

Summary
Background The phase I program of anticancer agents usually consists of multiple dose escalation studies to select a safe dose for various administration schedules. We hypothesized that pharmacokinetic and pharmacodynamic (PK–PD) modeling of an initial phase I study (stage 1) can be used for selection of an optimal starting dose for subsequent studies (stage 2) and that a post-hoc PK–PD analysis enhances the selection of a recommended dose for phase II evaluation. The aim of this analysis was to demonstrate that this two-stage model-based design, which does not interfere in the conduct of trials, is safe, efficient and effective. Methods PK and PD data of dose escalation studies were simulated for nine compounds and for five administration regimens (stage 1) for drugs with neutropenia as dose-limiting toxicity. PK–PD models were developed for each simulated study and were used to determine a starting dose for additional phase I studies (stage 2). The model-based design was compared to a conventional study design regarding safety (number of dose-limiting toxicities (DLTs)), efficiency (number of patients treated with a dose below the recommended dose) and effectiveness (precision of dose selection). Retrospective data of the investigational anticancer drug indisulam were used to show the applicability of the model-based design. Results The model-based design was as safe as the conventional design (median number of DLTs = 3) and resulted in a reduction of the number of patients who were treated with a dose below the recommended dose (−27%, power 89%). A post-hoc model-based determination of the recommended dose for future phase II studies was more precise than the conventional selection of the recommended dose (root mean squared error 8.3% versus 30%). Conclusions A two-stage model-based phase I design is safe for anticancer agents with dose-limiting myelosuppression and may enhance the efficiency of dose escalation studies by reducing the number of patients treated with a dose below the recommended dose and by increasing the precision of dose selection for phase II evaluation.
Keywords: Oncology, Phase I trial design, Optimal design, Dose escalation, Dose finding, Pharmacokinetics, Pharmacodynamics, Modeling and simulation
Introduction
The design of phase I dose escalation studies for experimental cytotoxic agents has extensively been discussed in the past decades. For safety issues, the initial dose level is usually far below the anticipated recommended dose. In the conventional algorithm-based design, which uses modified Fibonacci escalation and three patients per cohort, this may result in treatment of many patients with a dose below the recommended dose [1, 2]. A second disadvantage of the conventional design is the imprecise estimation of the recommended dose due to the limited number of patients at doses in the range of the recommended dose [3].
Various model-based modifications of the conventional method have been proposed, such as the continual reassessment method (CRM) [2], dose escalation with overdose control [4], the random walk rule [5] and decision theoretic approaches [6]. These model-based methods generally treat fewer patients at a dose level below the recommended dose and result in a more precise estimate of the recommended dose.
Although model-based approaches offer a promising alternative to the conventional phase I clinical trial designs in oncology, these methods are seldomly used in practice, which is largely due to operational difficulties and lack of prospective validation [7]. In order to avoid practical problems, He et al. [8] proposed a post-hoc model-based determination of the recommended dose using results from conventional Fibonacci dose escalation trials. This method allowed precise determination of the recommended dose without interfering in the conduct of the trial. Adversely, in their analysis, the number of patients treated at suboptimal dose levels was not optimized by this post-hoc analysis.
During phase I development, various administration regimens are commonly evaluated. The regimen with the best tolerability, convenience and highest dose intensity is selected for further development. Dose escalation studies are usually performed in parallel to select the appropriate dose for each administration regimen and to accelerate clinical development.
A two-stage model-based design is proposed for the phase I program of novel anticancer agents. The first stage of the model-based design is the conduct of a single phase I study using conventional modified Fibonacci-like dose escalation. During a model-based interim analysis, data from the first phase I trial are used to optimize the design of subsequent studies prior to their initiation. Optimization of the second stage of the design will be attained by (1) development of a pharmacokinetic and pharmacodynamic population model using data from the first stage and (2) a simulation study using the developed model, which will confer a safe dose to start dose escalation with alternative administration regimens. After the conduct of all dose escalation studies, the recommended doses for further clinical evaluation will be determined in a post-hoc analysis, which may enhance the effectiveness (precision of dose selection) of the phase I program.
The conduct of all dose escalation studies in this two-stage design is identical to the conduct of a classical dose escalation study. Only the starting doses during the second stage may be higher. The major advantage of the higher starting doses in the proposed two-stage model-based design is a potential reduction of the number of patients to be treated at a dose level below the recommended dose. In addition to the ethical benefit, this may also result in a reduction of cost and time to complete a dose escalation study.
The two-stage model-based design may be applied to investigational anticancer agents with well defined dose-limiting toxicity related to drug exposure. In the current analysis, the two-stage model-based design was applied to anticancer agents with dose-limiting myelosuppression. Neutropenia is a common dose-limiting toxicity for cytotoxic agents and may cause serious infections, which may be fatal. The neutrophil count over time can be related to drug exposure by a semi-physiological pharmacokinetic and pharmacodynamic (PK–PD) model [9]. The PK–PD characteristics of drugs with dose-limiting myelosuppression can thus be described by integrated PK–PD models. Therefore, anticancer agents with dose-limiting neutropenia were suitable compounds to demonstrate the potential of the proposed model-based analyses.
Simulation studies were performed to evaluate the two-stage model-based design. The objectives of this research were to demonstrate that (1) the number of patients treated at a dose level below the recommended dose can be reduced by the proposed model-based analysis and that (2) the two-stage model-based design can be safely used to optimize the design of dose escalation studies in oncology. The performance of the two-stage model-based design was retrospectively evaluated using clinical data of the phase I program of the investigational anticancer drug indisulam.
Methods
The design of the simulation study is shown in Fig. 1. Steps 1 to 5 are explained into detail below.
Fig. 1.

Flow-chart showing the design of this study and the corresponding outcome parameters
Simulation of pharmacokinetic and pharmacodynamic data (step 1)
PK and PD data were simulated for nine compounds with various pharmacokinetic profiles. (Figure 2) A three-compartment model was used with or without saturable elimination and with or without auto-induction. (Figure 3) Drug elimination was described by the Michaelis Menten parameters V max (maximal elimination rate) and K m (concentration related to half-maximal elimination). K m was relatively low (1 mg/L) or relatively high (100 mg/L) representing saturable and linear drug elimination, respectively. Auto-induction was described by an enzyme turnover model. Drug concentrations in the central compartment were related to an increased input rate into the enzyme compartment, resulting in an increased concentration of a hypothetical amount of enzyme and a proportional increase of the drug elimination rate. Drug elimination was maximally induced by 100% (I max), the effective concentration related to half-maximal induction (ECI50) was 1 mg/L and the mean turnover time of the enzyme (MTTenzyme) was 24 h. The distribution volumes of the peripheral compartments were relatively small (1 L) or relatively large (10 L) to vary the magnitude of drug distribution. Inter-individual and residual random effects were moderate or large. (Table 1) The differential equations of the pharmacokinetic models are given below.
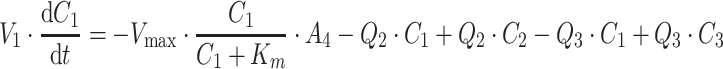 |
1 |
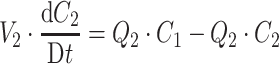 |
2 |
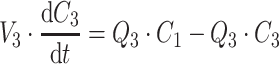 |
3 |
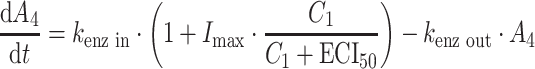 |
4 |
Fig. 2.

Typical pharmacokinetic profiles after treatment with a 1-h infusion of the highest (non-tolerated) dose of all virtual compounds. a compound 1; b compounds 2, 3, 4, 5; c compound 6, d compound 7; e compound 8; f compound 9
Fig. 3.

Structural pharmacokinetic model of the nine virtual compounds and the semi-physiological model describing the time profile of absolute neutrophil counts (ANC) after treatment with myelosuppressive chemotherapy
Table 1.
Pharmacokinetic and pharmacodynamic parameters of the nine virtual compounds
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Summary of PK–PD characteristics | ||||||||||
| Compartmentsa | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | |
| Saturable eliminationb | – | – | – | – | – | + | – | – | – | |
| Auto-induction | – | – | – | – | – | – | + | – | + | |
| IIV PK | Moderate | Moderate | Large | Moderate | Moderate | Moderate | Moderate | Moderate | Large | |
| IIV PD | Moderate | Moderate | Moderate | Large | Moderate | Moderate | Moderate | Moderate | Large | |
| Residual error PK | Moderate | Moderate | Moderate | Moderate | Large | Moderate | Moderate | Moderate | Moderate | |
| PK parameters | ||||||||||
| V 1 c | (L) | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| V max c | (mg/h) | 100 | 100 | 100 | 100 | 100 | 10 | 100 | 100 | 100 |
| K m | (mg/L) | 100 | 100 | 100 | 100 | 100 | 1 | 100 | 100 | 100 |
| V 2 c | (L) | 1 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Q 2 | (L/h) | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| V 3 c | (L) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 10 | 10 |
| Q 3 | (L/h) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| I max | (%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| ECI50 | (mg/L) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| MTTenz | (h) | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 |
| IIV V 1; V 2; V 3 d | (%) | 40 | 40 | 80 | 40 | 40 | 40 | 40 | 40 | 80 |
| IIV V max d | (%) | 30 | 30 | 60 | 30 | 30 | 30 | 30 | 30 | 60 |
| IIV Q 2; Q 3 d | (%) | 30 | 30 | 60 | 30 | 30 | 30 | 30 | 30 | 60 |
| IIV MTTenz d | (%) | 25 | 25 | 50 | 25 | 25 | 25 | 25 | 25 | 50 |
| Residual error PKe | (%) | 20 | 20 | 20 | 20 | 40 | 20 | 20 | 20 | 20 |
| PD parameters | ||||||||||
| Slope | (mg/L) | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
| MTT | (h) | 120 | 120 | 120 | 120 | 120 | 120 | 120 | 120 | 120 |
| Base | (109/L) | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 |
| Gamma | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | |
| IIV sloped | (%) | 45 | 45 | 45 | 90 | 45 | 45 | 45 | 45 | 90 |
| IIV MTTd | (%) | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| IIV based | (%) | 35 | 35 | 35 | 35 | 35 | 35 | 35 | 35 | 35 |
| Residual error PDe | (%) | 45 | 45 | 45 | 45 | 45 | 45 | 45 | 45 | 45 |
aAll data were simulated from three compartment models, but the contribution of one or both peripheral compartments was small for compounds 1–7
bAll data were simulated from a model with Michaelis Menten elimination, but saturation was only evident if K m was relatively low (1 mg/L)
cAllometric scaling was applied to V max ((WT/70)0.75) and to the volumes of distribution V 1, V 2, V 3 ((WT/70)1)
dIIV = inter-individual variability; geometric standard deviations are listed
eResidual errors were additive on a logarithmic scale; geometric standard deviations are listed
The dose-limiting toxicity of all virtual compounds was neutropenia. The time course of drug-related neutropenia was described by a semi-physiological population pharmacodynamic model that was developed by Friberg et al. [9] (Fig. 3). This model comprised a progenitor compartment of proliferating blood cells, three transit compartments representing the maturation chain in the bone marrow and a compartment corresponding to the central circulation. The model comprised two system related parameters: the mean transit time (MTT) and a feedback parameter gamma (γ). The MTT was the average time between neutrophil proliferation and completion of maturation and was related to the first order transition rate constant k tr (=4/MTT). The feedback parameter γ represented the induction of stem cell proliferation by endogenous growth factors and/or cytokines [10]. The baseline blood cell count was estimated from the observed baseline value and a residual error. The proliferation rate was reduced by drug exposure in the central pharmacokinetic compartment according to a linear function (Eq. 5).
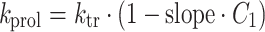 |
5 |
Using the PK–PD parameters from Table 1, data for dose escalation studies were simulated. The design of the simulated dose escalation studies was in accordance with the design of classical phase I studies [11]. Patients were treated in silico in three-patient cohorts, that were extended to six-patient cohorts if significant (≥CTC grade 2) toxicity was observed. Neutrophil counts were simulated with alternating 3 and 4 day intervals (twice weekly). A modified Fibonacci-like dose escalation strategy was used [1]. The dose was escalated by 100% if no or minimal (CTC grade 0 or 1) neutropenia was observed. If one or more patients had CTC grade 2 toxicity, the dose was escalated by 50% increments and if serious (CTC grade 3 or 4) neutropenia was observed, the dose was escalated by 25%. Dose-limiting neutropenia was defined as CTC grade 4 neutropenia (absolute neutrophil count ≤0.5 × 109 per liter) during at least two subsequent hematological measurements (∼1 week). If two or more patients in a six-patient cohort experienced dose-limiting neutropenia, this dose level was defined as the non-tolerated dose (NTD). The recommended dose for future studies was the dose level immediately below the NTD.
Dose escalation studies were simulated for five administration regimens:
A single 1-h infusion (D × 1) every 3 weeks
Daily 1-h infusions on days 1–5 (D × 5) every 3 weeks
Weekly 1-h infusions on days 1, 8, 15 and 22 (W × 4) every 6 weeks
A 120-h infusion (120 H) every 3 weeks
A continuous infusion during 3 weeks (CI)
Regimens 1–4 were evaluated in the phase I program of indisulam, that was used in this study as a real dataset for a retrospective evaluation of the two-stage model-based design [12]. These regimens comprised short and long infusions, single and multiple drug administrations and were therefore selected for the current study. Schedule 5, the continuous infusion, was added to cover the full range of short exposure to continuous exposure regimens.
Pharmacokinetic data were simulated according to rich sampling designs as commonly used in phase I trials. PK samples were taken during infusion at fixed time points. Additional PK samples were taken after infusion and were logarithmically spaced between the end of infusion and the last sampling point. The last sampling point corresponded to five times the mean residence time (MRT) after the end of infusion. To mimic the fact that limited knowledge may be available at the start of first in human studies, the true MRT was perturbed by a geometric standard deviation of 1.70 to account for prediction uncertainty. This standard deviation was derived from previous publications [13, 14]. The limit of quantitation (LOQ) was 50 ng/mL for all compounds. Data below the LOQ were excluded from data analysis.
Dose escalation studies with a modified Fibonacci-like escalation scheme (see above) were performed in silico for five administration regimens for each of the nine compounds. In total, 45 phase I studies were simulated using NONMEM (version VI, GloboMax, Hanover, MD, USA). Parameters with inter-individual variability were log-normally distributed. Data were logarithmically transformed and residual errors were additive on a logarithmic scale.
PK–PD model development (step 2)
Pharmacokinetic and pharmacodynamic models were developed for each of the 45 simulated data sets. The investigator was blinded to the ‘true’ PK–PD models that were defined by the parameters in Table 1.
Pharmacokinetic and pharmacodynamic data were analyzed sequentially. For PK–PD analysis, a data set was used that contained all dosing information and all pharmacokinetic and pharmacodynamic measurements. Pharmacokinetic parameters were fixed to the estimates of the pharmacokinetic analysis. This method has been described into more detail by Zhang et al. [15].
The estimation of the system parameters MTT and γ and the interindividual variability of MTT was supported by prior knowledge of the values of these parameters. The Bayesian priors were the geometric means of the estimates that were reported for various other anticancer agents by Friberg et al. (MTT = 116 h, γ = 0.167, IIV MTT = 22.4%). Prior uncertainty corresponded to the geometric standard deviation of these estimates (MTT 18%, γ 59%, IIV MTT 20%) [9].
All parameters (P) were assumed to be log-normally distributed in the study population. Consequently, interindividual variability was estimated using an additive function on a logarithmic scale (ln(P i) = ln(TVP) + η i) to describe the individual deviation (η i) from the population typical value (TVP). Differences between observed and individual predicted values were modeled as additive residual errors on a logarithmic scale. Data were analyzed with the first-order conditional method using NONMEM (version VI, GloboMax, Hanover, MD, USA). Discrimination between hierarchical models was based on the objective function value, goodness of fit plots and standard errors of parameter estimates.
Standard errors and measures of correlation between parameter estimates were obtained using the COVARIANCE option of NONMEM.
Prediction of safe starting dose (step 3)
The PK–PD models, that were developed in step 2 using data from a single in silico phase I study (simulated in step 1), were used to predict the outcome of subsequent dose escalation studies. For instance, the model that described the PK–PD data of compound 1 after a single 1-h infusion, was used to predict the recommended doses for the other four administration regimens for compound 1. Trial simulations were carried out to make these predictions. For each prediction, a dose escalation trial was simulated in 40,000-fold to account for uncertainty in the PK–PD parameter estimates and for variability between patients. A total number of 200 sets of PK–PD parameters from the final parameter estimates, their geometric standard errors and the correlation matrix were selected. For each set of PK–PD parameters, 200 trials were simulated with different random selections of patients.
One tenth of the predicted non-tolerated dose is conventionally considered a safe starting dose for dose escalation studies. Therefore, in order to determine the starting dose of a dose escalation trial, the non-tolerated dose of the previous trial was multiplied by 10% and perturbed using a geometric standard deviation of 1.54. The perturbation accounted for uncertainty in the predicted tolerability of anticancer agents prior to first in man studies. The geometric standard deviation was calculated from predicted and empirically determined non-tolerated doses of 21 anticancer drugs [16–19]. Patients were treated in three- or six-patient cohort and the modified Fibonacci-like dose escalation strategy was used (see above for details).
The median value of each set of 40,000 recommended doses was the predicted recommended dose. The five and 95 percentiles formed the 90% confidence intervals of the prediction. The lower boundary was considered the safe starting dose for an in vivo clinical dose escalation study.
Evaluation of the two-stage model-based design using simulated data (step 4)
Using the true PK–PD parameters (Table 1) and starting doses that were considered safe for clinical dose escalation studies, 1,000 replicate trials were simulated (two-stage model-based design). In addition, 1,000 replicate trials were simulated with starting doses perturbed around 10% of the recommended dose (conventional method).
The number of patients with dose-limiting neutropenia was assessed for both methods as a measure of safety of the two-stage model-based design. The median number of patients treated with a dose below the recommended dose was compared between the new and the conventional methods, for each virtual compound and for each administration regimen, as a measure of efficiency of the two-stage model-based design. The two-stage model-based design was considered successful if it did not result in an increased number of patients with dose-limiting neutropenia and if the number of patients treated with a dose below the recommended dose was reduced.
Post-hoc determination of recommended dose (step 5)
Based on the phase I program of a novel anticancer agent, a recommended dose must be defined for further testing in phase II clinical studies. In a conventional study design, the selection of the recommended dose is empirically based on clinical outcome and is defined as the highest dose level with more than two out of six patients with dose-limiting toxicity. In the two-stage model-based design, the recommended dose for phase II studies is based on a PK–PD analysis using all PK–PD data from phase I. This PK–PD analysis should be performed after finalization of the clinical phase I program and is therefore termed a post-hoc analysis.
To evaluate this part of the two-stage model-based design, the PK–PD models of each compound were updated using the data from all five phase I studies that were simulated in step 1 (Fig. 1). The updated models were used for the simulation of 40,000 dose escalation studies for each administration regimen. Parameter uncertainty and variability between patients were taken into account. The median value of the 40,000 simulated recommended doses was proposed for further testing in phase II studies. This method was used to select a recommended dose for all compounds (n = 9) and for all administration regimens (n = 5). The 45 selected recommended doses (D rec.,pred) were used to determine the precision of this strategy. The root mean squared relative prediction error (RMSE%) was calculated as a measure of precision (Eq. 6). The true recommended dose (D rec.,true) was defined as the median value of 1,000 simulated recommended doses using the true PK–PD model, conventional starting doses (i.e. the recommended dose of the previous trial multiplied by 10% and perturbed using a geometric standard deviation of 1.54) and the modified Fibonacci-like dose escalation strategy.
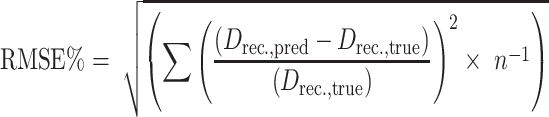 |
6 |
The precision of the two-stage model-based design was compared to the precision of the conventional design, where the recommended dose was based on the clinical outcome of a dose escalation study. The RMSE% of the conventional design was calculated for the 1,000 simulated recommended doses from the true PK–PD model (D rec.,pred) and their median value (D rec.,true).
The precision of the selection of the recommended dose for phase II evaluation was also assessed for the PK–PD models that were developed using data from a single phase I study.
Evaluation of the two-stage model-based design using clinical data of indisulam
The previously conducted phase I program of the investigational anticancer agent consisted of four dose escalation studies of four administration regimens: D × 1, D × 5, W × 4, 120 H [20–23]. Neutropenia was identified as the dose-limiting toxicity [20–23]. It was verified retrospectively if the number of patients treated with a dose below the recommended dose could have been reduced by the proposed two-stage model-based design. For each of the phase I studies, a PK–PD model was developed using indisulam plasma concentrations and absolute neutrophil counts that were measured during the first treatment cycle. The models were used to simulate 40,000 dose escalation studies for the alternative administration regimens, taking into account parameter uncertainty (×200) and variability between patients (×200). The median values were selected as the predicted recommended doses and the five percentiles were considered safe starting doses for the other three administration regimens. The selected starting doses were compared to the clinically determined recommended and non-tolerated doses. The number of patients treated at dose levels below the clinically determined recommended doses was assessed. In this retrospective evaluation, the two-stage model-based design was considered successful if all selected starting doses were below the highest administered dose levels and if the number of patients treated with at a dose level below the recommended dose was reduced by at least 10%.
Results
Simulation of pharmacokinetic and pharmacodynamic data (step 1)
In total, 45 phase I dose escalation studies were simulated. These studies each comprised five to 12 dose levels and 18–55 patients, and were considered representative for clinical phase I studies in oncology. In each study, two to seven patients experienced dose-limiting toxicity.
PK–PD model development (step 2)
For each simulated study, a pharmacokinetic model was successfully developed. One-, two- and three compartment models were selected with or without saturable elimination. Table 2 shows the structural PK models that were selected for all simulated data sets. Saturable elimination was correctly identified, irrespective of the administration schedule. Auto-induction could not be demonstrated for any of the simulated data sets (see “Discussion”). For compounds 8 and 9, three-compartment models were selected for the 1-h infusion schedules, but identification of the third compartment was less successful for the 120-h and continuous infusions (Table 2). The true PK model of compound 1 consisted of a single relevant compartment (Table 1), but the minor second compartment could be identified based on data from the W × 4 schedule (V 2 = 1.58 L and Q 2 = 0.87 L/h). Using data from the 120 H schedule, a second compartment could also be identified for compound 1, but the intercompartmental clearance was large and the model proved to be equivalent to a one-compartmental model (V 1 = 6.8 L, V 2 = 5.3 L and Q 2 = 19.9 L/h).
Table 2.
The structural pharmacokinetic models that were selected for each phase I study ( )
)

The compound numbers correspond to the numbers in Table 1. The black margins correspond to the “true” models that were used for data simulation
Prediction of safe starting dose (step 3)
This part of the study was most computer-time intensive with the simulation of 40,000 clinical trials for each scenario. In Fig. 4a, the distribution of the simulated recommended dose for the D × 1 regimen of compound 2 (n = 40,000) is shown for the PK–PD model that was developed based on data from a W × 4 phase I dose escalation study. The variability of this distribution is explained by variability between patients and by uncertainty of the PK–PD parameter estimates. Figure 4b shows the simulated recommended doses for one set of PK–PD parameter estimates (n = 200). The variability in Fig. 4b is explained by variability between patients. The large variability in Fig. 4b indicates that the uncertainty of PK–PD parameter estimates (3.4–60%) had a minor contribution to the overall variability of the simulated recommended doses. The five-percentiles were considered safe starting doses for stage 2 of the model-based design (step 4, Fig. 1). Regarding the scenario depicted in Fig. 4, 158 mg was selected as the starting dose for the D × 1 regimen of compound 2. This was 49% of the median recommended dose 320 mg. For all scenarios, the proposed starting doses were between 33% and 56% of the median recommended doses.
Fig. 4.

Simulated recommended doses of a 1-h infusion of compound 2, based on a PK–PD model that was developed using PK–PD data from a W × 4 dose escalation study. a The frequency distribution of all 40,000 simulated doses (variability comprises variability between patients and uncertainty of PK–PD parameter estimates). b The frequency distribution of 200 simulated doses for a single set of PK–PD parameter estimates (variability comprises variability between patients)
The median values and the five-percentiles of the simulated recommended doses are plotted in Fig. 5 for all scenarios. The doses in Fig. 5 are normalized to the true recommended dose, which was defined as the median value of 1,000 simulated recommended doses using the true PK–PD model. All starting doses that were proposed for stage 2 of the model-based design were below the true recommended dose.
Fig. 5.

The predicted recommended dose (median of 40,000 replicate values) and proposed starting dose (five-percentile of 40,000 replicate values) based on PK–PD models that were developed using data from a single phase I study, relative to the true recommended dose. Predicted recommended doses and starting doses are plotted for all compounds (1: □; 2: *; 3:+; 4: Δ; 5: ⁃; 6: ▬; 7: ✕; 8: ○; 9: ◊) and for all regimens (a D × 1; b D × 5; c W × 4; d 120 H; e CI)
Evaluation of the two-stage model-based design using simulated data (step 4)
Figure 6 shows the number of dose-limiting toxicities for the conventional design and for the various scenarios of the two-stage model-based design (median and ten to 90 percentile range). When the model-based starting doses were used in clinical trial simulations, the mean number of dose-limiting toxicities per dose escalation study ranged from 2.9 to 3.6 for the various scenarios. For the conventional design, the mean number of dose-limiting toxicities per study was between 3.0 and 3.6. The number of DLTs was relatively small for compound 6 with saturable elimination. This may reflect the more than proportional increase in drug exposure when the dose is escalated to the next dose level, which may result in a sharp increase in incidence of DLT at the first dose level above the recommended dose.
Fig. 6.

Median and ten to 90 percentiles ranges of the number of patients with dose-limiting toxicity for the conventional design (◊) and for stage 2 of the model-based design (stage 1: D × 1 ▪; D × 5 ▴; W × 4 ✕; 120 H *; CI ●) for all regimens and for all compounds
The number of patients who were treated at a dose level below the recommended dose, was on average 20.5 when a dose escalation study was started at a conventional starting dose level. The two-stage model-based design resulted in a reduction of this number to 13.0 patients. Figure 7 is a graphical representation of the number of patients treated at a dose level below the recommended dose (median and inter-quartile range). For all scenarios, this number was higher for the conventional study design (first plot of each sextet in Fig. 7) as compared to the two-stage model-based design.
Fig. 7.

Median and interquartile ranges of the number of patients treated with a dose below the recommended dose for the conventional design (◊) and for stage 2 of the model-based design (stage 1: D × 1 ▪; D × 5 ▴; W × 4 ✕; 120 H *; CI ●) for all regimens and for all compounds
For a complete phase I program, comprising of one conventional study at stage 1 followed by four studies with a model-based starting dose at stage 2, the reduction of the number of patients treated at a dose below the recommended dose was on average 27.2%. This was consistent for the various scenarios that were evaluated (range 12.2–39.8%). The power of the two-stage model-based design to include fewer patients at dose levels below the recommended dose was 88.6%.
The starting dose was not tolerated in 1.3% of the simulated conventional dose escalation studies. When the starting dose in stage 2 was based on data from stage 1, the initial dose level was not tolerated in only 0.3% of the simulated trials.
Post-hoc determination of recommended dose (step 5)
The PK–PD models were successfully updated using PK–PD data from all five dose escalation studies. More complex structural PK models could be selected for compounds 1, 3, 5 and 7 (Table 3). Figure 8 shows the recommended doses that were proposed for phase II evaluation, based on 40,000 simulated trials, relative to the ‘true’ recommended doses that were determined using the true PK–PD models. The precision of the selection of the recommended dose for phase II studies, expressed as the RMSE, was 8.3% for the post-hoc model-based dose selection. Conversely, the precision was only 30% for the conventional study design (ten to 90 percentile ranges are indicated in Fig. 8). If the recommended doses were determined by the post-hoc model-based method, using the PK–PD models that were based on data from a single phase I, the precision was 12% (Fig. 8).
Table 3.
The structural pharmacokinetic models that were selected for the combined data of all studies ( )
)

The compound numbers correspond to the numbers in Table 1. The black margins correspond to the “true” models that were used for data simulation
Fig. 8.

Visual representation of the precision of the selection of the recommended dose for the conventional method (ten to 90 percentile range of simulated empirically determined recommended doses, RMSE 30.0%) and for the two-stage model-based design (▴, RMSE 8.3%). The recommended doses that were predicted using data from a single phase I study, are also indicated (✕, RMSE 11.9%)
Evaluation of the two-stage model-based design using clinical data of indisulam
Pharmacokinetic data of indisulam (n = 3,327) were available from four phase I studies and from 143 patients [20–23]. The pharmacokinetic data for each phase I study were best described by three-compartment models with saturable elimination and saturable distribution. This model was described in detail in a previous publication from our group [12]. A model with a linear and a saturable elimination pathway adequately described the pharmacokinetic data from the D × 5, the W × 4 and the 120 H dose escalation studies. Conversely, the data from the D × 1 study did not contain sufficient information to identify the linear elimination pathway. Therefore, a reduced model without a linear elimination pathway was used to describe the data from the D × 1 study. Measurements of the absolute neutrophil count (n = 1,114) were available from 145 patients and could be adequately described by the semi-physiological model of neutropenia. (Figure 3) [24] The MTT of neutrophils (Fig. 3) was estimated at 191 h for the W × 4 regimen. For the other regimens, this parameter estimate was closer to the Bayesian prior (D × 1: 147 h; D × 5: 137 h; 120 H: 134 h).
In Fig. 9, the course of the dose escalation studies of indisulam is depicted for all administration regimens. The recommended doses that were determined in the clinical in vivo studies are indicated by a black dashed line [20–23]. The starting doses that were selected retrospectively by trial simulation using the four developed PK–PD models, were all below the highest administered dose and were all above the conventional starting dose.
Fig. 9.

The course of the dose escalation studies of indisulam. For each administration regimen (a D × 1; b D × 5; c W × 4 and d 120 H), the recommended dose (clinically determined during the phase I program  ; post-hoc estimate from final PK–PD model
; post-hoc estimate from final PK–PD model  ) and the proposed starting doses (
) and the proposed starting doses ( ) are indicated
) are indicated
The recommended doses that were predicted using the final PK–PD model, based on data from all four phase I studies, are indicated by ‘post-hoc’ in Fig. 9. The clinically determined recommended doses were lower than these post-hoc predictions for the D × 5 and 120 H infusion schedules and were higher for the D × 1 and W × 4 predictions. For indisulam, the ‘true’ PK–PD characteristics and the ‘true’ recommended dose are unknown. Therefore, it was not possible to determine the precision of the selection of the recommended dose for the conventional and the two-stage model-based design. Importantly, all retrospectively selected starting doses would have been safe for stage 2 of the model-based design.
For the complete phase I program consisting of four studies, the number of patients treated at a dose level below the recommended dose was 65 for the conventional design (17 (D × 1) + 18 (D × 5) + 18 (W × 4) + 12 (120 H); Fig. 9). For the D × 1 schedule, inclusion of the first seven patients would have been redundant if the starting dose would have been based on data from the D × 5 study. In addition, the two-stage model-based design would also have resulted in a reduction of six patients for the W × 4 schedule and nine patients for the 120 H schedule, if the D × 5 study was performed at stage 1. In total, 22 patients (34% of all patients treated at a dose level below the recommended dose) could have been treated at a higher dose level if the two-stage model-based design would have been applied to the phase I program of indisulam, starting with the D × 5 schedule. If one of the other schedules would have been selected for stage 1, the model-based design would have resulted in a reduction of the number of patients treated at a dose level below the recommended dose of 33 (51%), 28 (43%) or 14 (22%) for the D × 1, W × 4 and 120 H schedules, respectively.
Discussion
A two-stage model-based design for optimization of the phase I program of novel anticancer agents was proposed. This simulation study demonstrated that the proposed two-stage model-based design did not result in an increased frequency of dose-limiting toxicities. Furthermore, the two-stage model-based design resulted in a reduction of the number of patients treated at a dose level below the recommended dose. The precision of the selection of the recommended dose for future phase II evaluation was improved by a post-hoc model-based analysis.
The model-based design consists of two stages: an initial phase I study conducted in a conventional manner (stage 1) and subsequent phase I studies for alternative administration schedules, at starting doses that are based on advanced PK–PD analysis. The starting doses for stage 2 of the phase I program are determined by the five-percentiles of the simulated recommended doses. This strategy was considered safe, because the likelihood that the starting dose would be below the recommended dose, was close to 95%, provided that the correct structural PK–PD model was selected. In this study, we accounted for structural bias of the PK model, because various structural PK models were evaluated during model development. Structural model bias was not taken into account for the semi-physiological model of neutropenia. This was justified by previous validation of the semi-physiological model of neutropenia for a range of anticancer drugs with various mechanisms of action [9, 25–28].
The semi-physiological model comprised a linear relationship between the drug concentration and the proliferation rate of neutrophils. Nevertheless, the nadir neutrophil count was dependent to total drug exposure and the administration schedule in a non-linear manner. This was mainly due to the non-linear feedback mechanism in the model. The semi-physiological characteristics of this model allowed successful application to multiple administration schedules and dose levels.
In this study, the two-stage model-based design was evaluated for anticancer agents with dose-limiting neutropenia. The two-stage model-based design may also be applicable to drugs with other dose-limiting toxicities, provided that a PK–PD relationship can be identified and quantified. An important condition is, however, that the pharmacodynamic model should permit extrapolation of the concentration–effect relationship to higher or lower levels of drug exposure and to shorter or longer duration of drug exposure. Preferably, the pharmacodynamic model should show consistency between drugs and should be previously validated for similar compounds. Hematological toxicity has been well characterized in PK–PD models for multiple anticancer agents as opposed to other types of toxicity, such as neurotoxicity, hand-foot syndrome and diarrhea, and may therefore be the most suitable adverse event for a successful application of the two-stage model-based design.
Our aim was to optimize the efficiency and the efficacy of phase I dose escalation studies, without interfering in the conduct of the clinical trials. The dose escalation studies are carried out in a conventional manner, but the starting dose can be relatively higher as a result of optimal usage of all available clinical PK and PD data by the application of advanced PK–PD modeling. Contrary to CRM, this two-stage model-based design does not require data analysis during a dose escalation study. Consequently, it is expected that this two-stage model-based design will not involve operational or logistical difficulties.
Development of a PK–PD model using all phase I data, followed by post-hoc determination of a recommended dose for further evaluation in phase II clinical development was proposed to maximize the precision of the determination of the recommended dose for future studies. This study demonstrated that the precision was largely improved by the model-based analysis: the RMSE was 8.3% for the two-stage model-based design versus 30% for a conventional study design. The high imprecision of the conventional study design was also an important finding of this analysis. Due to wide inter-patient variability and the limited number of patients that is included in a phase I study, the outcome of a clinical dose escalation study is subject to large random variability.
A secondary goal of phase I clinical development is the assessment of the pharmacokinetics of a novel compound. When the two-stage model-based design is used, the number of patients treated at low dose levels is reduced. Depending on the schedule and the pharmacokinetic properties of a compound, this may compromise the characterization of the pharmacokinetics.
In this simulation study, auto-induction could not be demonstrated in the interim PK models (step 2) that were based on data from a single phase I study. Although the effect size was large (up to 100% increase of the elimination rate) in comparison to reported values for anticancer agents (e.g. 36% increase of the elimination rate of cyclophosphamide [29]), the effect of auto-induction was not obvious in the pharmacokinetic profiles (Fig. 2d, f). In addition, the value of mean turnover time of the inducible enzyme was realistic (24 versus 12 h for ifosfamide [30] and 45 h for cyclophosphamide [29]). It can therefore be concluded that the poor identification of auto-induction was not related to the PK parameters that were selected for the compounds in this study.
To confirm the recommended dose that is selected in a phase I trial, an enrichment phase may be added to the conventional study design, where additional patients are included at the recommended dose. The impact of an enrichment phase was not evaluated in this analysis. In addition, the conduct of a dose escalation study may be influenced by a steering team of investigators to select the dose escalation steps. The input of professionals is undoubtedly more subtle than the modified Fibonacci-like dose escalation strategy that was used in this simulation study. For these two reasons, the precision of the selection of a recommended dose for phase II studies may be underestimated in this analysis.
The results of the simulation studies were confirmed by retrospective evaluation using real clinical data. Indisulam was used as a model compound. This drug has a highly non-linear pharmacokinetic profile and indisulam-induced neutropenia showed wide inter-patient variability. For these PK–PD characteristics, indisulam was considered a suitable compound to challenge the proposed model-based. The two-stage model-based design resulted in the selection of starting doses for stage 2 of the phase I program that were all below the highest administered dose level and was therefore considered safe. The efficiency of the phase I studies could have been increased by application of the two-stage model-based design and the phase I program could have been conducted with 14 to 33 patients less. This might have resulted in a reduction of cost and time for the phase I development of indisulam.
This simulation study demonstrated that the proposed two-stage model-based design is safe for anticancer agents with dose-limiting myelosuppression. The two-stage model-based design enhanced the efficiency of dose escalation studies by an average 27% reduction of the number of patients included at a dose level below the recommended dose. The power of the proposed two-stage model-based design to reduce the number of patients treated with a dose below the recommended dose was 89%. The model-based design was also effective in the precise selection of the recommended dose for phase II evaluation. The good performance of the model-based design seemed to be irrespective of the administration schedule that was selected for the dose escalation study at stage 1. The proposed two-stage model-based design may enhance the early clinical development of investigational anticancer drugs.
Acknowledgments
The authors thank Nick Holford, Jakob Ribbing and Andrew Hooker for inspiring discussions.
This research was supported with grants from the Eisai network of companies, from the Swedish Cancer Society and from the Dutch Cancer Society.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Schneiderman MA (1967) Mouse to man: statistical problems in bringing a drug to clinical trial. Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability 4:855–866
- 2.O’Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. doi: 10.2307/2531628. [DOI] [PubMed] [Google Scholar]
- 3.Storer BE. Design and analysis of phase I clinical trials. Biometrics. 1989;45:925–937. doi: 10.2307/2531693. [DOI] [PubMed] [Google Scholar]
- 4.Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med. 1998;17:1103–1120. doi: 10.1002/(SICI)1097-0258(19980530)17:10<1103::AID-SIM793>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Durham SD, Flournoy N, Rosenberger WF. A random walk rule for phase I clinical trials. Biometrics. 1997;53:745–760. doi: 10.2307/2533975. [DOI] [PubMed] [Google Scholar]
- 6.Whitehead J, Brunier H. Bayesian decision procedures for dose determining experiments. Stat Med. 1995;14:885–893. doi: 10.1002/sim.4780140904. [DOI] [PubMed] [Google Scholar]
- 7.Dent SF, Eisenhauer EA. Phase I trial design: are new methodologies being put into practice? Ann Oncol. 1996;7:561–566. doi: 10.1093/oxfordjournals.annonc.a010671. [DOI] [PubMed] [Google Scholar]
- 8.He W, Liu J, Binkowitz B, Quan H. A model-based approach in the estimation of the maximum tolerated dose in phase I cancer clinical trials. Stat Med. 2006;25:2027–2042. doi: 10.1002/sim.2334. [DOI] [PubMed] [Google Scholar]
- 9.Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20:4713–4721. doi: 10.1200/JCO.2002.02.140. [DOI] [PubMed] [Google Scholar]
- 10.Testa NG, Dexter TM. The regulation of haematopoietic cell production. In: Hoffbrand AV, Lewis SM, Tuddenham EGD, editors. Postgraduate haematology. 4. Oxford: Butterworth-Heinemann; 1999. pp. 1–12. [Google Scholar]
- 11.Dodion P, Kenis Y, Staquet M. Phase I trials of single agents in adult solid tumours: preclinical and clinical aspects. Drugs Exp Clin Res. 1986;12:23–30. [PubMed] [Google Scholar]
- 12.van Kesteren C, Mathôt RAA, Raymond E, Armand JP, Dittrich C, Dumez H, Roche H, Droz JP, Punt CJA, Ravic M, Wanders J, Beijnen JH, Fumoleau P, Schellens JHM. Population pharmacokinetics of the novel anticancer agent E7070 during four phase I studies: model building and validation. J Clin Oncol. 2002;20:4065–4073. doi: 10.1200/JCO.2002.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. II. Volume of distribution and mean residence time. Drug Metab Dispos. 2004;32:612–619. doi: 10.1124/dmd.32.6.612. [DOI] [PubMed] [Google Scholar]
- 14.Ward KW, Erhardt P, Bachmann K. Application of simple mathematical expressions to relate the half-lives of xenobiotics in rats to values in humans. J Pharmacol Toxicol Methods. 2005;51:57–64. doi: 10.1016/j.vascn.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30:387–404. doi: 10.1023/B:JOPA.0000012998.04442.1f. [DOI] [PubMed] [Google Scholar]
- 16.Pessina A, Albella B, Bayo M, Bueren J, Brantom P, Casati S, Croera C, Gagliardi G, Foti P, Parchment R, Parent-Massin D, Schoeters G, Sibiril Y, Van den Heuvel R, Gribaldo L. Application of the CFU-GM assay to predict acute drug-induced neutropenia: an international blind trial to validate a prediction model for the maximum tolerated dose (MTD) of myelosuppressive xenobiotics. Toxicol Sci. 2003;75:355–367. doi: 10.1093/toxsci/kfg188. [DOI] [PubMed] [Google Scholar]
- 17.Gomez SG, Bueren JA, Faircloth G, Albella B. Use of CFU-GM assay for prediction of human maximum tolerated dose of a new antitumoral drug: Yondelis (ET-743) Toxicol In Vitro. 2003;17:671–674. doi: 10.1016/S0887-2333(03)00139-5. [DOI] [PubMed] [Google Scholar]
- 18.Masubuchi N, May RD, Atsumi R. A predictive model of human myelotoxicity using five camptothecin derivatives and the in vitro colony-forming unit granulocyte/macrophage assay. Clin Cancer Res. 2004;10:6722–6731. doi: 10.1158/1078-0432.CCR-04-0721. [DOI] [PubMed] [Google Scholar]
- 19.Moneta D, Geroni C, Valota O, Grossi P, de Jonge MJ, Brughera M, Colajori E, Ghielmini M, Sessa C. Predicting the maximum-tolerated dose of PNU-159548 (4-demethoxy-3′-deamino-3′-aziridinyl-4′-methylsulphonyl-daunorubicin) in humans using CFU-GM clonogenic assays and prospective validation. Eur J Cancer. 2003;39:675–683. doi: 10.1016/S0959-8049(02)00812-2. [DOI] [PubMed] [Google Scholar]
- 20.Raymond E, ten Bokkel Huinink WW, Taieb J, Beijnen JH, Faivre S, Wanders J, Ravic M, Fumoleau P, Armand JP, Schellens JHM. Phase I and pharmacokinetic study of E7070, a novel chloroindolyl sulfonamide cell-cycle inhibitor, administered as a one-hour infusion every three weeks in patients with advanced cancer. J Clin Oncol. 2002;20:3508–3521. doi: 10.1200/JCO.2002.09.030. [DOI] [PubMed] [Google Scholar]
- 21.Punt CJA, Fumoleau P, van de Walle B, Faber MN, Ravic M, Campone M. Phase I and pharmacokinetic study of E7070, a novel sulfonamide, given at a daily times five schedule in patients with solid tumors. A study by the EORTC-early clinical studies group (ECSG) Ann Oncol. 2001;12:1289–1293. doi: 10.1023/A:1012287111922. [DOI] [PubMed] [Google Scholar]
- 22.Dittrich C, Dumez H, Calvert H, Hanauske A, Faber M, Wanders J, Yule SM, Ravic M, Fumoleau P. Phase I and pharmacokinetic study of E7070, a chloroindolyl-sulfonamide anticancer agent, administered on a weekly schedule to patients with solid tumors. Clin Cancer Res. 2003;9:5195–5204. [PubMed] [Google Scholar]
- 23.Terret C, Zanetta S, Roche H, Schellens JHM, Faber MN, Wanders J, Ravic M, Droz JP. Phase I clinical and pharmacokinetic study of E7070, a novel sulfonamide given as a 5-day continuous infusion repeated every 3 weeks in patients with solid tumours. A study by the EORTC Early Clinical Study Group (ECSG) Eur J Cancer. 2003;39:1097–1104. doi: 10.1016/S0959-8049(03)00128-X. [DOI] [PubMed] [Google Scholar]
- 24.van Kesteren C, Zandvliet AS, Karlsson MO, Mathôt RAA, Punt CJA, Armand JP, Raymond E, Huitema ADR, Dittrich C, Dumez H, Roche H, Droz JP, Ravic M, Yule SM, Wanders J, Beijnen JH, Fumoleau P, Schellens JHM. Semi-physiological model describing the hematological toxicity of the anti-cancer agent indisulam. Invest New Drugs. 2005;23:225–234. doi: 10.1007/s10637-005-6730-3. [DOI] [PubMed] [Google Scholar]
- 25.Friberg LE, Brindley CJ, Karlsson MO, Devlin AJ. Models of schedule dependent haematological toxicity of 2′-deoxy-2′-methylidenecytidine (DMDC) Eur J Clin Pharmacol. 2000;56:567–574. doi: 10.1007/s002280000181. [DOI] [PubMed] [Google Scholar]
- 26.Troconiz IF, Garrido MJ, Segura C, Cendros JM, Principe P, Peraire C, Obach R. Phase I dose-finding study and a pharmacokinetic/pharmacodynamic analysis of the neutropenic response of intravenous diflomotecan in patients with advanced malignant tumours. Cancer Chemother Pharmacol. 2006;57:727–735. doi: 10.1007/s00280-005-0112-6. [DOI] [PubMed] [Google Scholar]
- 27.Sandstrom M, Lindman H, Nygren P, Lidbrink E, Bergh J, Karlsson MO. Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin–docetaxel regimen in breast cancer patients. J Clin Oncol. 2005;23:413–421. doi: 10.1200/JCO.2005.09.161. [DOI] [PubMed] [Google Scholar]
- 28.Sandstrom M, Lindman H, Nygren P, Johansson M, Bergh J, Karlsson MO. Population analysis of the pharmacokinetics and the haematological toxicity of the fluorouracil–epirubicin–cyclophosphamide regimen in breast cancer patients. Cancer Chemother Pharmacol. 2006;58:143–156. doi: 10.1007/s00280-005-0140-2. [DOI] [PubMed] [Google Scholar]
- 29.de Jonge ME, Huitema ADR, Rodenhuis S, Beijnen JH. Integrated Population Pharmacokinetic Model of both cyclophosphamide and thiotepa suggesting a mutual drug–drug interaction. J Pharmacokinet Pharmacodyn. 2004;31:135–156. doi: 10.1023/B:JOPA.0000034405.03895.c2. [DOI] [PubMed] [Google Scholar]
- 30.Kerbusch T, Mathot RAA, Keizer HJ, Kaijser GP, Schellens JHM, Beijnen JH. Influence of dose and infusion duration on pharmacokinetics of ifosfamide and metabolites. Drug Metab Dispos. 2001;29:967–975. [PubMed] [Google Scholar]