

Abstract
Although numerous studies have underlined the role of HDACs in breast physiology and tumorigenesis, little is known on the particular contribution of the various classes of HDACs in these processes. Using ERα-positive MCF-7 breast cancer cells, the effects of MC1575 and MC1568, two novel class II specific HDAC inhibitors (HDI), were analyzed on cell proliferation, apoptosis and estrogen signalling. The specificity of these HDIs was validated by measuring histone and α-tubulin acetylation and by the specific in vitro inhibition of recombinant HDAC4 using histone and non histone substrates, contrasting with the lack of inhibition of class I HDACs. In addition, MC1575 did not inhibit class I HDAC gene expression thus confirming the specific targeting of class II enzymes. Similar to TSA, MC1575 displayed a dose-dependent anti-proliferative effect and induced cell cycle arrest although this blockade occurred at a different level than TSA. Moreover, and in contrast to TSA, MC1575 had no effect on MCF-7 cells apoptosis. Interestingly, MC1575 was able to increase p2lwaf1/CIP1 mRNA levels but did not regulate the expression of other genes such as cyclin D1, p27, p14ARF, Bcl2, Baxα, Trail-R1 and -R2. Finally, MC1575 strongly induced ERβ gene expression but did not decrease ERα expression nor did it switch hydroxy-tamoxifen to an agonist activity. Altogether, these data suggest that the class II HDAC sub-family may exert specific roles in breast cancer progression and estrogen-dependence.
Keywords: Apoptosis; physiology; Breast Neoplasms; genetics; metabolism; pathology; Cell Cycle; physiology; Cell Division; physiology; Cell Line, Tumor; Estrogen Receptor alpha; metabolism; Gene Expression Regulation, Enzymologic; Gene Expression Regulation, Neoplastic; Histone Deacetylases; genetics; metabolism; Humans; Repressor Proteins; genetics; metabolism; Signal Transduction; physiology
Keywords: Histone deacetylase, histone deacetylase inhibitor, breast cancer, estrogen receptor, cell proliferation
Introduction
Human histone deacetylases (HDACs) form a large family of 18 members classified in four groups (I to IV) based on sequence homologies (1, 2). Class I enzymes, including HDAC1, 2, 3 and 8, are nuclear proteins with ubiquitous expression. Class II HDACs are divided in two classes: class IIa includes HDAC4, 5, 7 and 9 while class IIb is composed of HDAC6 and HDAC10. Class II HDACs have a tissue-specific pattern of expression and can shuttle between the nucleus and the cytoplasm depending on their phosphorylation status. HDAC6 and 10 form a particular group as they both contain two deacetylase domains and because HDAC6 can specifically deacetylate the cytoskeletal protein α-tubulin (3). Indeed, in addition to histones, HDACs have been shown to deacetylate various substrates including transcription factors, chaperones, as well as many regulators involved in DNA repair, cell signalling or metabolism (2). The diversity of HDACs also suggests differential roles for the various classes of enzymes depending on tissues or cell lines. For instance, recent studies have shown an essential role for HDAC6 in the clearance of ubiquitinated cellular protein aggregates (4) and class IIa HDACs have been involved in cardiac and vascular development, chondrocyte hypertrophy during skeletogenesis or thymocytes selection (5–8).
HDAC inhibitors (HDI) have shown in vitro and in vivo activities against various cancer types affecting cell cycle, programmed cell death, differentiation and angiogenesis (1, 9). HDI are thus considered as a new class of anticancer agents and are currently evaluated in several phase I and II clinical trials in patients with hematological and solid malignancies (10). Recently, one of them, Vorinostat, has been approved for the treatment of cutaneous T cell lymphoma (11, 12).
In breast tumor models, HDI have potent anti-proliferative effects in vitro and in vivo and interfere with estrogen signalling (13–17). Estrogens effects are mediated by two distinct estrogen receptors (ER) α and β, acting as transcriptional factors that belong to the nuclear receptor superfamily (18). We and others have shown that, in ERα-expressing breast cancer cells, HDI such as TSA strongly down-regulate ERα both at the mRNA and protein level while increasing ERβ gene expression (14, 19, 20). By contrast, in ERα-negative breast cancer cells, HDI and DNA methyltransferase inhibitors synergistically reactivate ERα gene expression by releasing various repressors from its promoter, including the class I enzyme HDAC1 (21–23). HDI such as TSA also increase ERα and ERβ transcriptional activity (14, 19) and, in MCF-7 cells, strongly stimulate the agonist activity of partial antiestrogens such as hydroxy-tamoxifen (OHTam) (14).
At present, little is known on the specific contribution of the various classes of HDACs in breast tumorigenesis or estrogen responsiveness. Studies using HDIs in breast cancer models have indeed used broad-range, non selective inhibitors such as TSA or SAHA, so that no information about the contribution of specific HDACs in biological pathways could be available. Recently, new HDI displaying specificity against class I, II or III HDACs have been described and used to identify the roles of these classes of HDACs in various cell responses (24–26). Using specific inhibitors of class II HDACs, the aim of this study was to define the particular contribution of this class of enzymes on cell proliferation, apoptosis, gene expression and ER signalling in ERα-expressing MCF-7 human breast cancer cells. Altogether, our data demonstrate that the class II HDAC sub-family specifically regulates these parameters and may thus exert a particular role in breast tumor progression and estrogen-dependence.
Results
Specificity of MC1575 and MC1568 on class II HDACs
The role of class II HDACs was investigated in ERα-expressing MCF-7 human breast cancer cells using MC1575 and MC1568, two inhibitory compounds displaying class II HDACs specificity (27, 28). HDAC specificity of MC1575 and MC1568 was first validated by measuring their effects on the levels of acetylated forms of H3 and H4 histones, and tubulin (Figure 1). HDAC blockade induced by MC1575 and MC1568 was confirmed by the accumulation of acetylated H3 and H4 histones in MCF-7 cells, although these compounds were found to be less potent than TSA, in agreement with their pharmacological properties (28). Moreover, MC1575, MC1568 and TSA, but not MS275, a specific inhibitor for class I HDACs, increased the levels of acetyl-tubulin, indicating their ability to inhibit HDAC6, a member of class II HDAC known to deacetylate α-tubulin (3).
Figure 1. Effects of MC1575 and MC1568 on deacetylase activity in MCF-7 cells.
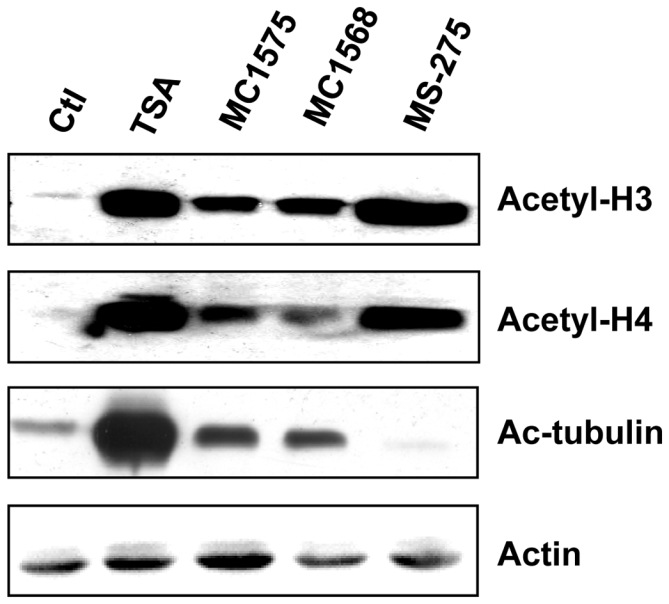
MCF-7 cells were treated for 6 h with TSA (1.7 μM), MC1575 (20 μM), MC1568 (20 μM) or MS-275 (1 μM). Cell lysates were analyzed by western immunoblotting using anti-acetylated histone H3 (Acetyl-H3), anti-acetylated histone H4 (Acetyl-H4) or anti-acetylated tubulin (Ac-tubulin) antibodies. Actin was used as a loading control.
We next measured the effects of MC1575 on purified class I HDAC1, 2, 3 and 8 and class II HDAC4 (Figure 2). MC1575, unlike SAHA, had a weak inhibitory effect on the enzymatic activity of class I HDAC1, 2, 3 and 8 even at the highest dose tested (20 μM - 10–30% inhibition) (Figure 2A). By contrast, at 20 μM, MC1575 displayed a 65% inhibitory effect on the activity of HDAC4, a representative member of class IIa HDAC (Figure 2B). Recently, Lahm et al. described trifluoroacetyl-lysine as a new non histone acetylated-lysine substrate specific for class IIa HDACs (29, 30). Using this substrate, MC1575 at 5 and 20 μM was found to efficiently inhibit HDAC4 enzymatic activity, at levels similar to SAHA (65% inhibition at 20 μM) (Figure 2B). Similar results were obtained using MC1568 (data not shown).
Figure 2. HDAC specificity of MC1575.
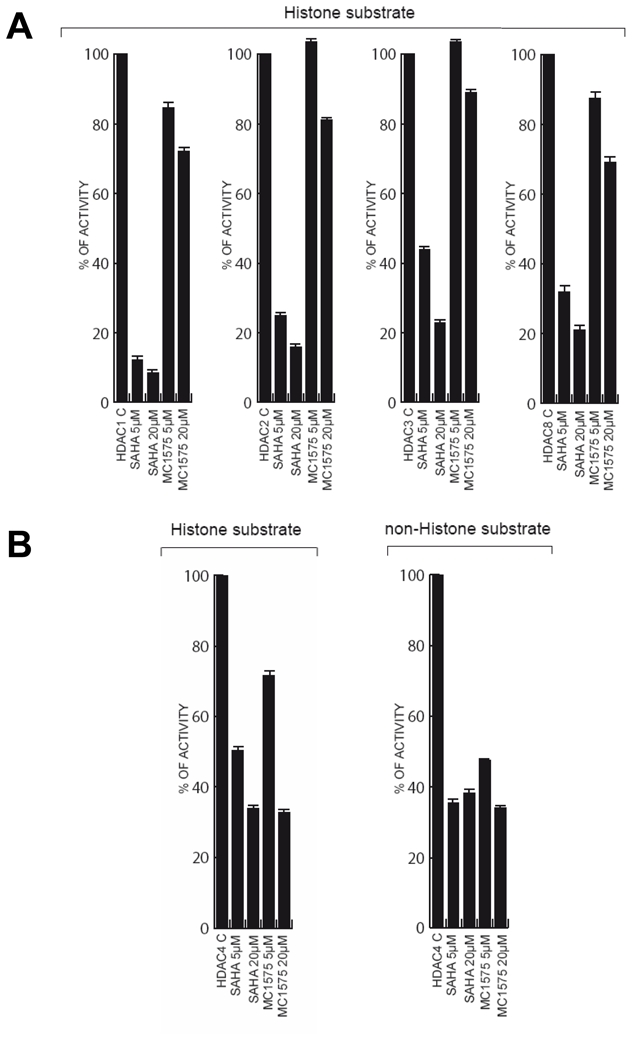
A- Histone deacetylase activity of class I enzymes (HDAC1, 2, 3 and 8). Recombinant HDAC1, 2, 3 and 8 were expressed and the deacetylase activity of purified HDAC was measured, using fluorescent substrates, in the absence (Control – C) or presence of SAHA or MC1575 (5 and 20 μM). The deacetylase activity of the various HDACs in presence of HDI is expressed as percentage of the control (HDAC alone). Results are expressed as mean and s.d. of triplicates.
B- Histone and non histone deacetylase activity of class IIa HDAC4. The histone deacetylase activity of HDAC4 was quantified as in A. The non histone deacetylase activity was measured using trifluoroacetyl-lysine as substrate.
Regulation of HDAC gene expression by HDIs
The cellular effects of HDIs are thought to rely on their ability to inhibit HDAC enzymatic activity. However, regulation of HDAC expression by HDIs may also play a role in the alterations of cell behaviour, as recently suggested by Dokmanovic et al. showing a strong down-regulation of HDAC7 expression by SAHA in various normal and tumor cell lines (31). We thus analysed the effects of TSA and MC1575 on the expression of class I, II and IV HDACs (HDAC1 to 11) by treating MCF-7 cells with either HDI and measuring the corresponding mRNA levels.
Of the eleven HDACs analysed, HDAC2, 3 and 8 (class I) and HDAC7 and 10 (class II) were found to be significantly regulated by TSA (Figure 3). Similar to the results obtained by Dokmanovic et al., TSA strongly inhibited HDAC7 gene expression (p = 0.0006 vs control cells). Although to a lesser extent, TSA was also found to significantly down-regulate mRNA levels of HDAC2, HDACS and HDAC10 (p = 0.0175, p = 0.0003 and p = 0.0017 vs control cells, respectively) and to increase that of HDACS (p < 0.0001 vs control cells). These regulations by TSA were specific since no variations of HDAC1 mRNA levels were noticed in the same conditions (Figure 3).
Figure 3. Regulation of HDAC gene expression by HDIs.
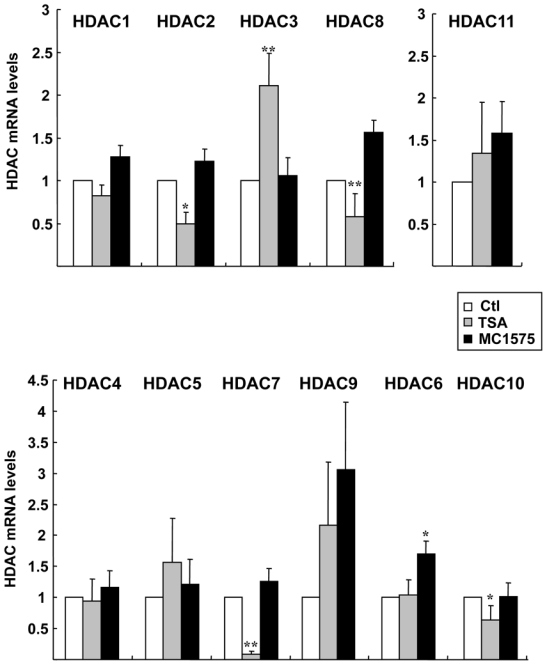
MCF-7 cells were treated with TSA (1.7 μM), MC1575 (20 μM) or vehicle alone (Control - Ctl) for 20 h and mRNA levels for the 11 HDACs were measured using RT-qPCR. Results are expressed relative to the TBP housekeeping gene and to the mRNA levels measured for the untreated control cells used as reference. Results represent mean and s.d of 8 (for TSA) or 4 (for MC1575) independent cell cultures. Raw data were used for statistical analysis. * p ≤ 0.01, ** p ≤ 0.001.
Interestingly, and in contrast to TSA, the class II specific inhibitor MC1575 did not modulate the mRNA levels of any of these HDACs but instead, induced the expression of class II HDAC6 (p = 0.0095 vs control cells) (Figure 3). We also observed a trend towards an increase in HDAC9 mRNA levels upon TSA and MC1575 treatment. However, because of the high variability in HDACS mRNA values due to the weak expression of its gene in MCF-7 cells, these variations upon HDI were not found to be statistically significant. Most importantly, none of the class I HDACs was down-regulated by MC1575, strengthening the specific targeting of class II enzymatic activity by this HDI.
Effects of class II HDIs on breast cancer cell proliferation, cell cycle and apoptosis
We then compared the effect of increasing concentrations of TSA and MC1575 on mammary tumor cell proliferation (Figure 4). As previously shown (15), TSA exhibited a potent anti-proliferative activity on MCF-7 cells which was less pronounced on the ERα-negative MDA-MB 231 breast cancer cell line. MC1575 was also found to have a dose-dependent growth-inhibitory activity on MCF-7 cells, albeit at higher concentrations (μM) than TSA (nM), which is consistent with their respective IC50 (28). Similar results were obtained using MC1568 (data not shown). Interestingly, ERα-negative MDA-MB 231 cells were also less sensitive to MC1575 treatment than ERα–positive MCF-7 cells (Figure 4).
Figure 4. Effects of class II HDI on breast tumor cells proliferation.
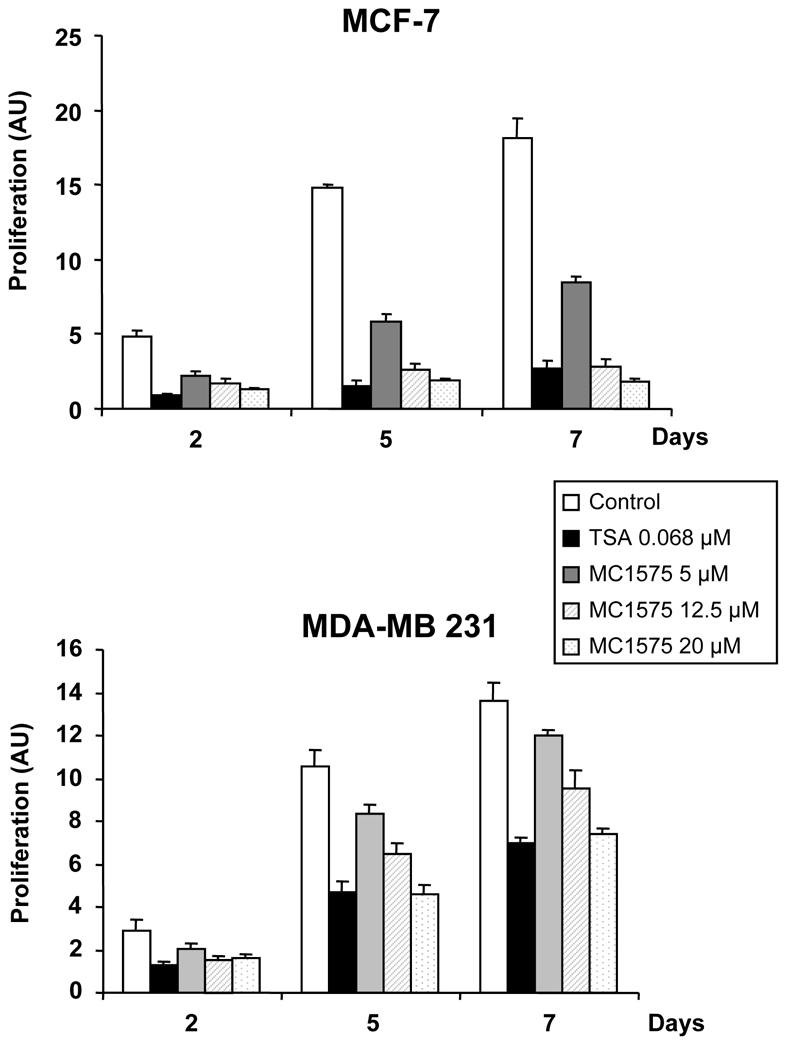
ERα-positive MCF-7 cells or ERα-negative MDA-MB 231 cells were treated with TSA (0.068 μM), MC1575 at 5, 12.5 and 20 μM or solvent alone (Control) and cell proliferation was measured by diaminobenzoic acid assay at day 2, 5 and 7. Results, expressed as arbitrary units (AU), represent mean and s.d. of triplicate wells and are representative of 4 independent experiments.
Flow cytometry analysis further confirmed the dose-dependent anti-proliferative effects of TSA and MC1575 on MCF-7 cells (Figure 5A). This effect was observed with the lowest concentration of MC1575 or MC1568 tested (5 μM). However, although both TSA and MC1575/1568 induced cell cycle arrest, a different profile was obtained with either HDI, showing accumulation of cells in the G2/M phase of the cell cycle for TSA, and in the G1 phase for MC1575 and MC1568.
Figure 5. Effects of class II HDI on cell cycle and apoptosis in MCF-7 cells.
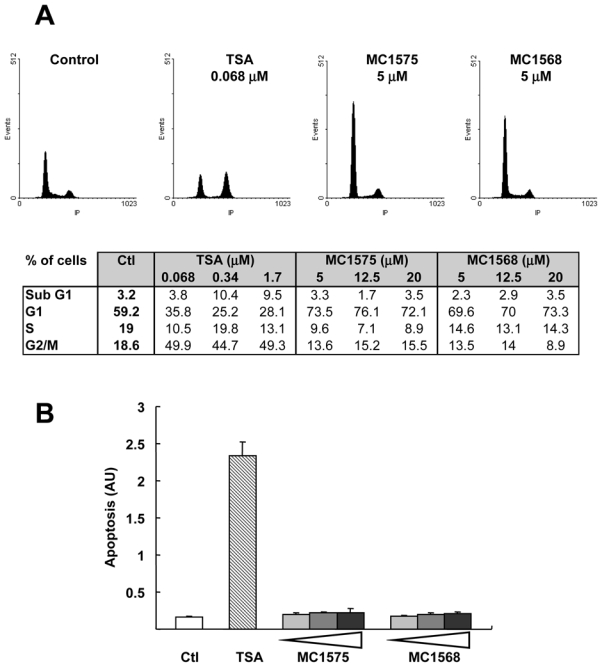
A- MCF-7 cells were treated for 20 h with increasing concentrations of TSA, MC1575, MC1568 or solvent alone (Control). Cell phase distribution was determined by PI staining and FACs analysis. Representative panels obtained for control cells or cells treated with TSA (0.068 μM), MC1575 (5 μM) or MC1568 (5 μM) are shown. In the table are presented the percentages of MCF-7 cells in the various phases of the cell cycle in response to HDI. B- MCF-7 cells were treated for 40 h with TSA (0.068 μM), increasing concentrations of MC1575 and MC1568 (5, 12.5 or 20 μM) or vehicle alone (Ctl) and apoptosis was measured using the Cell Death Detection ELISA kit. Results are expressed as arbitrary units and represent mean and s.d of 4 wells.
HDIs have been shown to induce apoptosis through various pathways in tumor cells, so we investigated whether MC1575 and MC1568 had the same effect in MCF-7 cells (Figure 5B). TSA was found to markedly induce MCF-7 cell apoptosis, whereas MC1575 and MC1568 had no effects even at the highest dose tested (20 μM). These results were consistent with those obtained using flow cytometry analysis as observed from the fraction of cells in the subG1 phase of the cell cycle, which increased upon TSA treatment but remained similar to the control upon MC1575 or MC1568 treatment (Figure 5A).
Inhibition of class II HDACs and expression of cell cycle and apoptosis regulators
Since HDIs impact MCF-7 cell proliferation and cell cycle progression, we next analyzed the expression of some cell cycle regulators in response to TSA or MC1575 treatment (Figure 6A). As previously shown for various HDIs, TSA and MC1575 were found to increase with equal potency the cell cycle inhibitor p21cip1/waf1 at the mRNA and protein levels. However, both HDI differently affected the expression of other cell cycle regulators: TSA markedly decreased the expression of cyclin D1 at the mRNA and protein levels, and decreased p27 and p14ARF gene expression, whereas MC1575 had no effect or a modest effect on these parameters.
Figure 6. Effects of class II HDI on the expression of cell cycle and apoptosis regulators.
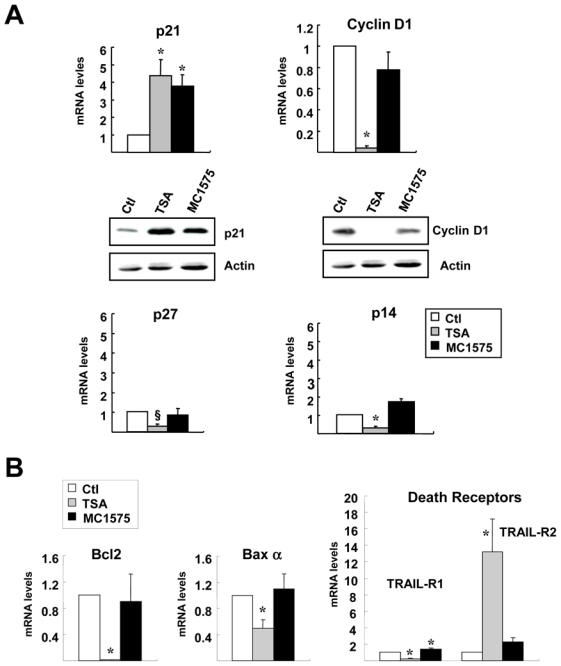
A- MCF-7 cells were treated with TSA (1.7 μM), MC1575 (20 μM) or vehicle alone (Control - Ctl) for 6 h (p27 and p14ARF) or 20 h (p21cip1/waf1 and cyclin D1) and mRNA levels for p21, cyclin D1, p27 and p14 genes were measured using RT-qPCR. Results are expressed relative to the TBP housekeeping gene and to the mRNA levels measured for the untreated control cells used as reference. Results represent mean and s.d of 4 independent cell cultures. For p21 and cyclin D1, western-blot analysis was performed in the same conditions, using actin as a loading control.
B- MCF-7 cells were treated for 20h with TSA (1.7 μM), MC1575 (20 μM) or vehicle alone (Control - Ctl) and Bcl2, Bax α, TRAIL-R1 and TRAIL-R2 mRNA levels were quantified using RT-qPCR. Results were expressed as in A and represent mean and s.d of 4 independent cell cultures. Raw data were used for statistical analysis, §p = 0.05, * p < 0.05 as compared to control cells.
When measuring mRNA levels for Bcl2 and Baxα, two members of the Bcl2 family involved in the intrinsic apoptotic pathway, we found that TSA had a strong inhibitory effect on the expression of the anti-apoptotic Bcl2 gene while modestly but significantly decreasing that of the pro-apoptotic Baxα regulator (Figure 6B). In addition, TSA was found to regulate the expression of members of the death receptors family involved in the extrinsic pathway, decreasing TRAIL-R1 expression and markedly inducing TRAIL-R2 mRNA accumulation (~ ×13). By contrast, MC1575 had no or only a very modest effect on these regulators, in accordance with its lack of regulation of MCF-7 apoptosis. Altogether, these results suggest that, in breast cancer cells, cell cycle and apoptosis regulators are specifically and differentially targeted by the various classes of HDACs.
Inhibition of class II HDACs and ER signalling
Several HDIs, including TSA, have been shown to differentially regulate ERα and ERβ expression and transcriptional activity in ERα-expressing mammary tumor cells (19, 20). These studies have been performed using non selective HDAC inhibitors, and we wanted to assess if a specific inhibition of class II HDACs would have similar effects in MCF-7 cells.
Consistent with previous data, TSA had marked effects on the expression of both ER isoforms in MCF-7 cells, down-regulating ERα both at the mRNA and protein levels, while increasing ERβ mRNA levels (Figures 7A and 7B). ERα and ERβ isoforms followed different kinetics upon TSA treatment, as ERα inhibition was detected at 6h and maintained after 20h, whereas ERβ induction was the strongest at 6h and decreased thereafter, suggesting different regulation pathways (Figure 7A and data not shown). Interestingly, MC1575 showed a different effect than TSA on these two parameters, as it had only a weak and not significant inhibitory effect on ERα expression (mRNA and protein), but a marked and stronger stimulatory effect on ERβ gene expression.
Figure 7. Effects of HDI on ERα and ERβ expression and activity in MCF-7 cells.
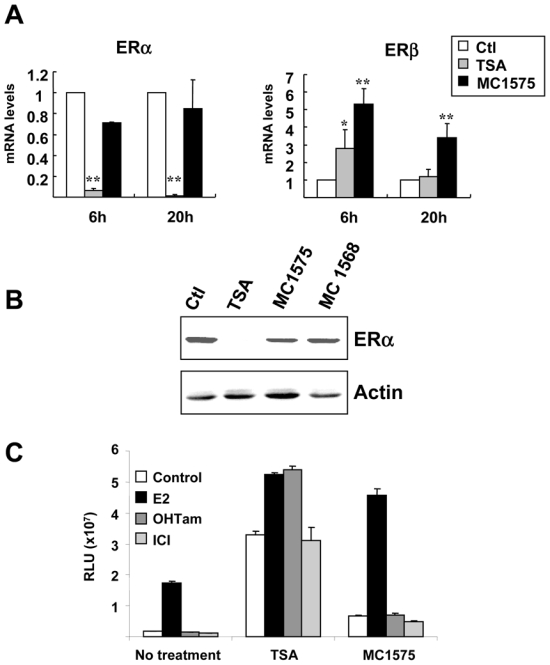
A- MCF-7 cells were treated for 6h or 20 h with TSA (1.7 μM), MC1575 (20 μM) or vehicle alone (Control - Ctl) and mRNA levels for ERα and ERβ were quantified using RT-qPCR. Results are expressed relative to the TBP housekeeping gene and to the mRNA levels measured for the control cells used as reference. Results represent mean and s.d of at least 4 independent cell cultures. Raw data were used for statistical analysis. * p < 0.05, ** p ≤ 0.01 as compared to control cells.
B- MCF-7 cells were treated for 20 h with TSA (1.7 μM), MC1575 (20 μM), MC1568 (20 μM) or vehicle alone (Control -Ctl) and ERα protein levels were analyzed by western immunoblotting. Actin was used as a loading control.
C- MELN cells were treated for 20h with control vehicle (Control), or 17β-estradiol (E2; 10−8M), OHTam (10−8M) or ICI (10−8M) in the absence or presence of TSA (1.7 μM) or MC1575 (20 μM) and luciferase activity was quantified. Results are expressed as relative luciferase units (RLU) and represent mean and s.d. of triplicate wells. These results are representative of 3 independent experiments.
We next studied the effects of TSA and MC1575 on ERα transcriptional activity (Figure 7C). Recent reports from our laboratory have shown that HDI, including TSA, enhanced the ligand dependent activity of ERα and ERβ and that partial antiestrogens such as OHTam switch their antagonist activity to an agonist one upon HDAC inhibition, this latter effect being related to ERα down-regulation (14, 19). In stably transfected bioluminescent MCF-7 cells (MELN clone), TSA strongly enhanced the transcriptional activity of ERα in presence or absence of ER ligands and switched OHTam to an agonist activity, without modifying the behaviour of the pure antagonist ICI 182,780. Inhibition of class II HDACs using MC1575 weakly enhanced ERα transcriptional activity as compared to TSA, but in contrast to TSA did not switch the partial antiestrogen OHTam to an agonist, in accordance with its lack of effect on ERα expression in MCF-7 cells.
Discussion
Several studies have underlined the role of HDACs in breast physiology and tumorigenesis (13, 15–17, 32, 33). Some studies have focused on particular HDACs (HDAC1, 3 and 6) and their roles in breast carcinoma (34–36). However, little is known on the specific contribution of the various classes of HDACs in mammary tumorigenesis, which is an important issue from a cognitive point of view and in the context of HDI development as promising anticancer therapies. The aim of this study was thus to investigate this issue in a model of estrogen responsive breast tumor cell line using HDAC inhibitors specifically targeting the class II HDAC sub-family.
MC1575 and MC1568 are newly designed synthetic inhibitors of class II HDACs (27, 28). Although these HDI are less potent in inhibiting HDACs than TSA, their selectivity against class II HDACs has been shown using various models (24, 25, 27, 28). We confirmed this selectivity in MCF-7 cells and by means of recombinant HDACs, showing that MC1575 and MC1568 had no or a weak effect on the histone deacetylase activity of class I enzymes (Figures 1 and 2).
Class IIa HDACs expressed in mammalian cells have been shown to recruit class I HDACs, which display high deacetylase activity, and several reports have questioned whether class IIa HDACs by themselves had an intrinsic deacetylase activity. Recently, Lahm et al. showed that the low catalytic activity of mammalian class IIa HDACs was linked to the presence of a unique histidine residue in the catalytic domain of these enzymes in place of the tyrosine residue observed in the conserved active site of class I HDACs (30). Despite this structural particularity, class IIa HDACs were shown to possess a weak but measurable enzymatic activity in vitro against acetyllysine histone substrates. We also found that recombinant HDAC4, which is representative of class IIa HDACs family according to sequence and structure homologies, had histone deacetylase activity in vitro and showed that MC1575 efficiently inhibited this activity, being as efficient as SAHA at the highest dose tested. Interestingly, by screening a panel of acetylated lysine-like molecules, Lahm et al. also identified trifluoroacetyl-lysine as a substrate specific for class IIa HDACs, on which these enzymes were highly active, suggesting that mammalian class IIa HDACs may have additional biological substrates and activities, different from canonical histone deacetylation (29, 30). Using this specific substrate, we also showed that MC1575 and MC1568 were potent inhibitors of HDAC4 catalytic activity.
In addition to the well-documented in vitro and in vivo inhibition of HDAC enzymatic activity, HDI may also impact global acetylation levels by controlling HDAC expression. Using the non selective HDI TSA, we observed that class II HDAC7 was markedly down-regulated at the mRNA level in various breast cancer cell lines (Figure 3 and data not shown), thus confirming previously published data (31). We also found that TSA could significantly modulate the expression of other class I and class II HDACs (down-regulation of HDAC2, 8 and 10 and increase of HDAC3). Interestingly, none of these negative regulations was observed using the class II specific HDI MC1575. Altogether, these results, along with previous studies (25, 28), confirm that MC1575 and MC1568 are specific class II HDI, as they do not inhibit class I HDAC activity or expression.
Using these specific inhibitors, we first addressed the effect of class II HDAC inhibition on human breast cancer cell proliferation. Non selective HDI have been shown to display anti-proliferative effects in various tumor models, including breast tumors, and this effect has been linked to cell cycle arrest at the G1/S and/or G2/M check-points (1, 9). Among the few genes regulated by HDI, the cell cycle inhibitor p2lcip1/waf1 is consistently up-regulated by these compounds, which may explain in part their anti-proliferative effects (37). Studies based on HDAC1-deficient cells or siRNA approaches suggest a predominant role of class I HDACs, and more particularly HDAC1 and HDAC3, in the control of cell proliferation and cell cycle (38–40). For instance, Lagger et al. found that HDAC1-deficient embryonic stem cells presented reduced proliferation rates along with an increased expression of the cell cycle inhibitors p21waf1 and p27KIP1(39). Similarly, Glaser et al. using a siRNA approach, showed that inhibiting the expression of HDAC1 or HDAC3 in HeLa cells induced morphological changes and reduced their proliferation, in contrast to inhibition of class II HDAC4 and HDAC7, which had no effects (38). Finally, analysis of HDAC5, 9 and 4 null-mice phenotypes suggest that class II HDACs are mainly involved in tissue-specific growth and differentiation, rather than in cell proliferation (5, 7).
Our results indicate that in addition to class I HDACs, class II enzymes may also be involved in cell proliferation and cell cycle control, at least in breast cancer cells. First, we showed that MC1575 and MC1568 were both able to inhibit MCF-7 and MDA-MB 231 cell proliferation in a dose-dependent manner. Moreover, as observed with TSA and other non selective HDI, those compounds strongly induced P21waf1 gene and protein expression. Interestingly, MC1575 and MC1568 induced different effects on cell cycle and cyclin D1 expression as compared to TSA, suggesting that class I and II HDACs may control cell cycle at specific levels.
HDIs have also been shown to induce apoptosis in tumor cell models through various molecular pathways, including regulation of the expression of members of the Bcl2 family, up-regulation of death receptors or induction of oxidative injury (41, 42). As previously shown, TSA was found to strongly induce apoptosis in MCF-7 cells, to decrease the expression of Bcl2 gene and markedly induce the expression of the death receptor TRAIL-R2 (43, 44). By contrast, our results clearly showed that inhibition of class II HDACs was not involved in the pro-apoptotic effects of HDI in MCF-7 cells. The absence of apoptotic effects upon MC1575 and MC1568 treatment was consistent with the weak effects of these compounds on the expression of Bcl2, Baxα and death receptors TRAIL-R1 and TRAIL-R2 genes. Similarly, Inoue et al. found in another tumor model, that inhibition of class I but not class II HDACs was critical for sensitization of cells to TRAIL-induced apoptosis, suggesting that our observation in MCF-7 cells may be a more general phenomenon (25).
Finally, we and others have shown that TSA differentially regulated ERα and ERβ in ERα-positive human breast and ovarian cell lines, leading to a strong decrease in ERα accumulation, contrasting with an increase in ERβ expression (14, 19, 20). In the present study, we confirmed these results and found that the regulation of ERα and ERβ expression upon HDI treatment followed different kinetics, suggesting that various molecular pathways are involved. Moreover, specific inhibition of class II HDACs led to a different profile of regulation for both ER isoforms than TSA, as ERα was weakly altered whereas ERβ was still strongly induced. The use of MS-275, a class I specific HDAC inhibitor, further suggested that ERα down-regulation was predominantly linked to class I HDAC inhibition (data not shown). In contrast, ERβ gene expression was induced by all HDIs, indicating that various classes of HDACs could be involved in its regulation. Whatever the underlying molecular mechanisms, the effects of MC1575 and MC1568 on ERα and ERβ isoforms in MCF-7 cells potentially represent an interesting physiological condition whereby ERβ expression is induced while that of ERα remains unchanged.
As previously shown (14), ERα transcriptional activity was strongly induced by TSA. Although MC1575 also increased ERα transcriptional activity, this effect was weaker than that observed with TSA suggesting that inhibition of class II HDACs was probably not the only factor involved in this regulation. The switch of the partial antiestrogen OHTam to an agonistic transcriptional activity upon HDI treatment has been linked to ERα down-regulation (14). Our results are consistent with this hypothesis since upon MC1575 treatment, MCF-7 cells displayed high levels of ERα protein together with an antagonist ER activity for OHTam.
From a clinical point of view, it has been suggested that combined therapies associating HDI and antiestrogen could be helpful for the treatment of patients with breast carcinomas expressing or not ERα. HDAC inhibition has indeed been shown not only to enhance the anti-proliferative action of antiestrogens on ERα-positive breast cancer cells (45) but also to sensitize ERα-negative breast cancer cells to tamoxifen after reactivation of ERβ expression (20). In this context, our data highlighting the antiproliferative activity of MC1575 and its effects on ER expression and activity (i.e higher levels of ERs than in the presence of TSA and persistence of an antagonistic activity for OHTam) suggest that such a drug could be of potential interest for future therapeutical approaches in combination with antiestrogens.
In conclusion, our results evidence major differential effects of class II HDAC inhibition on cell cycle progression, apoptosis, gene expression and ER signalling in mammary tumor cells, strengthening the notion that the different HDAC sub-classes could play specific roles in breast tumorigenesis. It should be stressed that, when considering the different sets of genes analyzed in this study (HDACs, cell cycle, apoptosis regulators or ERs), MC1575 always failed to reproduce the negative regulations of gene expression observed upon TSA treatment, while recapitulating most of its positive effects. Further studies using additional HDIs and siRNAs will be needed to precise the role of individual HDAC, especially class II enzymes, on the regulation of gene expression and cellular processes in breast tumor cells. The development of such approaches that specifically target HDAC isotypes will be critically important in the future for a better comprehension of the roles of these proteins in physiological or pathological processes and to propose the most ideal therapies with greatest efficacy and least unintended side effects.
Materials and Methods
Reagents
Estradiol-17β (E2) and trichostatin A (TSA) were from Sigma-Aldrich (St Quentin Fallavier, France). Hydroxy-tamoxifen (OHTam) and ICI 182,780 (ICI) were kind gifts of Sanofi-Aventis and Astrazeneca, respectively. MC1575 and MC1568 were synthesized as previously described (27, 28), dissolved in DMSO and stored at −20°C before use. MS-275 was obtained from Calbiochem (VWR, Strasbourg, France).
Cell culture
MCF-7 cells were grown in Ham’s F-12/Dulbecco’s modified Eagle’s medium (1:1) (F12/DMEM) supplemented with 10% fetal calf serum (FCS) (Invitrogen) and antibiotics. Before hormonal treatment, cells were stripped of endogenous steroids by passage in medium without phenol red containing 3% charcoal-stripped FCS (DCC medium). For experiments using estrogen ligands or HDIs, control cells were grown in medium complemented with vehicle alone (ethanol or DMSO). The MELN cell line was derived from MCF-7 cells stably transfected with the ERE-βGlob-Luc-SVneo plasmid (46).
RNA extraction and real-time quantitative PCR
Total RNA was extracted using RNeasy minikit (Qiagen, Courtaboeuf, France) according to the manufacturer’s conditions. For RT-PCR, 1.5 μg of total RNA was subjected to reverse transcription using the Omniscript Reverse Transcriptase kit (Qiagen, Courtaboeuf, France). Real-time PCR quantification was then performed using a SYBR Green technology (Light Cycler, Roche). For each sample, mRNA levels of specific genes were corrected for TBP mRNA levels used as a reference gene and normalized to a calibrator sample (untreated MCF-7 cells). The primers for ERα, ERβ, cyclin D1, TBP, p21 and p27 genes have been described elsewhere (47–49). Primers for the other genes are depicted in Table I.
Table I.
Primers sequences used for qPCR.
| Primers | Gene accession no. | PCR size product (nt) | |
|---|---|---|---|
| HDAC 1 |
F-5′-CCTGAGGAGAGTGGCGATGA-3′ R-5′-GTTTGTCAGAGGAGCAGATCGA-3′ |
NM 004964 | 69 |
| HDAC 2 |
F-5′-GCTCTCAACTGGCGGTTCAG-3′ R-5′-AGCCCAATTAACAGCCATATCAG-3′ |
NM 001527 | 75 |
| HDAC 3 |
F-5′-CCCAGACTTCACACTTCATCCA-3′ R-5′-GGTCCAGATACTGGCGTGAGTT-3′ |
NM 003883 | 70 |
| HDAC 4 |
F-5′-GACCTGACCGCCATTTGC-3′ R-5′-GGGAGAGGATCAAGCTCGTTT-3′ |
NM 006037 | 73 |
| HDAC 5 |
F-5′-CAACGAGTCGGATGGGATGT-3′ R-5′-GGGATGCTGTGCAGAGAAGTC-3′ |
NM 005474 | 74 |
| HDAC 6 |
F-5′-TGCCTCTGGGATGACAGCTT-3′ R-5′-CCTGGATCAGTTGCTCCTTGA-3′ |
NM 006044 | 69 |
| HDAC 7 |
F-5′-AGCAGCTTTTTGCCTCCTGTT-3′ R-5′-TCTTGCGCAGAGGGAAGTG-3′ |
NM 015401 | 66 |
| HDAC 8 |
F-5′-CGGCCAGACCGCAATG-3′ R-5′-CACATGCTTCAGATTCCCTTT-3′ |
NM 018486 | 56 |
| HDAC 9 |
F-5′-AGGCTCTCCTGCAGCATTTATT-3′ R-5′-AAGGGAACTCCACCAGCTACAA-3′ |
NM 014707 | 75 |
| HDAC 10 |
F-5′-ATGACCCCAGCGTCCTTTACT-3′ R-5′-CGCAGGAAAGGCCAGAAG-3′ |
NM 032019 | 66 |
| HDAC 11 |
F-5′-CCCCTTGGTCATGGGATTT-3′ R-5′-CATCCACACCAGTGCCTATAGC-3′ |
NM 024827 | 68 |
| Bcl 2 |
F-5′-GGTGCCACCTGTGGTCCACCTG-3′ R-5′-CTTCACTTGTGGCCCAGATAGG-3′ |
NM 633.2 | 459 |
| Bax α |
F-5′-ATGGACGGGTCCGGGGAGGAGC-3′ R-5′-CCCCAGTTGAAGTTGCCGTCAG-3′ |
NM 138761.2 | 323 |
| P14ARF |
F-5′-GGTTTTCGTGGTTCACATCC-3′ R-5′-CCTCAGTAGCATCAGCACGA-3′ |
NM 058195 | 91 |
| TRAIL-R1 (TNFRS10A) |
F-5′-TGCTTCCAACAATTTGTTTGCT-3′ R-5′-CGTGGTGCAGGGACTTCTCT-3′ |
NM 003844.2 | 79 |
| TRAIL-R2 (TNFRS10B) |
F-5′-GGTTCCAGCAAATGAAGGTGAT-3′ R-5′-AAGGGCACCAAGTCTGCAAA-3′ |
NM 003842.3 | 75 |
Western-blot analysis
Whole-cell extracts were prepared in high-salt lysis buffer (HSB) containing 500 mM NaCl, 50 mM Tris (pH 8), 1% NP40, 1 mM DTT, and proteases inhibitors (Roche Diagnostics). Proteins were quantified using the Bradford assay (Bio-Rad Laboratories), and 60 μg were usually loaded on SDS-PAGE and transferred to nitrocellulose membrane. Blots were saturated in TEST buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% Tween 20 (v/v), 5% non-fat dehydrated milk (w/v)), incubated with specific primary antibodies for cyclin D1 (clone sc-718, Tebu), ERα (clone sc-543, Tebu), p21WAF1/CIP1 (Cell Signaling), acetyl-Histone 3, acetyl-Histone 4 (Upstate Biotechnology), acetyl-tubulin (clone 6–11B-1, Sigma) or actin (Sigma), and probed with the appropriate secondary antibody (Sigma). Detection was done using the Chemiluminescence Reagent Plus kit (Perkin-Elmer Life Science).
Deacetylase assays
Full-length HDAC1, 2, 3, 8 and 4 with C-terminal His tag were expressed using baculovirus expression systems. For histone deacetylase activity assays, purified HDACs (100 ng for class I HDACs; 250 ng for HDAC4 on histone substrate; 20 ng of HDAC4 on non histone substrate) were pre-incubated or not with HDI for 15 min and used in the HDAC fluorescent activity assay performed according to the supplier’s instructions (BIOMOL, Palatine House, Matford Court, UK). Fluorescence was quantified using a TECAN Infinite M200 station. For measurement of HDAC4 activity against non histone substrate, the trifluoroacetyl-lysine substrate, specific for class IIa HDACs, was synthesized and used as previously described (29, 30). Assays were carried out in triplicates.
Cell proliferation
For proliferation studies, cells were seeded at 25.103 cells/well in 24-well dishes in F12/DMEM medium supplemented with 10% FCS. After 24 h, cells were treated with either vehicle alone or HDIs at various concentrations and total cell DNA was quantified by diaminobenzoic acid assay at day 2, 5 and 7 as previously described (19). During cell proliferation assay, treatment with HDI or vehicle alone was renewed every two days.
Cell cycle analysis
For cell cycle analysis, 2.106 cells were seeded in 25 cm2 flasks. After 24 h, cells were treated for 20 h with either solvent alone or HDI at various concentrations. Cells were then pelleted, washed and incubated in a staining solution containing 10 μg/ml propidium iodide (PI). The cellular suspension was passed through a FACS Vantage flow cytometer (Becton Dickinson). For each sample, at least 2.104 events were acquired and analyzed using CellQuest 3.3 (Becton Dickinson) and ModFit LT 3.1 softwares.
Apoptosis measurement
MCF-7 cells were plated in 6-well plates (5.104 cells/well) and treated or not with TSA, MC1575 or MC1568 for 40 h. Apoptosis was quantified 24 h later using the Cell Death Detection ELISA (Roche Molecular Biochemicals), according to the manufacturer’s conditions. Values from absorbance measurements at 405 nm were corrected using DNA quantification in separate wells treated in parallel.
Luciferase assays
For measurement of ERα-dependent transactivation assays, MELN cells were plated in 6-well plates (5.105 cells per well). Cells were lysed at 4°C for 10 min in 400μl of lysis buffer (25mM Tris pH 7.8, 2mM EDTA, 10% glycerol, 1% Triton X-100) and luciferase activity was measured as previously described (50).
Statistical analysis
Results are expressed as mean ± standard deviations (s.d.). Statistical analysis was performed on raw data using Student’s t test or Mann-Whitney U test for comparison of two groups. A probability level of 0.05 was chosen for statistical significance. Statistical analysis was performed using GraphPad InStat version 3.06 (GraphPad Software, San Diego, CA, USA).
Acknowledgments
Grant support: This work was supported by the “Institut National de la Santé et de la Recherche Médicale”, the University of Montpellier I, the “Ligue Nationale centre le Cancer”, the “Association pour la Recherche sur le Cancer” and the Centre Hospitalo-Universitaire de Montpellier, HEALTH-F4-2007-200767, LSHC-CT2005-518417, PRIN 2006 and AIRC 2007. V.D. was a recipient of fellowships from the French Minister of Research and the “Association pour la Recherche sur le Cancer”. P.O.H was supported by the Ligue Nationale Centre le Cancer.
The authors wish to thank Stephan Jalaguier for critical reading of the manuscript.
References
- 1.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 2.Yang X-J, Seto E. HATs and HDACs: from structure, function and regulation to novel stratégies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 3.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature Rev Cancer. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 4.Boyault C, Zhang Y, Fritah S, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson AN. Histone deacetylase 5 and 9 govern responsiveness of the heart to a subset of stress signals ans play redundant roles in the heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dequiedt F, Kasler H, Fischle W, et al. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 2003;18:687–698. doi: 10.1016/s1074-7613(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 7.Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–5467. doi: 10.1038/sj.onc.1210613. [DOI] [PubMed] [Google Scholar]
- 8.Vega RB, Matsuda K, Oh J, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 9.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Rev Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 10.Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treatment Rev. 2006;32:157–165. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111–1120. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 12.Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 13.Davis T, Kennedy C, Chiew Y-E, Clarke CL, deFazio A. Histone deacetylase inhibitors decrease proliferation and modulate cell cycle gene expression in normal mammary epithelial cells. Clin Cancer Res. 2000;6:4334–4342. [PubMed] [Google Scholar]
- 14.Margueron R, Duong V, Bonnet S, et al. Histone deacetylase inhibition and estrogen receptor α levels modulate the transcriptional activity of partial antiestrogens. J Mol Endocrinol. 2004;32:583–594. doi: 10.1677/jme.0.0320583. [DOI] [PubMed] [Google Scholar]
- 15.Margueron R, Licznar A, Lazennec G, Vignon F, Cavailles V. Oestrogen receptor α increases p21 waf1/Cip1 gene expression and the antiproliferative activity of histone deacetylase inhibitors in human breast cancer cells. J Endocrinol. 2003;179:41–53. doi: 10.1677/joe.0.1790041. [DOI] [PubMed] [Google Scholar]
- 16.Vigushin DM, Ali S, Pace PE, et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7:971–976. [PubMed] [Google Scholar]
- 17.Yang X, Ferguson AT, Mass SJ, et al. Transcriptional activation of estrogen receptor α in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000;60:6890–6894. [PubMed] [Google Scholar]
- 18.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 19.Duong V, Licznar A, Margueron R, et al. ERα and ERβ expression and transcriptional activity are differentially regulated by HDAC inhibitors. Oncogene. 2006;25:1799–1806. doi: 10.1038/sj.onc.1209102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang ER, Lim S-J, Lee ES, et al. The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor α-negative breast cancer cells to tamoxifen. Oncogene. 2004;23:1724–1736. doi: 10.1038/sj.onc.1207315. [DOI] [PubMed] [Google Scholar]
- 21.Kenn JC, Van L, Mack KM, et al. A novel histone deacetylase inhibitor, Scriptaid, enhances expression of functional estrogen receptor α (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat. 2003;81:177–186. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- 22.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Release of methyl CpG binding proteins and histone deacetylase 1 from the estrogen receptor α (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol Endocrinol. 2005;19:1740–1751. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q, Ataja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biology and Therapy. 2007;6:64–69. doi: 10.4161/cbt.6.1.3549. [DOI] [PubMed] [Google Scholar]
- 24.Illi B, Dello Russo C, Colussi C, et al. Nitric Oxide modulates chromatin folding in human endothelial cells via PP2A activation and class II HDACs nuclear shuttling. Circ Res. 2008;102:51–58. doi: 10.1161/CIRCRESAHA.107.157305. [DOI] [PubMed] [Google Scholar]
- 25.Inoue S, Mai A, Dyer MJS, Cohen GM. Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2006;66:6785–6792. doi: 10.1158/0008-5472.CAN-05-4563. [DOI] [PubMed] [Google Scholar]
- 26.Kim IA, Shin JH, Kim IH, et al. Histone deacetylase inhibitor-mediated radiosensitization of human cancer cells: class differences and the potential influence of p53. Clin Cancer Res. 2006;12:940–949. doi: 10.1158/1078-0432.CCR-05-1230. [DOI] [PubMed] [Google Scholar]
- 27.Mai A, Massa S, Pezzi R, Rotili D, Loidl P, Brosch G. Discovery of (aryloxopropenyl)pyrrolyl hydroxyamides as selective inhibitors of class IIa histone deacetylase homologue HD1-A. J Med Chem. 2003;46:4826–4829. doi: 10.1021/jm034167p. [DOI] [PubMed] [Google Scholar]
- 28.Mai A, Massa S, Pezzi R, et al. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (Aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem. 2005;48:3344–3353. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 29.Jones P, Altamura S, De Francesco R, et al. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18:1814–1819. doi: 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 30.Lahm A, Paolini C, Pallaoro M, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acd Sci. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dokmanovic M, Perez G, Xu W, et al. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther. 2007;6:2525–2534. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- 32.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor α. Int J Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 33.Varshochi R, Halim F, Sunters A, et al. ICI182.780 induces p21Waf1 gene transcription through releasing histone deacetylase 1 and estrogen receptor a from Sp1 sites to induce cell cycle arrest in MCF-7 breast cancer cell line. J Biol Chem. 2005;280:3185–3196. doi: 10.1074/jbc.M408063200. [DOI] [PubMed] [Google Scholar]
- 34.Krusche CA, Wülfing P, Kersting C, et al. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat. 2005;90:15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 35.Saji S, Kawakami M, Hayashi S, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005;24:4531–4539. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, Yamashita H, Toyama T, et al. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10:6962–6968. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- 37.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–1374. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK. Role of class I and class II histone deacetylase in carcinoma cells using siRNA. Biochem Biophys Res Commun. 2003;310:529–536. doi: 10.1016/j.bbrc.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 39.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senese S, Zaragoza K, Minardi S, et al. Role for histone deacetylase 1 in human cell proliferation. Mol Cell Biol. 2007;27:4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–6271. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 42.Rosato RR, Grant S. Histone deacetylase inhibitors: insight into mechanisms of lethality. Expert Opin Ther Targets. 2005;9:809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- 43.Chopin V, Slomianny C, Hondermarck H, Le Bourhis X. Synergistic induction of apotosis in breast cancer cells by cotreatment with butyrate and TNF-alpha, TRAIL, or anti-Fas agonist antibody involves enhancement of death receptors’ signaling and requires p21waf1. Exp Cell Res. 2004;298:560–573. doi: 10.1016/j.yexcr.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 44.Chopin V, Toillon RA, Jouy N, Le Bourhis X. p21waf1/cip1 is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in breast cancer cells. Oncogene. 2004;23:21–29. doi: 10.1038/sj.onc.1207020. [DOI] [PubMed] [Google Scholar]
- 45.Hodges-Gallagher L, Valentine CD, El Bader S, et al. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res Treat. 2007;105:297–309. doi: 10.1007/s10549-006-9459-6. [DOI] [PubMed] [Google Scholar]
- 46.Balaguer P, François F, Comunale F, et al. Reporter cell lines to study the estrogenic effects of xenoestrogens. Sci Total Environ. 1999;233:47–56. doi: 10.1016/s0048-9697(99)00178-3. [DOI] [PubMed] [Google Scholar]
- 47.Anicotte J-S, lankova I, Miard S, et al. Peroxisome Proliferator-Activated Receptor γ regulates E-cadherin expression and inhibits growth and invasion of prostate cancer. Mol Cell Biol. 2006;26:7561–7574. doi: 10.1128/MCB.00605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y-X, Fang J-Y, Zhu H-Y, Lu R, Cheng Z-H, Qiu D-K. Histone acetylation regulates p21WAF1 expression in human colon cancer cell lines. World J Gastroenterol. 2004;10:2643–2646. doi: 10.3748/wjg.v10.i18.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delfour C, Roger P, Bret C, et al. RCL2, a new fixative, preserves morphology and nucleic acid integrity in paraffin-embedded breast carcinoma and microdissected breast tumor cells. J Mol Diagnostics. 2006;8:157–169. doi: 10.2353/jmoldx.2006.050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duong V, Boulle N, Daujat S, et al. Differential regulation of estrogen receptor α turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. 2007;67:5513–5521. doi: 10.1158/0008-5472.CAN-07-0967. [DOI] [PubMed] [Google Scholar]