

Abstract
The transcription factor NF-κB activates a number of genes whose protein products are proinflammatory. In quiescent cells, NF-κB exists in a latent form and is activated via a signal-dependent proteolytic mechanism in which the inhibitory protein IκB is degraded by the ubiquitin–proteasome pathway. Consequently, inhibition of the proteasome suppresses activation of NF-κB. This suppression should therefore decrease transcription of many genes encoding proinflammatory proteins and should ultimately have an anti-inflammatory effect. To this end, a series of peptide boronic acid inhibitors of the proteasome, exemplified herein by PS-341, were developed. The proteasome is the large multimeric protease that catalyzes the final proteolytic step of the ubiquitin–proteasome pathway. PS-341, a potent, competitive inhibitor of the proteasome, readily entered cells and inhibited the activation of NF-κB and the subsequent transcription of genes that are regulated by NF-κB. Significantly, PS-341 displayed similar effects in vivo. Oral administration of PS-341 had anti-inflammatory effects in a model of Streptococcal cell wall-induced polyarthritis and liver inflammation in rats. The attenuation of inflammation in this model was associated with an inhibition of IκBα degradation and NF-κB-dependent gene expression. These experiments clearly demonstrate that the ubiquitin–proteasome pathway and NF-κB play important roles in regulating chronic inflammation and that, as predicted, proteasome inhibition has an anti-inflammatory effect.
The pain, swelling, and tissue destruction that accompanies inflammatory disease results from a cascade of events that is initiated and propagated by the production of cytokines and chemokines and the cell surface expression of cell adhesion molecules (1). NF-κB is a key transcription factor that is required for the expression of many of these proinflammatory mediators (1).
NF-κB is a member of the Rel family of proteins and is typically a heterodimer composed of p50 and p65 (RelA) subunits (2). In quiescent cells, NF-κB resides in the cytosol in latent form, bound to an inhibitor protein IκB (2). Stimulation of these cells with various cytokines, lipopolysaccharide (LPS), viruses, antigens, or oxidants triggers a series of signaling events that ultimately leads to the phosphorylation and proteolytic degradation of IκB and liberation of NF-κB (2, 3). NF-κB then translocates into the nucleus, binds to specific DNA sequences in the promoters of target genes, and stimulates transcription. The protein products of these genes include cytokines, chemokines, and cell adhesion molecules and together play an important role in the immune and inflammatory response by controlling leukocyte trafficking and activation (2, 4, 5). Significantly, many of these proinflammatory proteins also are able to act in an autocrine or paracrine fashion to further stimulate activation of NF-κB (1). If activation of NF-κB is inhibited, production of this array of proinflammatory molecules is predicted to be suppressed (2, 6–9). Thus, inhibitors of NF-κB activation should be anti-inflammatory. To discover such inhibitors, we targeted the proteolytic system responsible for the degradation of the inhibitor protein IκB.
Studies in several laboratories have now established that the regulated proteolysis of IκB is mediated by the ubiquitin–proteasome pathway of protein degradation (6, 10–12). This is the principal pathway for intracellular protein turnover (13, 14) and for the degradation of important intracellular regulatory proteins, including cyclins (15), c-fos (16), c-jun (17), and the tumor suppressor protein p53 (18). Protein substrates that enter the ubiquitin–proteasome pathway are first “marked” for degradation by their covalent ligation to poly-ubiquitin chains. Ubiquitinated proteins are then recognized, bound, and degraded by the 26S proteasome complex to small peptides and monomeric ubiquitin (13, 14).
Degradation of certain proteins is subject to a further level of regulation by phosphorylation, which targets the substrate protein for recognition by specific ubiquitin-conjugating enzymes. For example, in the case of IκBα, a specific kinase first phosphorylates Ser-32 and Ser-36 of IκBα in response to various extracellular stimuli (10, 12, 19, 20). Phosphorylation targets IκBα for ubiquitination, and the ubiquitinated IκBα is then selectively recognized and degraded by the 26S proteasome (10).
Taken together, it is clear that inhibitors of enzymes of the ubiquitin–proteasome pathway should suppress the activation of NF-κB by stabilizing the inhibitor IκB, thereby reducing levels of multiple proinflammatory proteins and, ultimately, providing therapeutic anti-inflammatory effects. From among the enzymes of this pathway, we focused our initial drug discovery efforts on the enzyme that catalyzes the final, common step of the pathway, the proteasome.
The proteasome is highly conserved and may exist in different forms in the cytosol of eukaryotic cells. At the core of these forms is the 20S proteasome, a large (≈700 kDa) multi-subunit structure that possesses several peptidolytic activities (21, 22). However, the 20S proteasome is unable to degrade ubiquitinated proteins. The form of the proteasome that can hydrolyze these substrates is the 26S proteasome, a species which forms in an ATP-dependent manner from combination of one 20S particle with two 19S regulatory complexes (21, 22).
Several investigators have recently shown that both aldehyde inhibitors of the proteasome and the specific proteasome inhibitor lactacystin can inhibit the degradation of IκB and block the activation of NF-κB in cells (6, 10, 21, 23). In this report, we describe the anti-inflammatory activity of a new series of potent proteasome inhibitors. These inhibitors are dipeptide boronic acids, exemplified herein by PS-341. We show that PS-341 can enter mammalian cells and inhibit NF-κB activation and NF-κB-dependent gene expression. In addition, because NF-κB-driven cytokine, cell adhesion molecule, and enzyme expression have been implicated in the pathogenesis of chronic arthritis (24, 25), we examined the effect of PS-341 treatment on the chronic polyarthritis and liver inflammation induced by an i.p. injection of bacterial cell wall peptidoglycan/polysaccharide (PG/PS) in vivo. We demonstrate that PS-341 is efficacious in this chronic rodent model of inflammation and derives its biological activity through a mechanism involving proteasome inhibition.
MATERIALS AND METHODS
Electrophoretic Mobility Shift Assay and Western Blot Analysis.
Primary human umbilical vein endothelial (HUVE) cells (pooled, Cell Systems) were grown to confluence in 6-well plates. Confluent monolayers were refed with growth medium (Cell Systems) containing PS-341 or DMSO (0.1% final concentration) for a 1-hr preincubation. Tumor necrosis factor (TNF)α (1,000 units/ml; 10 ng/ml) was then added for the times indicated. The cells were washed with PBS, and whole cell extracts were prepared and analyzed by Electrophoretic mobility shift assay and Western blot for the presence of NF-κB and IκBα, respectively, as previously described (6). Western analysis of liver and synovial tissue also were performed to measure IκBα and inducible NO synthase (iNOS) protein levels.
Cell Surface Fluorescent Immunoassay.
Primary HUVE cells (pooled, Cell Systems) were prepared as described above and preincubated with either PS-341 or DMSO for 1 hr. TNFα (1,000 units/ml; 10 ng/ml) was then added for 4 or 15 hr. The cells were then processed for fluorescent immunoassay as described (26). E-Selectin, intercellular-adhesion molecule (ICAM)-1, vascular cell adhesion molecule 1 (VCAM-1) (Becton Dickinson), and endoglin (p96) (Tucker Collins and Michael Gimbrone, Harvard Medical School) antibodies were used as primary antibodies. A nonbinding IgG1 antibody (Becton Dickinson) was included in the assay as a control. The results are expressed as the amount of fluorescence (minus the background fluorescence using the control antibody) ± SEM.
Streptococcal Antigen-Induced Arthritis.
Chronic severe erosive polyarthritis was induced in female Lewis rats (Harlan–Sprague–Dawley) (135–145 g) by a single i.p. injection of group A Streptococcal cell wall PG/PS (Lee Laboratories) at a dose of 25 μg of rhamnose/gram bodyweight (27). Control animals received saline alone. Rats were dosed once daily by oral gavage (0.2 ml) for either 28 or 21 days beginning on day 0 or day 7 after arthritis induction, respectively, with 0.3 mg/kg PS-341 in 0.5% methylcellulose (n = 7) or with methylcellulose vehicle alone (n = 7). Saline-treated control animals did not receive PS-341. Changes in rear paw volume were quantified daily by plethysmography using a Buxco Edema Table (Buxco Electronics, Sharon, CT). Joints also were scored daily for symptoms of arthritis by using a standard scoring system modified from Dabbagh et al. (28). Each joint could receive a maximum score of 4 or a maximum score of 16 for each animal. In addition, the appearance of liver and spleen nodules and adhesions were noted and scored after completion of the study at necropsy (day 28).
Histological inspection of the joints also was performed at necropsy. Joints from the hind paws were obtained and decalcified in an extraction buffer [100 mM Tris/274 mM disodium EDTA/7.5% polyvinylpyrrolidone (Mr 40,000), pH 6.95] at 4°C for 21–28 days. After extraction, the tissues were washed in PBS (154 mM NaCl/1.5 mM KH2PO4/2.7 mM Na2HPO4, pH 7.2), fixed for 2–4 hr in 2.5% paraformaldehyde in PBS, rinsed in PBS, dehydrated in graded ethanol series (50–95% at 4°C), and embedded in Immunobed or JB-4 Plastic (Polysciences). Serial sections were cut at 5 μm parallel to the long axis of the joint and stained with Mallory’s trichrome stain (Sigma).
NO Metabolite and Cytokine Measurements.
The stable decomposition products of NO, nitrate (NO3−), and nitrite (NO2−) were quantified by first reducing all NO3− to NO2− by using Aspergillus nitrate reductase (Boehringer Mannheim) followed by addition of the Griess reagent (29). Serum IL-1α/β was quantified by using the A375.S2 melanoma cell cytotoxicity assay (30) and serum IL-6 was determined by using the IL-6 dependent 7TD1 cell bioassay (31). Cell viability was determined by using an MTT assay (32).
Statistical Analyses.
Statistical differences were identified by using one-way analysis of variance, and multiple comparisons were performed by using Newman Keuls’ post hoc analysis.
RESULTS AND DISCUSSION
Peptide aldehydes based on calpain inhibitor I, Ac-Leu-Leu-Norleu-H, constitute the first class of proteasome inhibitors examined. One member of this series, MG-132 (Z-Leu-Leu-Leu-H; Z = carbobenzoxoyl), is a 4-nM reversible inhibitor of the chymotryptic activity of the proteasome, and it inhibits a range of proteasome-dependent activities in cell culture (21, 33). Subsequently, the natural product lactacystin was shown to be a selective, irreversible inhibitor of the proteasome (34). In addition, chemical modifications revealed that peptide boronic acids were also inhibitors of the proteasome. These compounds are much more potent and more selective than their corresponding aldehydes (21, 35). Furthermore, dipeptide boronic acids are reversible and are more stable than lactacystin [T1/2 in aqueous buffer of >3 mo and 13 min, respectively (36)]. This increase in potency, selectivity, and stability allowed us to develop a series of low molecular weight dipeptide boronic acids, exemplified herein by PS-341 (Pyz-Phe-boroLeu; Pyz, 2, 5-pyrazinecarboxylic acid). 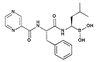
PS-341 inhibits several activities of purified mammalian proteasomes. The compound is a slow-binding inhibitor of the chymotryptic activity of the 20S proteasome with an inhibitory kinetic constant: Ki = 6 × 10−10 M (35, 37). Both the hydrolysis of polypeptide substrates (e.g., insulin B chain and casein) by the 20S proteasome and the hydrolysis of ubiquitinated proteins by the 26S proteasome are inhibited by PS-341 (data not shown). Importantly, these compounds are selective inhibitors of the proteasome with only modest activity toward serine and thiol proteases (35).
In addition to these activities against purified proteasomes, PS-341 can enter cells and inhibit proteasome-mediated intracellular proteolysis. Importantly, PS-341 inhibits the TNFα-stimulated activation of NF-κB in primary HUVE cells by blocking the degradation of the inhibitor IκBα (Fig. 1 A and B). PS-341 inhibits NF-κB activation with an IC50 of ≈0.5 μM. PS-341 does not block the phosphorylation of IκBα, because the phosphorylated form of IκBα can be detected in the presence of drug (Fig. 1A). Similar results also were observed in HeLa cells (data not shown).
Figure 1.

(A) PS-341 inhibits the activation of NF-κB in TNFα-treated HUVE cells. Different concentrations of PS-341 were added to HUVE cells (lanes 3–6) 1 hr before treating with TNFα (1,000 units/ml; 10 ng/ml) for 30 min. In lanes 1 and 2, the cells were pretreated with the inhibitor diluent DMSO. TNFα was added to the cells in lanes 2–6. Electrophoretic mobility shift assay was performed as described in Materials and Methods. NS is a nonspecific DNA-binding complex. (B) PS-341 inhibits the degradation of IκBα in TNFα-treated HUVE cells. HUVE cells were pretreated with PS-341 (5 μM) for 1 hr before the addition of TNFα (1,000 units/ml; 10 ng/ml) for different periods of time. IκBα protein levels were measured by Western analysis as described in Materials and Methods. ∗, phosphorylated IκBα.
Because PS-341 inhibits NF-κB activation, we next examined whether it could suppress the expression of genes whose transcription is regulated by NF-κB. It has been shown that NF-κB is required for the inducible expression of various cell adhesion molecules and cytokines (2, 4, 5, 26). In addition, the proteasome inhibitors MG-132 and lactacystin have been shown to inhibit the activation of NF-κB and the expression of cell adhesion molecules and cytokines in cultured cells (21, 23, 26, 33). In Fig. 2, we demonstrate that PS-341 inhibits TNFα-induced expression of the cell surface adhesion molecules E-Selectin, ICAM-1, and VCAM-1 on HUVE cells. VCAM-1 is especially sensitive to PS-341 with an IC50 value that is <10 nM. Northern blot analysis demonstrates that this inhibition is at the level of gene expression (data not shown). No cytotoxicity was observed with PS-341 at the concentrations used in the experiment (not shown). As a control, PS-341 had no effect on the expression of the NF-κB-independent, constitutively expressed, cell surface protein endoglin (p96; a low affinity transforming growth factor (TGF) type β receptor) (26). Consistent with the inhibition of cell adhesion molecule expression, PS-341 inhibits adhesion of human monocytic U937 cells to TNFα-activated HUVE cells (not shown).
Figure 2.

PS-341 inhibits the expression of cell adhesion molecules on HUVE cells. HUVE cells were pretreated with different concentrations of PS-341 for 1 hr. TNFα (1,000 units/ml; 10 ng/ml) was then added for 4 hr (A and B) or 15 hr (C). E-Selectin, ICAM-1, and VCAM-1 cell surface expression was measured by fluorescent immunoassay as described in Materials and Methods.
Interestingly, the concentration of PS-341 that completely suppresses adhesion molecule expression (Fig. 2) is ≈10-fold lower than that needed to inhibit NF-κB DNA binding (Fig. 1A). This observation has been observed before and is discussed extensively by Read et al. (26). A threshold level of active NF-κB is probably required for optimal cell adhesion molecule expression and because NF-κB works with other transcription factors to stimulate adhesion molecule expression, a small decrease in the level of activated NF-κB can lead to a profound change in the level of gene transcription (26).
PS-341 also inhibits the production of cytokines whose transcription is NF-κB dependent. For example, IL-2 and IL-6 production by phorbol 12-myristate 13-acetate/ionomycin-stimulated Jurkat T cells and LPS-stimulated whole blood, respectively, were inhibited by PS-341 (IC50 ≈0.1 μM) (data not shown). This inhibition is at the level of gene expression (not shown). Among the transcription factors that regulate IL-2 gene expression, which include NF-AT, AP-1, NF-κB, and Oct-1, only NF-κB activation is inhibited by PS-341 (Z. Chen and L. Parent, unpublished observations). We have not examined the effect of PS-341 on other transcription factors involved in the inflammatory response (e.g., STAT proteins), however aldehyde proteasome inhibitors do not have an effect on STAT1 activation (6).
Rheumatoid arthritis is associated with the up-regulation of a variety of proinflammatory mediators such as TNFα (38), IL-1 (39), IL-6 (40), iNOS (41), and endothelial cell adhesion molecules, including VCAM-1 (42). As described above, the expression of these gene products are regulated by NF-κB (2). Moreover, immunolocalization studies have demonstrated nuclear translocation of NF-κB, which is indicative of IκB degradation, in the vascular endothelium and type A synovial lining cells in synovial tissue from patients with rheumatoid arthritis (24). Inhibitors of NF-κB would therefore be expected to block the expression of proinflammatory mediators and to have an anti-inflammatory effect.
We have shown that PS-341 inhibits the activation of NF-κB and the expression of proinflammatory cytokines and cell adhesion molecules in vitro (Figs. 1 and 2). In addition, we recently reported that peptide boronic acid proteasome inhibitors are active in vivo and can inhibit the inflammation associated with antigen-induced colitis in rats (43). Consequently, we investigated the anti-inflammatory properties of PS-341 in an animal model of rheumatoid arthritis.
In this arthritis model, the disease is initiated by an i.p. injection of group A Streptococcal cell wall (PG/PS) into female Lewis rats (27, 44, 45). Typically, this model exhibits a peripheral and symmetrical, biphasic polyarthritis with cycles of exacerbated recurrence and remission and is clinically and histologically similar to rheumatoid arthritis (27). An acute T cell-independent arthritis develops within 24–48 hr, which persists for 4–5 days, and then partially resolves. This acute, neutrophil-predominant, inflammatory response is then followed by a spontaneously reactivating chronic inflammation at approximately day 15, which develops into a chronic T cell-dependent, progressive, erosive synovitis. In addition to polyarthritis, this PG/PS model also induces chronic granulomatous inflammation of the liver and spleen.
In the first experiment, rats were dosed daily with 0.3 mg/kg PS-341 by oral gavage for 28 days with the first dose administered on the day of the PG/PS injection. During the 28 days, inflammation was measured subjectively as the Total Arthritic Index, which is a score based on redness, swelling, and deformity of the joint and is similar to the scoring system used in the clinic (28). The results from such an experiment are shown in Fig. 3A and demonstrate that PS-341 attenuates the acute phase and markedly inhibits the chronic phase of the inflammatory response.
Figure 3.

PS-341 suppresses PG/PS-induced polyarthritis in rats. Polyarthritis was induced in female Lewis rats by a single i.p. injection of PG/PS. Rats were dosed once daily by oral gavage for 28 (A) or 22 (B) days with 0.3 mg/kg PS-341 in 0.5% methylcellulose (○) or with methylcellulose vehicle alone (•). (B) PS-341 was first administered 7 days after the initial PG/PS injection. The Total Arthritis Index was scored daily according to a validated assessment protocol described in Materials and Methods. Each point in A and B represents mean ± SEM (n = 7).
Significantly, PS-341 is equally efficacious when therapeutically administered after the onset of arthritis. Oral administration of PS-341 beginning 7 days after PG/PS injection significantly attenuated the progression of chronic polyarthritis (Fig. 3B). Animals receiving PS-341 from day 7–28 showed a significant inhibition in the Total Arthritis Index, which continued throughout the experimental period. These changes in arthritic indices were mirrored by comparable inhibition of the PG/PS-induced increase in average hind paw volume (ml), or joint swelling, on day 28. The average hind paw volume of the vehicle-treated, PG/PS- and PS-341-treated, PG/PS rats was 1.32 ± 0.10 ml and 0.41 ± 0.06 ml, respectively. In addition, after an initial reduction in body weight during the acute inflammatory episode, PS-341-treated animals gained significantly more body weight than vehicle-treated arthritic animals (data not shown), indicating that this dose of PS-341 is well-tolerated and not toxic. The increased bodyweight also is probably due to the fact that the animals could access their food more readily because joint swelling was much less in the PS-341-treated rats. Furthermore, treatment of the arthritic rats with PS-341 (0.3 mg/kg) significantly reduced 20S proteasome activity in circulating leukocytes, 4 hr after treatment, by ≈55% when compared with animals receiving methylcellulose vehicle only (data not shown). This result demonstrates that PS-341 is inhibiting its biochemical target, the proteasome, in vivo.
Histological examination of the joints obtained from the day 7–28, drug-treated (PS-341) animals revealed major differences in joint pathology when compared with joints from vehicle-treated arthritic animals and the saline controls (Fig. 4). We found that PG/PS induces massive synovial thickening and pannus formation over the surface of the articular cartilage with extensive inflammatory cell infiltrate and edema. This outcome was accompanied by cartilage destruction and subchondral bone erosion. Bone erosion was also evident in the medullary sinuses of the subchondral bone (Fig. 4B). Importantly, treatment with PS-341 attenuated the cellular infiltration and joint degradation induced by PG/PS (Fig. 4C). Pannus formation was still evident in the joints from PS-341-treated animals; however, the extensive cellular infiltrate observed in the vehicle-treated arthritic rats was diminished considerably. In the presence of PS-341, degradation of the articular cartilage was minimal and confined mainly to the surface. The extensive subchondral bone erosion seen at the articular surface and in the medullary canals also was markedly attenuated.
Figure 4.

Effect of PS-341 on PG/PS-induced alterations in the histologic appearance of the joint, 4 weeks after the induction of arthritis. (A) Normal saline control. Typical appearance of the articular cartilage (c), the subchondral bone (b), and the synovial villus and membrane (s). (B) Arthritic joint (vehicle-treated, PG/PS). Dramatic expansion and development of the synovial membrane and synovial villi into the erosive pannus (p), which is associated with the degradation of the articular cartilage (c) and subchondral bone (b). Inflammatory cell infiltration (arrows) is evident in the pannus and in the medullary canals (m) of the subchondral bone. Cellular infiltrate and fibrin (i) is seen in the articular space. (C) PS-341- (0.3 mg/kg/day) treated, PG/PS-induced arthritis. The pannus (p) remains present with the synovial membrane(s) thickened but exhibits a marked reduction in the number of inflammatory cells. Degradation of the articular cartilage (c) is attenuated and confined to the cartilage surface (arrow). Little or no erosion of the subchondral bone (b) is apparent, and there is minimal erosive activity and inflammatory cell infiltration in the medullary canals (m) of the subchondral bone. (Magnification, ×20; scale bar, 50 μm).
The chronic phase of PG/PS-induced arthritis is associated with an increase in NF-κB dependent proinflammatory cytokine production and cell adhesion molecule expression. A significant increase in serum IL-1, IL-6, and also nitrate and nitrite levels were observed in the PG/PS-treated animals (Fig. 5). IL-6 and NO metabolites were inhibited by PS-341 treatment (0.3 mg/kg/day, days 7–28). PS-341 treatment did not however inhibit the production of serum IL-1 (Fig. 5). The reason for this lack of inhibition is unknown because the IL-1β gene has NF-κB-binding sites in its promoter (46). Interestingly, recent in vitro experiments have shown that LPS-induced TNFα production is blocked by the aldehyde proteasome inhibitor calpain inhibitor I, whereas the inhibition of LPS-stimulated IL-1β production is weak (47). It may be possible that different cell types, stimuli, and Rel family members in conjunction with other transcription factors, all play an important role in ultimately determining what is necessary for optimal transcription of these genes.
Figure 5.

Effect of PS-341 on PG/PS-induced IL-1, IL-6, and NO metabolite production. PS-341 was administered orally at a dose of 0.3 mg/kg/day beginning 7 days after the induction of arthritis and continuing through day 28. IL-1, IL-6, and NO metabolites were measured on day 28 as described in Materials and Methods. Each bar represents mean ± SEM; ∗, P < 0.05 compared with saline-treated controls; §, P < 0.05 compared with vehicle-treated, PG/PS rats.
The PG/PS-induced polyarthritis that is observed in this model is accompanied by formation of granulomatous lesions of both the liver and spleen. At the experimental endpoint, the effect of PS-341 on both liver and spleen inflammation was examined. Therapeutic administration of PS-341 blocked the formation of PG/PS-induced lesions in the liver, but had a much weaker effect on splenic inflammation (data not shown). The reasons for the lack of effect of PS-341 in the spleen is not known at the present time. Significantly, we found enhanced levels of inducible iNOS protein (Fig. 6A), and iNOS and VCAM-1 mRNA (not shown) in liver tissue from vehicle-treated arthritic rats, which was abolished by treatment with PS-341. The expression of these gene products is regulated by NF-κB (41, 42). Similar results were also observed for iNOS protein expression in synovial tissue (data not shown).
Figure 6.

(A) PS-341 inhibits iNOS protein expression in PG/PS-treated rats. Western blot analysis of iNOS was performed on extracts from liver tissue samples from control (n = 1); vehicle-treated, PG/PS (n = 2); and PS-341-treated (0.3 mg/kg/day, days 7–28), PG/PS rats (n = 2). Tissue samples were prepared at the time of sacrifice on day 28. Equal amounts of protein (75 μg) were loaded in each lane. (B) Effect of PS-341 on liver IκBα protein levels in PG/PS-treated rats. Western blot analysis of IκBα was performed on extracts from liver tissue samples from control (n = 1); vehicle-treated, PG/PS (n = 2); and PS-341-treated (0.3 mg/kg/day, days 7–28), PG/PS rats (n = 2). Liver samples were prepared at the time of sacrifice on day 28. Equal amounts of protein (100 μg) were loaded in each lane.
These observations suggest that PS-341 is inhibiting proinflammatory mediators by blocking the degradation of IκB and the subsequent activation of NF-κB. To examine this directly, we measured the levels of IκBα protein in liver tissue from control, vehicle-treated, and PS-341-treated animals. The data presented in Fig. 6B demonstrates that PG/PS-induced inflammation of the liver (day 28) is accompanied by a decrease in the amount of IκBα protein. Significantly, this decrease is inhibited in animals treated with PS-341. Thus, the proteasome inhibitor PS-341 stabilizes the IκBα protein in vivo.
In summary, we have developed a class of potent and selective inhibitors of the 26S proteasome. Previous studies have demonstrated that lactacystin and peptide aldehyde inhibitors of the proteasome block the activation of NF-κB in cell culture (6, 10, 21, 23). We show that the proteasome inhibitor PS-341 prevents the activation of NF-κB and has significant anti-inflammatory properties in an in vivo model of disease. Thus, NF-κB is a critical regulator of the inflammatory response in vivo. Moreover, proteasome inhibitors such as PS-341 also may prevent cell cycle progression in vivo by blocking the temporal degradation of key cell cycle regulatory proteins (15). Because synovial hyperproliferation and T cell clonal expansion play a role in arthritis, PS-341 also may function to block this proliferation, thereby affecting inflammation and joint function. In any case, the ubiquitin–proteasome pathway represents an important drug discovery target for the treatment of inflammatory diseases.
Acknowledgments
We thank Dr. Margaret Read for critically reading the manuscript. This work was supported in part by a grant from the Arthritis Center of Excellence at LSU Medical Center in Shreveport (to M.B.G.).
ABBREVIATIONS
- LPS
lipopolysaccharide
- PG/PS
peptidoglycan/polysaccharide
- TNF
tumor necrosis factor
- iNOS
inducible NO synthase
- HUVE
human umbilical vein endothelial
- VCAM-1
vascular cell adhesion molecule 1
- ICAM-1
intercellular-adhesion molecule 1
References
- 1.Barnes P J, Karin M. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin A S. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 3.Thanos D, Maniatis T. Cell. 1995;80:1–20. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 4.Grilli M, Chiu J J-S, Lenardo M J. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle P A, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 6.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–786. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 7.Auphan N, DiDonato J, Rosette C, Helmbertg A, Karin M. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 8.Scheinman R, Cogswell P, Lofquist A, Baldwin A. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 9.Suto M J, Ransone L J. Curr Pharm Design. 1997;3:515–528. [Google Scholar]
- 10.Chen Z, Hagler J, Palombella V J, Melandri F, Scherer D, Ballard D, Maniatis T. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 11.Scherer D C, Brockman J A, Chen Z, Maniatis T, Ballard D W. Proc Natl Acad Sci USA. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiDonato J, Mercurio F, Rosette C, Wuli J, Suyang H, Ghosh S, Karin M. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochstrasser M. Curr Opin Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 14.Ciechanover A, Schwartz A L. Proc Natl Acad Sci USA. 1998;95:2727–2730. doi: 10.1073/pnas.95.6.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King R W, Deshaies R J, Peters J-M, Kirschner M W. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 16.Stancovski I, Gonen H, Orian A, Schwartz A L, Ciechanover A. Mol Cell Biol. 1995;15:7106–7116. doi: 10.1128/mcb.15.12.7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Treler M, Staszewski L M, Bohmann D. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 18.Scheffner M, Huibregste J M, Vierstra R D, Howley P M. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 19.Maniatis T. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 20.Stancovski I, Baltimore D. Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 21.Adams J, Stein R. Annu Rep Med Chem. 1996;31:279–288. [Google Scholar]
- 22.Baumeister W, Walz J, Zühl F, Seemüller E. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 23.Bellas R E, FitzGerald M J, Fausto N, Sonenshein G E. Am J Pathol. 1997;151:891–896. [PMC free article] [PubMed] [Google Scholar]
- 24.Marok R, Winyard P G, Coumbe A, Kus M L, Gaffney K, Blades S, Mapp P I, Morris C J, Blake D R, Kaltschmidt C, et al. Arthritis Rheum. 1996;39:583–591. doi: 10.1002/art.1780390407. [DOI] [PubMed] [Google Scholar]
- 25.Handel M L, McMorrow L B, Gravallese E M. Arthritis Rheum. 1995;38:1762–1770. doi: 10.1002/art.1780381209. [DOI] [PubMed] [Google Scholar]
- 26.Read M A, Neish A S, Luscinskas F W, Palombella V J, Maniatis T, Collins T. Immunity. 1995;2:493–506. doi: 10.1016/1074-7613(95)90030-6. [DOI] [PubMed] [Google Scholar]
- 27.Cromartie W J, Craddock J C, Schwab J H, Anderie S K, Yang C H. J Exp Med. 1977;146:1585–1602. doi: 10.1084/jem.146.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dabbagh A J, Blake D R, Morris C J. Ann Rheum Dis. 1992;51:516–521. doi: 10.1136/ard.51.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grisham M B, Johnson G G, Gautreaux M D, Berg R D. Methods Enzymol. 1995;7:84–90. [Google Scholar]
- 30.Nakai S, Mizumo K, Kanesta M, Yoshikatsu H. Biochem Biophys Res Commun. 1988;154:1189–1196. doi: 10.1016/0006-291x(88)90266-5. [DOI] [PubMed] [Google Scholar]
- 31.Frei K, Leist T P, Meager A, Gallo P, Leppert D, Zinkernagel R M, Fontana A. J Exp Med. 1988;168:449–453. doi: 10.1084/jem.168.1.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosmann T. J Immunol. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg A L, Stein R L, Adams J. Curr Biol. 1995;2:503–508. doi: 10.1016/1074-5521(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 34.Fenteany G, Standaert R F, Lane W S, Choi S, Corey E J, Schreiber S L. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 35.Adams J, Behnke M, Chen S, Cruickshank A A, Dick L R, Grenier L, Klunder J M, Ma Y-T, Plamondon L, Stein R. Bioorg Med Chem Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 36.Dick L R, Cruikshank A A, Grenier L, Melandri F D, Nunes S L, Stein R L. J Biol Chem. 1996;271:7273–7276. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- 37.Stein R L, Melandri F, Dick L. Biochemistry. 1996;35:3899–3908. doi: 10.1021/bi952262x. [DOI] [PubMed] [Google Scholar]
- 38.Yocum D E, Esparza L, Dubry S, Benjamin J B, Volz R, Scuderi P. Cell Immunol. 1989;122:131–145. doi: 10.1016/0008-8749(89)90154-8. [DOI] [PubMed] [Google Scholar]
- 39.Fontana A, Hengartner H, Weber E, Fehr K, Grob P J, Cohen G. Rheumatol Int. 1982;2:49–53. doi: 10.1007/BF00541245. [DOI] [PubMed] [Google Scholar]
- 40.Houssiau F A, Devogelaer J P, Van Damme J, De Deuxchaisnes C N, Van Snick J. Arthritis Rheum. 1988;31:784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- 41.Sakurai H, Kohsaka H, Liu M-F, Higashiyama H, Hirata Y, Kanno K, Saito I, Miyasaka N. J Clin Invest. 1995;96:2357–2363. doi: 10.1172/JCI118292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wahl S M, Allen J B, Hines K L, Imamichi T, Wahl A M, Furcht L T, McCarthy J B. J Clin Invest. 1994;94:655–662. doi: 10.1172/JCI117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conner E M, Brand S, Davis J M, Laroux S, Palombella V J, Fuseler J W, Kang D Y, Wolf R E, Grisham M B. J Pharmacol Exp Ther. 1997;282:1615–1622. [PubMed] [Google Scholar]
- 44.Wilder R L, Allen J B, Wahl L H, Calandra G B, Walh S M. Arthritis Rheum. 1983;26:1442–1451. doi: 10.1002/art.1780261205. [DOI] [PubMed] [Google Scholar]
- 45.Clark R L, Cuttino J T, Anderle S K, Cromartie W J, Schwab J H. Arthritis Rheum. 1979;22:25–35. doi: 10.1002/art.1780220105. [DOI] [PubMed] [Google Scholar]
- 46.Pan Z K, Ye R D, Christiansen S C, Jagels M A, Bokoch G M, Zuraw B L. J Immunol. 1998;160:3038–3045. [PubMed] [Google Scholar]
- 47.Haas M, Page S, Page M, Neumann F J, Marx N, Adam M, Ziegler-Heitbrock H W, Neumeier D, Brand K. J Leukocyte Biol. 1998;63:395–404. doi: 10.1002/jlb.63.3.395. [DOI] [PubMed] [Google Scholar]