consumption
Oxygen consumption measurements were carried out using an XF24 extracellular flux analyzer (Seahorse Biosciences). This device uses fluorescence-based optical sensors and custom multi-well plates to make repeated oxygen consumption measurements of intact cells growing as monolayers. Drug was added automatically during measurement. After establishing baseline oxygen consumption rates, DCA was introduced (five wells per treatment) and measurements continued for 60 to 80 min. Data was acquired from three replicate plates per cell line.
PET imaging
PET imaging was carried out using a micro-PET R4 rodent model scanner (CTI Concorde Microsystems). Tumor-bearing mice (250-1,000 mm3) were injected i.v. with 9.25 MBq (250 μCi) of 18F-fluoroazomycin arabinoside (18F-FAZA) or 7.4 MBq (200 μCi) 18F-fluorodeoxyglucose (18F-FDG) via the tail vein. Three hours after injection (18F-FAZA) or 1 h after injection (18F-FDG), mice were anaesthetized using isofluorane inhalation and positioned in the PET scanner. Scans were acquired and the data was reconstructed using the two-dimensional ordered-subsets expectation maximization algorithm. Quantitation was carried out using ASIPro VM software (CTI Concorde Microsystems). Tumor volumes were manually delineated, and the signal was measured relative to a 5-mm diameter region in the thorax. 18F-FDG and 18F-FAZA were produced at the Stanford Cyclotron Facility, using established protocols.
Data analysis
Changes were analyzed by ANOVA, followed by pair-wise comparisons using a two-tailed Student’s t test. For FAZA uptake, in which animals served as their own controls, paired t tests were done. The χ2 test was used to determine the difference in the proportion of complete responses to therapy. For all data, P < 0.05 were considered significant. Error bars represent the SEM.
Results
To quantify the hypoxic selectivity of PR-104A in RKO cells, and to determine the influence of HIF1 function on toxicity, colony formation was measured using RKO and RKOshHIF cells exposed to PR-104A under normoxic or hypoxic conditions. RKOshHIF cells have been stably transfected with a plasmid expressing short hairpin RNA against HIF1α, which reduces >90% of the HIF1α protein in control cells (9, 11). RKO cells do not have detectable HIF2α protein (data not shown). Figure 1 shows that at either time point, hypoxic cells were far more sensitive to the drug, and that HIF1 function had no effect on toxicity. The hypoxic cytotoxicity ratio (the amount of drug required to kill an equivalent proportion of cells under normoxic conditions relative to hypoxic conditions) was approximately 10, which is consistent with reported data (7).
Fig. 1.

Hypoxic cytotoxicity of PR-104A in RKO and RKOshHIF cells. Clonogenic survival curves for sparsely plated RKO (A) and RKOshHIF (B) cells exposed to increasing concentrations of PR-104A for either 4 or 24 h under normoxic or hypoxic (<0.02% O2) conditions.
DCA acts as a pyruvate analogue to inhibit the PDK enzymes with a range of efficiencies, depending on the particular PDK isoform (14, 15). This causes an increase in pyruvate dehydrogenase activity and carbohydrate metabolism in the mitochondria (14). We examined the effect of DCA on oxygen consumption in vitro using the Seahorse Biosciences XF24 device. This instrument allows for repeated measurements of oxygen consumption in intact cell monolayers. DCA transiently increased oxygen consumption in a dose-dependent manner in RKO and RKOshHIF cells (Fig. 2). Although the experiments were carried out under normoxic conditions, the effect of DCA was less pronounced in the RKOshHIF cells.
Fig. 2.
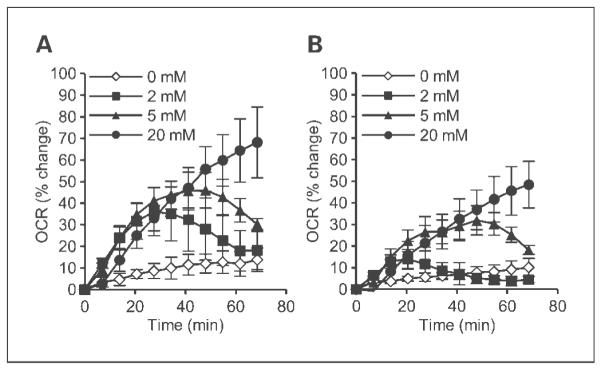
DCA causes a dose-dependent increase in oxygen consumption of tumor cells in vitro. The oxygen consumption rate (OCR) was measured using the XF24 extracellular flux analyzer following the addition of increasing concentrations of DCA in RKO (A) and RKOshHIF (B) tumor cells growing as monolayers.
To determine the effect of DCA administration on tumor oxygenation, we used the hypoxia-sensitive PET tracer 18F-FAZA to noninvasively measure tumor hypoxia. 18F-FAZA is reduced under hypoxic conditions to a reactive species that binds to intra-cellular macromolecules and provides a quantitative measure of viable hypoxic tissue (16). To establish the sensitivity of this technique, animals bearing RKO and RKOshHIF tumors were exposed to a reduced oxygen environment. On day 1, baseline 18F-FAZA measurements were made on all tumors under normoxic breathing conditions. The following day, repeat 18F-FAZA measurements were made on the same tumors while the mice were breathing a defined gas mixture of 7% O2. Figure 3A shows that low oxygen breathing significantly increased the 18F-FAZA signal in the tumors but also in the normal tissue. This is consistent with previous measurements showing that this breathing protocol lowers both tumor and normal tissue pO2 (17). To determine the effect of DCA on FAZA uptake, a similar set of animals were imaged. On the first day, baseline 18F-FAZA uptake was measured. On the following day, the same tumors were measured 1 h after injection of 50 mg/kg of DCA i.p. The mean 18F-FAZA intensity of the tumors was determined relative to a control region in the thorax (Fig. 3B and C). DCA caused a significant increase in tumor hypoxia that was comparable to breathing 7% oxygen, and was more pronounced in the RKO tumors. There was no significant difference between the RKO and RKOshHIF tumors after DCA treatment at the time point investigated. After 1 wk, animals were imaged again, and tumor hypoxia had returned to baseline levels. A second DCA treatment at this time point produced a similar increase in tumor hypoxia (Supplementary Fig. S1). Animals were also imaged using standard 18F-FDG PET to assess glucose uptake, which might be expected to decrease when cells shift their carbohydrate metabolism towards oxidative phosphorylation. We observed that RKO tumors had a slightly higher baseline 18F-FDG uptake, and that DCA treatment produced a small decrease in this signal (Supplementary Fig. S2).
Fig. 3.

DCA increases tumor hypoxia in vivo. A, mean 18F-FAZA activity in RKO and RKOshHIF tumors and thorax (normal tissue control) on two consecutive days. During tracer uptake, mice were placed in normoxic environment (day 1) or hypoxic (7% O2) environment (day 2; n = 5). B, mean tumor/background (thorax) 18F-FAZA signal in RKO and RKOshHIF tumors. On day 1, mice were treated with PBS as control. On day 2, mice were treated with 50 mg/kg of DCA 1 h prior to tracer injection (n = 11). C, representative 18F-FAZA PET image showing a transverse section of a tumor-bearing mouse imaged after PBS injection (left), and DCA injection (right). *, statistically significant differences.
To test the hypothesis that the increase in tumor hypoxia as measured by PET would lead to increased efficacy of the hypoxic cytotoxin PR-104, we used a standard growth delay assay to evaluate this drug combination. DCA treatment prior to PR-104 administration significantly increased the tumor growth delay in RKO tumors compared with either drug alone (Fig. 4A). Some animals showed a complete response, and this was also significantly increased by pretreatment with DCA (Fig. 4B). There was no effect of DCA on the activity of PR-104 against RKOshHIF tumors, consistent with the HIF-dependent suppression of oxygen consumption via upregulation of PDK enzymes (Fig. 4C and D). One dose-limiting toxicity for hypoxic cytotoxins is the bone marrow (7). Therefore, we tested whether DCA pretreatment further decreased the WBC counts in experimental mice. Figure 4E shows that PR-104 alone caused a decrease in WBCs. DCA treatment did not increase either bone marrow toxicity or FAZA uptake in normal tissue, suggesting that its effects were primarily in hypoxic tumor tissue, and/or that normal tissue vasculature could respond with increased oxygen delivery when needed.
Fig. 4.

DCA pretreatment potentiates PR-104 antitumor activity without increasing normal tissue toxicity. RKO (A and B) and RKOshHIF (C and D) tumor-bearing mice were treated with 50 mg/kg of DCA i.p., 308 mg/kg of PR-104 i.p., 50 mg/kg of DCA followed 2 h later by 308 mg/kg of PR-104, or PBS control q4dx3 (n = 6-10). A and C, tumor growth over time; B and D, animal survival (tripling of initial tumor volume) following treatment. *, P < 0.05 for the DCA + PR-104 group compared with the PR-104 alone group (A). P = 0.051 based on a χ2 test of the proportion of mice surviving in the DCA + PR-104 group compared with the PR-104 alone group (B). E, WBC counts of mice treated on the same schedule as above.
Discussion
Hypoxic cytotoxins represent a class of therapeutics designed to take advantage of the unique physiology of solid tumors. By targeting the hypoxic regions of tumors that are most resistant to radiation and chemotherapies, these drugs should produce greater than additive benefits when combined with standard therapies. PR-104 is a new hypoxic cytotoxin (7) with different characteristics relative to tirapazamine (3). PR-104 has modest activity in normoxia, and some effect against model tumors as a single agent.
There are opportunities to manipulate tumor physiology to increase the effectiveness of these drugs. Here, we have shown one strategy for improving the effectiveness of PR-104, and potentially other similar agents. Consistent with our previous work (11), we show here that thwarting the HIF-dependent adaptive response using the PDK inhibitor DCA causes a transient increase in oxygen consumption and results in a temporary increase in tumor hypoxia. We now show that this increase in hypoxia can be measured by the clinically tested hypoxic radio-tracer 18F-FAZA, and this leads to improved effectiveness of PR-104. Importantly, we saw no evidence of increased FAZA uptake in the normal tissues of the animals, nor did we see clinical evidence of increased normal tissue toxicity (Fig. 4). DCA is a clinically approved drug with very few side effects (18), and established transient pharmacokinetics. Additionally, other reports suggest that DCA may have direct antitumor effects in other models (19), making it a promising therapeutic candidate.
It has become clear during the clinical testing of tirapazamine, that patient selection will be critical to evaluate the maximal benefit of hypoxic cytotoxins. Hypoxic radiotracer uptake in humans (6) and animal tumors (20) can identify the stochastic group of subjects that benefit from the addition of tirapazamine. Identification of patients with hypoxic tumors will also likely be important to identify those that will benefit from the addition of PR-104. We show here that DCA treatment can be used to transiently shift tumors into the “high hypoxic radiotracer uptake group.” This change in tumor oxygenation translates into meaningful differences in tumor response without increased normal tissue toxicity. As we understand the molecular mechanisms that control the unique metabolism of solid tumors in greater detail, there may be additional opportunities for similar strategies that alter the tumor microenvironment to sensitize tumor cells and overcome barriers to effective therapy.
Translational Relevance.
This translational project is based on the preclinical finding that the hypoxia-inducible factor 1 transcription factor is responsible for downregulating mitochondrial oxygen consumption through the induction of the pyruvate dehydrogenase kinase 1 (PDK1) gene. We have used this in vitro finding to develop a novel drug combination based on the hypothesis that blocking this adaptive response with the small molecule dicholoroacetate will make tumor hypoxia more severe. The increased hypoxia can be imaged with a clinically developed positron emission tomography radiotracer (18F-fluoroazomycin arabinoside), and could potentiate the antitumor effect of a clinically relevant drug (PR-104). The translation of basic biology to a preclinical model lays the foundation for the development of a clinical trial, complete with a surrogate marker of efficacy.
Supplementary Material
Acknowledgments
Grantsupport: Funding from the National Cancer Institute (N.C. Denko). R.A. Cairns was a Research Fellow of The Terry Fox Foundation through an award from the National Cancer Institute of Canada.
Footnotes
Disclosure of Potential Conflicts of Interest A.J. Giaccia is a consultant to POroacta Corp., Auckland, New Zealand.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol. 2004;14:198–206. doi: 10.1016/j.semradonc.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Cairns R, Papandreou I, Denko N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol Cancer Res. 2006;4:61–70. doi: 10.1158/1541-7786.MCR-06-0002. [DOI] [PubMed] [Google Scholar]
- 3.Brown JM. The hypoxic cell: a target for selective cancer therapy–eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999;59:5863–70. [PubMed] [Google Scholar]
- 4.McKeown SR, Cowen RL, Williams KJ. Bioreductive drugs: from concept to clinic. Clin Oncol (R Coll Radiol) 2007;19:427–42. doi: 10.1016/j.clon.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys. 1986;12:1239–42. doi: 10.1016/0360-3016(86)90267-1. [DOI] [PubMed] [Google Scholar]
- 6.Rischin D, Hicks RJ, Fisher R, et al. Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol. 2006;24:2098–104. doi: 10.1200/JCO.2005.05.2878. [DOI] [PubMed] [Google Scholar]
- 7.Patterson AV, Ferry DM, Edmunds SJ, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res. 2007;13:3922–32. doi: 10.1158/1078-0432.CCR-07-0478. [DOI] [PubMed] [Google Scholar]
- 8.Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283:28106–14. doi: 10.1074/jbc.M803508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci U S A. 2007;104:9445–50. doi: 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Secomb TW, Hsu R, Ong ET, Gross JF, Dewhirst MW. Analysis of the effects of oxygen supply and demand on hypoxic fraction in tumors. Acta Oncol. 1995;34:313–6. doi: 10.3109/02841869509093981. [DOI] [PubMed] [Google Scholar]
- 13.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 14.Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem. 2000;275:15773–81. doi: 10.1074/jbc.M909488199. [DOI] [PubMed] [Google Scholar]
- 15.Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15:992–1004. doi: 10.1016/j.str.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piert M, Machulla HJ, Picchio M, et al. Hypoxia-specific tumor imaging with 18F-fluoroazomycin arabinoside. J Nucl Med. 2005;46:106–13. [PubMed] [Google Scholar]
- 17.Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHTmurine tumors. Cancer Res. 2001;61:8903–8. [PubMed] [Google Scholar]
- 18.Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478–87. doi: 10.1016/j.addr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Beck R, Roper B, Carlsen JM, et al. Pretreatment 18F-FAZA PETpredicts success of hypoxia-directed radiochemotherapy using tirapazamine. J Nucl Med. 2007;48:973–80. doi: 10.2967/jnumed.106.038570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.