

Abstract
ERK/MAPK pathway activity is regulated by the antagonist function of activating kinases and inactivating protein phosphatases. Sustained ERK pathway activity is commonly observed in human malignancies, however the mechanisms by which the pathway is protected from phosphatase-mediated inactivation in the tumor tissue remain obscure. Here we show that methylesterase PME-1-mediated inhibition of the protein phosphatase 2A (PP2A) promotes basal ERK pathway activity, and is required for efficient growth factor response. Mechanistically PME-1 is shown to support ERK pathway signaling upstream of Raf, but downstream of growth factor receptors and PKC. In malignant glioblastoma, PME-1 expression levels correlate with both ERK activity and cell proliferation in vivo. Moreover, PME-1 expression significantly correlates with disease progression in human astrocytic gliomas (N=222). Together, these observations identify PME-1 expression as one mechanism by which ERK pathway activity is maintained in cancer cells, and suggest important functional role for PME-1 in the disease progression of human astrocytic gliomas.
Keywords: PP2A, PPME-1, Ras/Raf/MEK/ERK, Malignant glioblastoma, tumor suppressor
Introduction
Protein phosphatase 2A (PP2A) is a human tumor suppressor that inhibits cellular transformation by regulating activity of several signaling proteins critical for malignant cell behaviour (reviewed in (1, 2)). Recent studies have indicated that phosphorylation and methylation of the C-terminal tail of the catalytic PP2A subunit (PP2Ac) play an important role in the regulation of both catalytic activity of PP2Ac, and recruitment of different substrate specific B-subunits to the PP2A complex (Reviewed in (3)). The nature of PP2Ac C-terminal kinase(s) remain elusive, but the methylation status of PP2Ac leucine 309 is regulated by a methylating enzyme (LCMT) and methylesterase PME-1 (3-6). Recent structural analysis of PME-1-PP2A complex revealed that, in addition to the previously identified role of PME-1 as PP2Ac leucine 309 methylesterase (4, 5), PME-1 inhibits PP2A activity by directly binding to the active site of PP2Ac (6). However, PP2A target proteins regulated by PME-1-mediated PP2A inhibition have not been elucidated.
Ras proteins (H-Ras, K-Ras and N-Ras) are small GTPases that mediate signals from activated receptor tyrosine kinases (RTKs) to intracellular signaling pathways. In addition, activating Ras mutations and/or increased Ras activity is detected in the majority of human malignant tumors (7). Although Ras activates several cellular signaling pathways that promote malignant cell behavior, recent studies have demonstrated that Ras-mediated activation of two of its effector cascades, ERK and RalA, is required for transformation of some types of human cells (8, 9), while simultaneous activation of the ERK and PI3K-Akt pathway is sufficient to replace activated H-Ras in other cell types (10). Interestingly, all Ras effector pathways described above (ERK, RalA, PI3K-Akt) are targeted by PP2A tumor suppressor activity (2, 11). Although a few cancer-relevant mechanisms of inhibiting PP2A tumor suppressor activity have been reported (2, 11-14), cancer-associated mechanisms preventing PP2A-mediated inactivation of the ERK pathway remain elusive.
The ERK pathway is a classical three-tiered mitogen activated protein kinase (MAPK) cascade consisting of Raf, MEK and ERK proteins. This pathway mediates the effects of activated Ras in the regulation of transcription, cell survival, proliferation and differentiation via ERK-mediated target protein phosphorylation (reviewed in (15, 16). As elucidated above, PP2A has been demonstrated to be a key negative regulator of ERK pathway activity (17, 18). Importantly, PP2A inhibition results in sustained ERK pathway activation, and prolongs transient TPA-elicited ERK phosphorylation (19, 20). Moreover, increased ERK pathway activity in brain tissue was observed in transgenic mice overexpressing a dominant-negative form of the PP2A catalytic subunit C (21). Altogether, these results suggest that PP2A inhibition is required for sustained ERK pathway activity; a phenomenon observed in several human malignancies in vivo (7, 22).
Here we demonstrate that PME-1 promotes both basal and growth factor induced ERK pathway activity in malignant cells. Moreover, in human glioblastoma tissues PME-1 expression correlates with both ERK pathway activity and with proliferation. In addition, PME-1 expression correlates with progression of low-grade astrocytic gliomas to malignant glioblastomas. Thus, results of this study identify an in vivo mechanism by which the ERK pathway activity is shielded from PP2A-mediated inactivation in human malignant glioma.
Materials and Methods
Cell culture and siRNA transfections
HeLa, HT-1080, U118-MG NIH-3T3, and HEK293(Phoenix™) cells were cultured in DMEM (Sigma-Aldrich Co., St. Louis, MO) and T98G glioma cells in Eagle's minimum Essential medium (BioWhittaker, Lonza) supplemented with 10% heat-inactivated fetal calf serum (FCS) and penicillin (100 units/ml)-streptomycin (100 ug/ml). HeLa, HT-1080, NIH-3T3, and T98G cells were obtained from ATCC and U118-MG cells were kind gift from Dr. N. Nupponen (University of Helsinki). HEK-TER cells (overexpressing RasV12) and HEK-TEmA cells (overexpressing either B-RafE600, or MEKDD together with myr-Akt) have been described in (10). siRNA transfections were performed by transfecting scrambled (5′GUAACAAUGAGAGCACGG3′) or PME-1 (5′GGUACAGCUAUGGAUGCAC3′) specific double stranded siRNA with Oligofectamine™ or Lipofectamine™RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions. For TPA and serum activation experiments, cells were serum starved (0.5% serum) for 8 hours prior treatments.
Viral infections
Stable PME-1 shRNA cell lines were generated by infecting cells with shPME-1-expressing lentivirus. The pLKO.1-Scr-Puro and pLKO.1-puro vectors containing five different shRNAs specific for PME-1 (shPME-1) were provided by the RNAi Consortium (Broad Institute of Harvard and MIT) (23). Following sequences were used in shPME-1 siRNAs. PME-1.1: 5′GCAGCGATTATTAGTAGAGTT3′, PME-1.2: 5′GTACAGCTATGGATGCACTTA3′, PME-1.3: 5′CTGGTGTTGATAGATTGGATA3′, PME-1.4: 5′CCCAGGTTAAATACAGCCCAT3′, PME-1.5: 5′GCTTATCCAATCTCTTTCTTA3′. Plasmids were transfected into 293FT cells with packing plasmid and envelope plasmid. Supernatants were harvested after transfection and sterile filtered. Cells were infected with viral supernatant at MOI 1000 and selected with puromycin (Sigma-Aldrich Co., St. Louis, MO).
Western blotting and antibodies
Samples for Western blotting were collected in to SDS-PAGE sample buffer(1× SDS Sample Buffer: 62.5 mM Tris-HCl (pH 6.8 at 25°C), 2% w/v SDS, 10% glycerol, 50 mM DTT, 0.01% w/v bromophenol blue) and boiled for 5 min, and centrifuged for 10 min at 14,000 × g to remove insoluble material. After SDS-PAGE proteins were transferred on to a nitrocellulose membrane (Protran, Schleicher and Schuell). Primary antibodies used for immunoblotting are described in Supplemental materials and methods.
Proliferation assays
For Soft-agar assays HeLa cells were seeded on 3 cm plates 72 hr after siRNA transfection. Agar assays were performed in medium containing 10% fetal bovine serum as described in (13) and colonies were counted after 14 days. Anchorage-independent colonies were classified according to a number between 200—10,000 pixels. For foci formation assays HeLa cells were treated as above and seeded on 6-well plate and methanol/crystal violet stained colonies were counted after 8 days. The number and size of colonies were analysed from microscopy images (10× magnification) using ImageJ 1.33u software. For proliferation assays, U118-MG, HeLa or HT-1080 cells were plated in duplicates or triplicates day prior to transfection and transfected with scrambled or PME-1 specific siRNAs for 48 or 72 hours. Transfected cells were left untreated or treated with 10 μM of UO126 for 48 or 72 hours. 1×104 HEK TER cells overexpressing H-RasV12 and HEK-TE cells overexpressing B-RafE600, or MEKDD were plated in triplicates for 6 days. Number of viable cells was determined using a Z2 Particle Count and Size Analyzer (Beckman-Coulter, Miami, FL).
Immunohistochemistry
The expression of PME-1, p-MEK and p-Elk-1 proteins were studied immunohistochemically from 222 grade 2-4 astrocytic gliomas. Sections from (thickness 5 μm) routinely processed tumour microarray paraffin blocks were cut and mounted on SuperFrost Plus slides and dried overnight at 37°C. The sections were then dewaxed and rehydrated. High temperature antigen retrieval was carried out in 10nM Tris-HCl / 1mM EDTA buffer (pH 9.0). Immunostainings were done with the TechMate staining automate using the EnVision detection system (Dako Ltd.Cambridgeshire, UK). The sections were incubated with antibodies against PME-1, p-MEK1/2 (Cell Signaling Technology, Inc. USA) and p-Elk-1 (Santa Cruz Biotechnology) using dilutions 1:100, 1:50 and 1:50 respectively for 30 min at room temperature. Immunostaining present in cytoplasm or nuclei were scored negative (-), positive (+) or strongly positive (++) based on the highest intensity observed in the sample. Scores were given independently by two different observers.
Results
PME-1 regulates PP2Ac leucine 309 methylation in human cancer cells
In order to identify mechanism(s) regulating PP2A activity in cancer, proteins that interact with PR65α, the scaffolding subunit of PP2A, were affinity-purified from HT-1080 fibrosarcoma cells using the tandem affinity purification (TAP) strategy (13). Tandem affinity purification from cytoplasmic extracts of either mock transfected or PR65TAP expressing HT-1080 cells revealed several proteins that co-purified only with the PR65TAP (Fig. 1A, Supplemental table 1). Importantly, B55α, eIF4A, PME-1 and PP2Ac were recently identified as PR65-associating proteins also by using Strep-tag purification strategy (24). One of the PR65 interacting proteins was PME-1, which has originally been identified to preferentially interact with inactive PP2Ac (5). The interaction between PME-1 and PP2A complex was further confirmed here by co-precipitation of endogenous PP2Ac and PR65 with overexpressed PME-1-Strep protein in Streptactin pull-down experiment (Fig. 1B).
Figure 1. PME-1 functions as a PP2A C-terminal methylesterase in cultured cells.
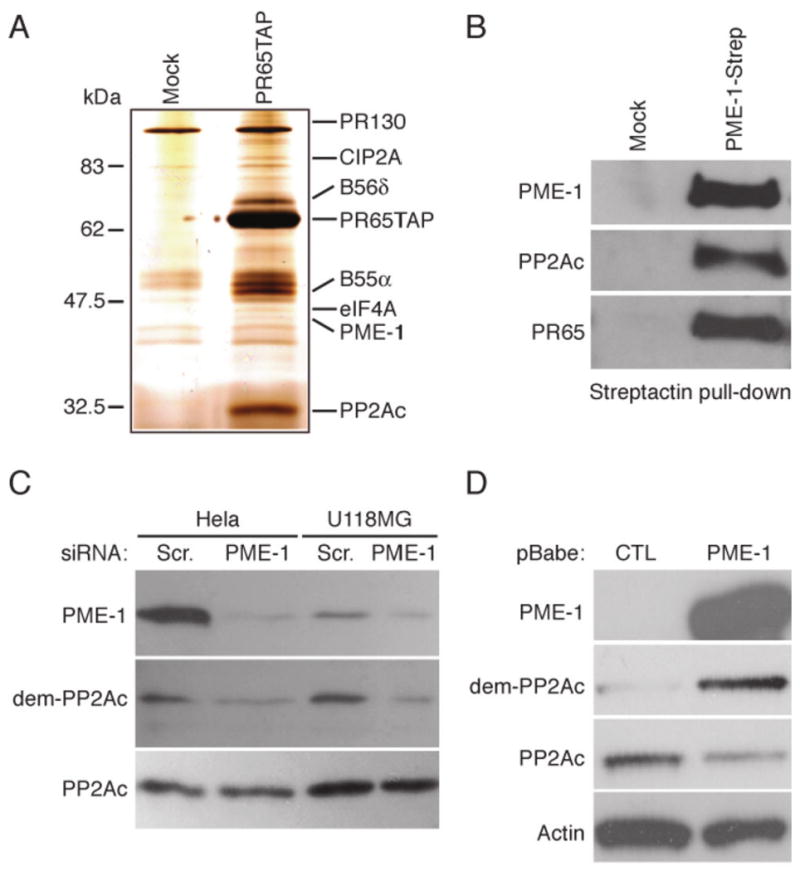
(A) Silver stain analysis of proteins co-purifying with TAP-tagged PR65 protein from HT-1080 cells. Proteins excusively identified in PR65TAP eluates were excised from silver stained gel and identified by mass-spectrometric peptide sequencing. (B) Endogenous PP2Ac and PR65 co-precipitates with PME-1-Strep in Streptactin pull-down experiment from HT-1080 cells. (C) HeLa and U118-MG cells were transfected with scrambled (Scr,) or with PME-1 specific siRNA for 72 hours, and methylation status of the catalytic subunit of PP2A (PP2Ac) was analysed using an antibody that specifically recognizes demethylated leucine 309 of PP2Ac (dem-PP2Ac). (D) Retroviral overexpression of PME-1 results in demethylation of PP2Ac in NIH-3T3 cells. (B-D) Shown are representative data from 2-3 independent experiments with similar results.
PME-1 yeast homologue PPE1 has been demonstrated to inhibit catalytic activity of PP2Ac in yeast cells (25). However, the cellular target proteins for PME-1 regulated PP2A activity have not been identified as yet. To characterize the cellular function of PME-1 in human cancer cells, we first verified that PME-1 regulates methylation of PP2Ac leucine 309 in cancer cells. For this purpose we used an antibody that specifically recognizes the de-methylated form of PP2Ac leucine 309 (dem-PP2A)(5). We found that PME-1 depletion resulted in an increase of PP2Ac leucine 309 methylation in HeLa, U-118MG and HT-1080 cancer cell lines (Fig. 1C and data not shown). PP2Ac demethylation was not altered between untransfected and scrambled siRNA transfected cells, demonstrating that PP2Ac leucine 309 methylation is not subjected to regulation by siRNA response (Supplementary Fig. 1). In addition, retroviral-mediated overexpression of PME-1 in NIH-3T3 fibroblasts caused a decrease in PP2Ac leucine 309 methylation (Fig. 1D). Together, these results verify the role of PME-1 as the PP2A C-terminal methylesterase in cultured cells.
PME-1 supports both basal and growth factor-stimulated MEK-ERK activity
To determine whether PME-1 regulates the phosphorylation/activity of the PP2A target proteins, we analyzed the phosphorylation status of different established PP2A target proteins in HeLa cells transfected with either scrambled or PME-1-specific siRNA. Among the MAPK proteins, PME-1 depletion was found to inhibit ERK phosphorylation without any notable effects on p38 or JNK MAPK phosphorylation (Fig. 2A and data not shown). In addition to HeLa cells, PME-1 depletion inhibited ERK phosphorylation in HT-1080 and U118MG cells (Fig. 2B).
Figure 2. PME-1 supports ERK pathway activity.
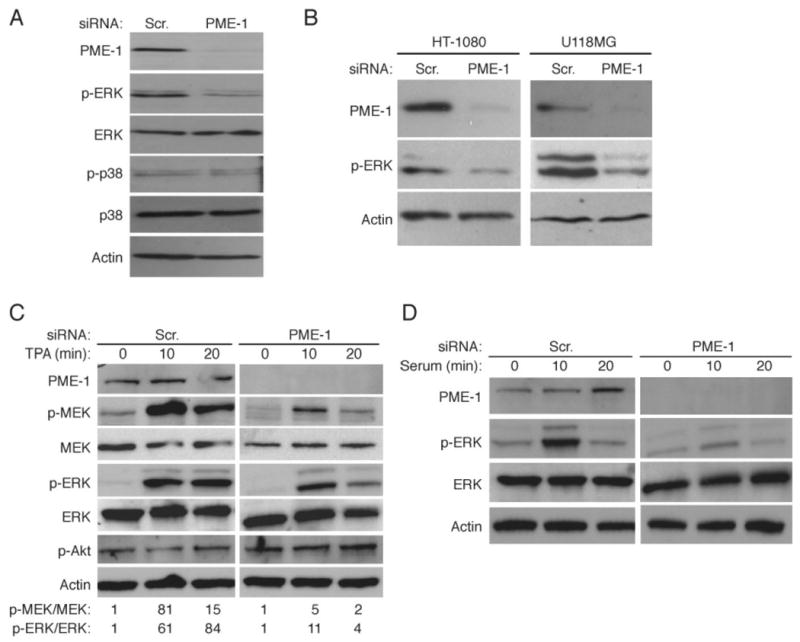
(A, B) Western blot analysis of ERK and p38 phosphorylation in HeLa cells transfected with scrambled or PME-1 siRNA for 72 hours. Equal loading was confirmed with anti-ERK, anti-p38 and anti-actin antibodies. (C) PME-1 supports phorbol ester TPA-elicited ERK pathway activation. Scambled and PME-1 siRNA transfected cells were serum-starved and thereafter exposed to 0.4 μg/ml of TPA for indicated periods of time. Quantification of relative phosphorylation of MEK and ERK proteins is shown below the panels. (D) PME-1 supports serum-elicited ERK pathway activation. (A-D) Shown are representatives of 2-3 independent experiments with similar results.
These observations demonstrate that PME-1 depletion inhibits basal ERK pathway activity. Phorbol ester TPA treatment activates the ERK pathway via protein kinase C (PKC) activation, in Ras-independent but Raf-dependent manner. To study whether PME-1 supports ERK signaling downstream of PKC, TPA-elicited activation of MEK and ERK was compared in cells transfected with either scrambled or PME-1-specific siRNA. We found that TPA treatment resulted in robust MEK and ERK activation after 10 minutes in scrambled siRNA transfected cells, whereas Akt serine 308 phosphorylation was not altered (Fig. 2C). However, as compared to scrambled siRNA transfected cells, PME-1 depleted cells displayed clearly diminished MEK and ERK activation in response to TPA (Fig. 2C). In addition, PME-1 depletion inhibited serum-induced ERK phosphorylation (Fig. 2D). Taken together, these results demonstrate that PME-1 supports basal, as well as growth factor-stimulated ERK activity downstream of growth factor receptors and PKC, but upstream of MEK.
Inhibition of the ERK pathway activity by PME-1 depletion is mediated by PP2A activity
Viral oncoprotein SV40 small-t inhibits PP2A activity and stimulates ERK signaling at the level upstream of MEK activation (18, 26). To question whether PME-1 supports ERK signaling via its effects on PP2A activity, MEK phosphorylation was analyzed in PME-1-depleted human embryonic kidney epithelial cells (HEK-TER) expressing SV40 large T antigen, hTERT and HRAS (10), and either SV40 small-t or a control vector. As shown in Fig. 3A, PME-1 depletion by various short hairpin RNAs (shRNA) correlated with MEK inactivation in control cells, whereas MEK phosphorylation is unaltered in response to PME-1 depletion in small-t expressing cells. These results show that PME-1 depletion does not inhibit MEK activity in cells in where PP2A activity is suppressed.
Figure 3. PME-1 promotes Ras-mediated ERK pathway activity upstream of Raf.
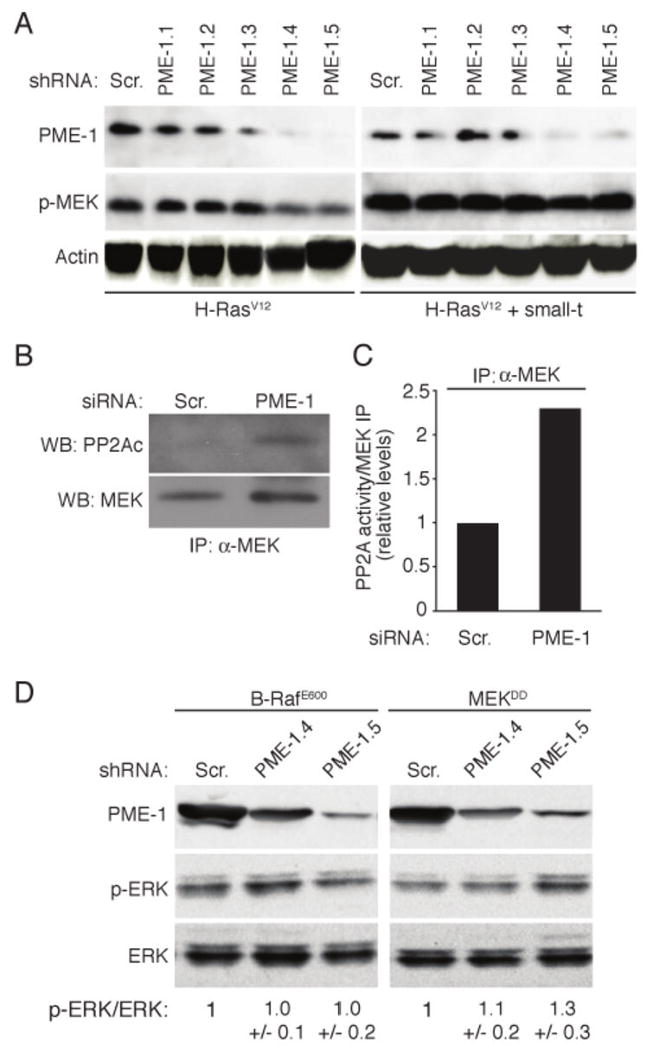
(A) PME-1 depletion fails to inhibit MEK phosphorylation in small-t expressing cells. Immunoblot analysis of MEK phosphorylation in HEK-TER cells infected with indicated PME-1 or Scr shRNAs. Small-t expression in cell clones is indicated below the panels. (B) PME-1 depletion increases PP2A recruitment to MEK complex. Interaction of PP2A with MEK was analysed by anti-MEK antibody immunoprecipitation (IP) of lysates from Scr and PME-1 transfected HeLa cells. (C) Fractions of MEK immunoprecipitates shown in (B) were subjected to an in vitro PP2A assay using DiFMUP (6,8-difluoro-4-methylumbelliferyl phosphate) as a substrate in conditions selective for PP2A activity measurement. (D) Immunoblot analysis of phospho-ERK levels in HEK-TEmA cells expressing B-RafE600, or MEKDD and indicated PME-1 or Scr shRNAs. Quantification of relative phosphorylation of ERK is shown below the panels. Shown is mean +/- S.D. of two experiments.
Ras/Raf-mediated activation of MEK occurs in signaling protein complexes that involve several regulatory and scaffold proteins critical for signal propagation (15, 16). In order to further confirm that PME-1 regulates PP2A activity directed towards the Ras/Raf/MEK signaling complex, we analyzed MEK complex-associated PP2A activity in PME-1 or scrambled siRNA transfected cells. MEK antibody was used for co-immunoprecipitation in order to avoid risk of immunoprecipitating Ras or Raf isoforms that are not involved in MEK activation in HeLa cells. As shown in Fig. 3B, an increase in co-immunoprecipitation of the catalytic PP2Ac subunit with MEK immunocomplex was observed in PME-1 depleted cells. Accordingly, PME-1 siRNA transfected cells demonstrated an increase in MEK complex-associated PP2A activity in an in vitro PP2A assay using 6,8-difluoro-4-methylumbelliferyl phosphate as the substrate (Fig. 3C).
Our data thus far suggest that PME-1 depletion inhibits ERK pathway activity at the level downstream of Ras, but upstream of MEK. To further pinpoint the level on the ERK pathway where PME-1 functions, the effect of PME-1 depletion on ERK phosphorylation was studied in HEK-TEmA clones in which the pathway activity was stimulated, rather than with active H-Ras mutant (Fig. 3A), by constitutively active alleles of either B-Raf (B-RafE600), or MEK (MEKDD)(10). In addition, these cells express activated Akt (10). We found that in contrast to H-RasV12 expressing cells (Fig. 3A), PME-1 depletion did not inhibit ERK phosphorylation in either B-RafE600 or MEKDD expressing cells (Fig. 3D). Taken together these results demonstrate that PME-1 protects the ERK pathway from PP2A-mediated inactivation. Moreover, these findings indicate that PME-1 functions upstream of Raf to promote ERK pathway activity.
PME-1 supports malignant cell growth
MEK-ERK signaling promotes cancer cell proliferation and survival (7, 15). To ascertain the functional role of PME-1, HeLa cells depleted of PME-1 were assessed for their ability to form dense foci on a monolayer. We found that PME-1 depletion resulted in a significant inhibition of colony growth (Fig. 4A). To study whether PME-1 contributes to malignant cell growth, PME-1 depleted HeLa cells were analyzed for their capacity to grow in an anchorage-independent manner. As shown in Fig. 4B, PME-1 depletion significantly inhibited anchorage-independent proliferation of HeLa cells. In addition to HeLa cells, PME-1 depletion inhibited proliferation also in HT-1080 cells (data not shown). Importantly, no signs of cleavage of the caspase substrate protein PARP were observed in PME-1 depleted cells 72h hours after siRNA transfection (Supplementary Fig. 2), suggesting that inhibition of malignant cell growth by PME-1 depletion is not caused by induction of programmed cell death.
Figure 4. PME-1 supports malignant cell growth.
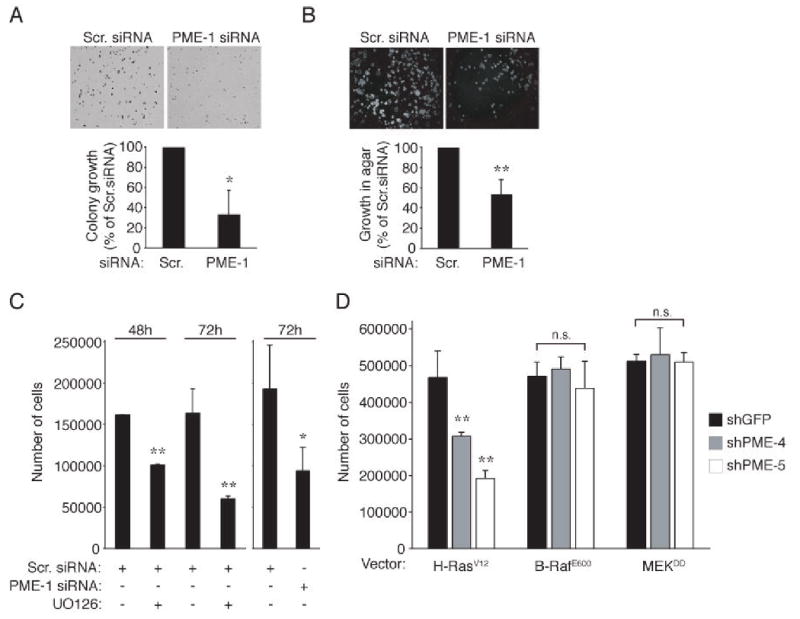
(A) Dense foci formation of HeLa cells transfected with scrambled or PME-1 siRNAs. (B) Anchorage-independent growth of HeLa cells transfected with scrambled or PME-1 siRNA. (A, B) Colony numbers were analysed by Image J software. Data is presented as percentual change of number of colonies as compared to Scr. siRNA transfected cells which were set in each experiment as 100%. Shown is mean + S.D. of 4-5 experiments. *p= 0.001, *p = 0.002, Student's t test. (C) Proliferation of HeLa cells transfected with scrambled or PME-1 siRNA or treated with UO126 MEK inhibitor for 48-72 hours. Shown is a mean value +S.D. of 2 replicate plates. Experiment was performed 3 times with similar results. *p<0.05, **p<0.01, Student's t test. (D) Effect of PME-1 depletion on proliferation rates of indicated HEK-TE cells. The data is presented as mean + S.D. of 3 independent experiments. **p< 0.01, n.s.= not significant.
To examine whether PME-1 affects cell growth through regulation of the ERK pathway, we analyzed the correlation of proliferation rates between cells treated either with a specific MEK inhibitor, UO126 or with PME-1 siRNA. We found that inhibition of MEK activity with UO126 resulted in approximately 40% reduction in cell number after 48 hours, and 60% inhibition after 72 h (Fig. 4C). Instead, a 50% reduction in cell number was observed in PME-1 depleted cells 72 h after siRNA transfection (Fig. 4C). These results demonstrate that maximal proliferation of HeLa cells is dependent on both PME-1 expression and ERK pathway activity. To determine if inhibition of cell proliferation induced by PME-1 depletion correlates with ERK pathway inactivation, we analyzed the effect of PME-1 shRNA on proliferation of cell clones in which ERK pathway activity was activated either by H-RasV12, B-RafE600, or MEKDD overexpression (10). We found that whereas PME-1 shRNAs inhibited both MEK phosphorylation and proliferation of Ha-RasV12 infected cells (Fig. 3A and 4D), PME-1 depletion failed to inhibit either of these properties in cells infected with B-RafD600 or MEKDD (Fig. 3D and 4D). These observations demonstrate that PME-1 promotes cell proliferation, while inhibition of PME-1 partially suppresses the tumorigenic phenotype of cancer cells. Our results also indicate that PME-1 enhances cell proliferation, at least in part, through its ability to maintain ERK pathway activity.
PME-1 promotes ERK pathway activity in human malignant glioblastomas
Mitogenic signaling through the ERK pathway has been proposed to contribute to the progression of low-grade astrocytic gliomas to malignant glioblastomas (27-29). In order to study if PME-1 positively regulates the ERK pathway in cells of human glioblastoma origin, PME-1 was depleted from U-118MG and T98G glioblastoma cells. We found that, in addition to inhibition of ERK phosphorylation, PME-1 depletion potently inhibited phosphorylation of its downstream target Elk-1 (Fig. 5A), a transcription factor implicated in glioblastoma cell proliferation (28). Accordingly, inhibition of Elk-1 phosphorylation by PME-1 depletion manifested in a significant reduction of proliferation in both of the examined glioblastoma cell lines (Supplementary Fig. 3). Importantly, inhibition of MEK activity with UO126 also resulted in a significant inhibition of glioblastoma cell proliferation (Supplementary Fig. 3).
Figure 5. PME-1 promotes ERK activity and proliferation in human malignant glioblastoma.
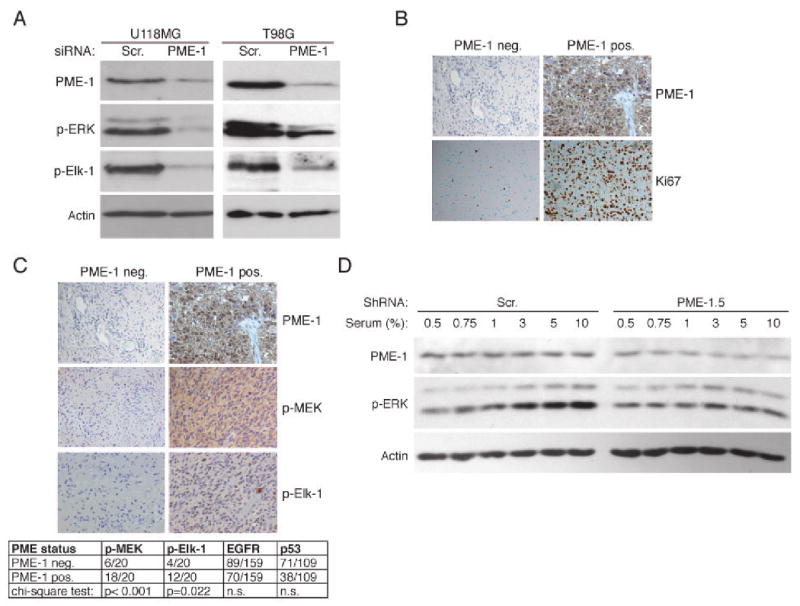
(A) Western blot analysis of ERK pathway activity in U118-MG and T98G glioma cells transfected with Scr, or PME-1 siRNA for 72 hours. (B) Expression of PME-1 and Ki-67 proliferation index was studied immunohistochemically from 222 grade 2-4 astrocytic glioma samples. Shown are representative Ki-67 staining of samples negative or positive for PME-1 expression. (C) Correlation of PME-1 expression and phosphorylation of MEK and Elk-1 was studied immunohistochemically from PME-1 positive and negative glioblastoma samples (each N=20). Statistical analysis of the correlation of PME-1 status to MEK and Elk-1 activity, EGFR amplification status and p53 immunopositivity in glioblastomas is shown below the stainings. n.s. = not significant. (D) Western blot analysis of serum-induced ERK activation in U118-MG glioma cells infected with indicated PME-1 or Scr shRNAs. Shown is representative of three independent experiments using two different shScr and shPME-1 clones.
Identification of PME-1 as a protein that supports proliferation of human glioblastoma cells prompted us to study the clinical significance of PME-1 in human astrocytic gliomas. The specificity of the PME-1 antibody used for immunohistochemical stainings of glioma samples was verified by siRNA approach (Supplementary Fig. 4). Altogether 98 samples out of 222 grade 2–4 astrocytic gliomas were found positive for PME-1 protein expression. The representative samples of PME-1 positive tumors are shown in Fig. 5B and in Supplementary Fig. 5. Importantly, increased PME-1 expression was found to correlate with malignant progression of astrocytic gliomas: PME-1 immunopositivity correlated with increasing malignancy grade (p = 0.021)(Supplementary Table 2). Moreover, PME-1 immunopositivity very significantly correlated with increasing Ki-67 proliferation index in the total tumor material (p < 0.001)(Supplementary Table 3; Fig. 5B). The difference in cell proliferation was significant even when the comparison between PME-1 positive and negative tumors was made separately within grade 2 gliomas (N = 62) and glioblastomas of grade 4 (N = 160)(Supplementary Table 3). Importantly, neither EGFR amplification nor p53 expression status of the tumors corresponded with the very tight correlation of PME-1 expression with glioblastoma proliferation (Supplementary Table 3). EGFR and p53 status of tumors was also unrelated to PME-1 expression, as were the age and the gender of the patients (Supplementary Table 2). Representative glioma tissue samples exhibiting low and high PME-1, Ki-67 or 53 immunopositivity, in addition to EGFR amplification are shown in Fig. 5B and in the supplementary Fig. 6.
ERK phosphorylation was recently demonstrated as an independent prognostic factor in malignant glioblastoma (29). Since we found that PME-1 supports ERK phosphorylation and activity in glioblastoma cells in culture (Fig. 5A), we investigated p-MEK and p–Elk levels in glioblastoma tissue samples that do or do not express PME-1. In gliomas that express PME-1, we found significantly more p-Elk-1 (p = 0.022) and p-MEK immunopositivity (p < 0.001) when compared to PME-1 negative tumors (Fig. 5C). Increased ERK pathway activity in human glioblastoma has been proposed to result from increased expression of growth factors and their receptors in the tumor tissue (27, 29). To investigate whether, in addition to receptor signaling, PME-1 contributes to growth factor response of glioblastoma cells, we generated U-118MG cell clones in which PME-1 levels were stably suppressed by expressing PME-1-specific shRNA. As shown in Fig. 5D, cells expressing a scrambled shRNA showed induction of ERK phosphorylation in 3% serum, whereas exposure to higher levels of serum failed to induce ERK activity in cells expressing a PME-1 shRNA. Together with the data shown in Fig. 2D, these results indicate that cancer cell growth factor response is in part dependent on the ability of PME-1 to inhibit ERK pathway-directed PP2A activity.
Discussion
The ERK pathway is an important mediator of the oncogenic activity of receptor tyrosine kinases (RTKs) and Ras GTPases (16), as well as oncogenic alleles of Ras (8). However, the mechanisms that sustain RTK and Ras-elicited ERK pathway activity in malignant cells have remained elusive. In this work we demonstrate that PME-1 protects ERK pathway activity from PP2A-mediated inactivation in human malignant glioma. Our data show that PME-1 depletion results in increased methylation of PP2Ac leucine 309 and dephosphorylation of MEK, ERK and Elk-1 proteins. PME-1 depletion inhibited ERK pathway activity induced by RasV12, whereas cells expressing constitutively active alleles of either B-Raf or MEK, were found resistant to PME-1 shRNA (Fig. 3A, 3D). Together, these observations suggest that PME-1 supports ERK pathway activity at the level upstream of Raf. As PP2A-mediated dephosphorylation of Raf has been shown to stimulate rather than inhibit Raf kinase activity (17, 30), the target for PME-1 regulated PP2A activity could be a protein involved in Raf activation. Candidates for such proteins could be scaffolds involved in linking the RTK proteins to Raf/MEK/ERK complex (15, 16). However, it is clear that future studies are needed to identify the molecular target for PME-1 regulated PP2A activity. Also, based on the data presented here we cannot exclude the possibility that, in addition to its role in supporting ERK activity, PME-1 would in part support malignant cell growth by regulating other PP2A target pathways.
Interestingly, although the role of RTK activation in response to growth factors is well studied, the role of phosphatases in this process remain incompletely understood. Here we show that PME-1 augments the cellular growth factor response (Fig. 2D and 5D). Based on these results, it is tempting to speculate that in conditions of limited growth factor supply in tumor tissues, cancer cells with high PME-1 expression levels may retain ERK pathway activity, and thereby maintain their proliferative capacity. In addition to results of our cell culture experiments, this notion is clearly supported by in vivo correlation of PME-1 expression, proliferation and ERK pathway activity in human malignant glioblastoma.
Human glioblastoma patients display extremely poor survival, and there are currently no effective treatment regimes for this disease (27). We have found that PME-1 expression correlates with ERK pathway activity in human malignant glioblastoma (Fig. 5C) and supports ERK activity in glioblastoma cells (Fig. 5A). These results indicate that PME-1 could represent a potential diagnostic marker to identify subgroups of patients that would benefit from treatment with small molecule Raf/MAPK pathway inhibitors, currently in clinical trials for glioblastoma (27). In addition, as expression of phosphorylated ERK was recently shown to be associated with increased radiation resistance of malignant glioblastoma (29), inhibition of PME-1 could sensitize glioblastoma cells to radiotherapy. In this regard, identification of small molecule inhibitors of PME-1 methylesterase activity, or PME-1-PP2Ac interaction (6, 31), would be important to probe potential suitability of PME-1 as a drug target in malignant glioblastoma.
These observations identify PME-1 expression as a hitherto unrecognized mechanism supporting ERK pathway activity in cancer cells. Importantly, our data suggests that in addition to growth factors and their receptors, PP2A functions as an important modulator of the cellular growth factor response. Considering the established role for growth factor-elicited ERK signaling in several human malignancies (7, 22), we postulate that the importance of these results reaches beyond the demonstrated role of PME-1 in human glioblastomas. Moreover, this first identification of a cellular role for PME-1 in the regulation of cellular signaling further highlights the novelty of the presented results. Together with other recently published data (12-14), these findings further emphasizes the relevance of identifying mechanisms that regulate the tumor suppressor function of PP2A in human malignancies. Finally, the results of this work may open novel opportunities for the treatment and diagnosis of human cancers addicted to oncogenic ERK pathway activity (32).
Supplementary Material
Acknowledgments
We thank Dr. Nina Nupponen for glioblastoma cell lines. Paula Kosonen, Merja Lehtinen, Taina Kalevo-Mattila and Raisa Vuorinen are thanked for their excellent technical assistance and Minna Niemelä for participation on experiments. This work was supported by grants from the Academy of Finland (project 1121413), Competitive Research Funding of the Pirkanmaa Hospital District (project 9J151), Emil Aaltonen Foundation, Helsinki University Central Hospital Research Funds, Sigrid Jusélius Foundation and the Finnish Cancer Society.
References
- 1.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et biophysica acta. 2008 doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends in molecular medicine. 2008;14(4):152–60. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends in biochemical sciences. 2008 doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Longin S, Jordens J, Martens E, et al. An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem J. 2004;380(Pt 1):111–9. doi: 10.1042/BJ20031643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogris E, Du X, Nelson KC, et al. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem. 1999;274(20):14382–91. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133(1):154–63. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 7.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 8.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6(2):171–83. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Zhao JJ, Roberts TM, Hahn WC. Functional genetics and experimental models of human cancer. Trends in molecular medicine. 2004;10(7):344–50. doi: 10.1016/j.molmed.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Boehm JS, Zhao JJ, Yao J, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 11.Mumby M. PP2A: Unveiling a Reluctant Tumor Suppressor. Cell. 2007;130(1):21–4. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65(18):8183–92. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 13.Junttila MR, Puustinen P, Niemela M, et al. CIP2A Inhibits PP2A in Human Malignancies. Cell. 2007;130(1):51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 14.Sablina AA, Chen W, Arroyo JD, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell. 2007;129(5):969–82. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6(11):827–37. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 16.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26(22):3113–21. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 17.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. Faseb J. 2008;22(4):954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 18.Sontag E, Sontag JM, Garcia A. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-kappaB activation. Embo J. 1997;16(18):5662–71. doi: 10.1093/emboj/16.18.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amaral MC, Casillas AM, Nel AE. Contrasting effects of two tumour promoters, phorbol myristate acetate and okadaic acid, on T-cell responses and activation of p42 MAP-kinase/ERK-2. Immunology. 1993;79(1):24–31. [PMC free article] [PubMed] [Google Scholar]
- 20.Braconi Quintaje SB, Church DJ, Rebsamen M, Valloton MB, Hemmings BA, Lang U. Role of protein phosphatase 2A in the regulation of mitogen-activated protein kinase activity in ventricular cardiomyocytes. Biochem Biophys Res Commun. 1996;221(3):539–47. doi: 10.1006/bbrc.1996.0632. [DOI] [PubMed] [Google Scholar]
- 21.Kins S, Kurosinski P, Nitsch RM, Gotz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol. 2003;163(3):833–43. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 23.Moffat J, Grueneberg DA, Yang X, et al. A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell. 2006;124(6):1283–98. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 24.Junttila MR, Saarinen S, Schmidt T, Kast J, Westermarck J. Single-step Strep-tag purification for the isolation and identification of protein complexes from mammalian cells. Proteomics. 2005;5(5):1199–203. doi: 10.1002/pmic.200400991. [DOI] [PubMed] [Google Scholar]
- 25.Hombauer H, Weismann D, Mudrak I, et al. Generation of Active Protein Phosphatase 2A Is Coupled to Holoenzyme Assembly. PLoS biology. 2007;5(6):e155. doi: 10.1371/journal.pbio.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell. 1993;75(5):887–97. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- 27.Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110(1):13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- 28.Uht RM, Amos S, Martin PM, Riggan AE, Hussaini IM. The protein kinase C-eta isoform induces proliferation in glioblastoma cell lines through an ERK/Elk-1 pathway. Oncogene. 2007;26(20):2885–93. doi: 10.1038/sj.onc.1210090. [DOI] [PubMed] [Google Scholar]
- 29.Pelloski CE, Lin E, Zhang L, et al. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin Cancer Res. 2006;12(13):3935–41. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 30.Dougherty MK, Muller J, Ritt DA, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17(2):215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 31.Longin S, Zwaenepoel K, Martens E, et al. Spatial control of protein phosphatase 2A (de)methylation. Exp Cell Res. 2008;314(1):68–81. doi: 10.1016/j.yexcr.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 32.Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10(5):425–35. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.