

Abstract
There has been a considerable amount of interest in the immunological community about new phenotypic subsets of CD4+ T cells, particularly cells that produce the cytokine interleukin (IL)-17 [named T helper type 17 (Th17) cells]. While the initial discovery of Th17 cells and the pathways that controlled their development was in the mouse, recent attention has shifted to the existence of these cells and the relevant upstream cytokine signals in humans. While it is clear that CD4+ T cells producing IL-17 exist in vivo, their relevance to disease pathogenesis is only just being understood. In this paper, we review the data regarding the generation of human Th17 cells in vitro and the evidence that this effector population is important in human disease states.
Keywords: autoimmunity, cytokines, differentiation, T cells, transcription factors
Introduction
A first line of defence against the continuous threat of pathogenic microbes that invade our body is formed by innate immunity, which responds to danger immediately with antigen-non-specific events without inducing classical immunological memory. In a second stage, dendritic cells (DC) orchestrate immunity and tolerance by initiating adaptive antigen-specific immunity by stimulating naive T cells in the tissue draining lymphoid organs with an antigenic peptide complexed to a major histocompatibility molecule (signal 1). In addition, DC decide between immunity and tolerance by expressing various patterns of T cell co-stimulatory or inhibitory molecules (signal 2). Finally, dendritic cells promote protective effector T cell responses adapted to the invading class of pathogen by expressing variable sets of T cell-polarizing molecules (signal 3) [1]. Effector CD4+ T helper cells are highly heterogeneous and comprise distinct subsets characterized by different profiles of cytokine production (Fig. 1). CD4+ T helper type 1 (Th1) cells develop from naive T cells upon the induction of expression of tissue-specific transcription factor T-bet [2,3], which mediates the production of interferon (IFN)-γ which, in turn, is instrumental in the induction of cellular immunity against intracellular pathogens such as viruses, certain types of (myco)bacteria and protozoa. Th1 responses regulate the activation of CD8+ T cells and influx of macrophages. CD4+ Th2 cells are generated from naive CD4+ Th cells upon induction of the transcription factor GATA3 [4], which drives the production of interleukin (IL)-4, IL-5 and IL-13. These cytokines are critical in the control of nematode infections. Th2 responses are associated with production of immunoglobulin (Ig)E antibodies and recruitment of eosinophils. Recently, it was established that CD4+ T cells that produce IL-17A and IL-17F preferentially could be generated and that they seem to form a separate lineage of Th17 cells [5,6]. These cells express retinoic acid-related orphan receptor gamma-t (RORγt) as a key transcription factor for their differentiation [7]. In addition to IL-17, these cells may also produce IL-22 and IL-21 [8].
Fig. 1.
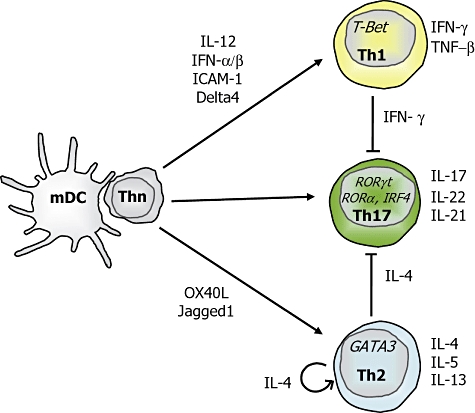
Dendritic cells (DC) promote the development of distinct T helper cell subsets.
Function of IL-17 and Th17 cells
IL-17 was first described in 1995 as a glycoprotein of approximately 20 kDa (155 amino acids) with a close sequence homology to murine IL-17 and with an open reading frame of the T lymphotropic herpesvirus Saimiri [9]. IL-17 is secreted as a 32 kDa homodimer that binds the IL-17 receptor (IL-17R), a type I transmembrane protein that exhibits a broad tissue distribution [10]. The analysis of the three-dimensional crystal structure of two members of the IL-17 family of cytokines suggests that they form cystein knot structures as those of nerve growth factor (NGF) or platelet-derived growth factor cytokines (PDGF) [11]. Apart from IL-17(A), five additional IL-17 members have been described, termed IL-17B, C, D, E (or IL-25) and F, all of which have conserved residues in their c-terminal region and also form homodimers. CD4+ T cells produce both IL-17A and F [12], whereas IL-25 is produced mainly by Th2 cells [13], and IL-17B, C and D are expressed more broadly. CD4+ T cells may produce three different dimeric forms of IL-17, consisting of IL-17F/F, IL-17F/A or IL-17A/A [14]. In naive murine cells IL-17F/F is produced in the highest quantity, followed by IL-17F/A and IL-17 A/A. However, IL-17A/A is more potent then IL-17F/F, with IL-17A/F having an intermediate potency. IL-17 is an important mediator in tissue inflammation as it has pleiotropic effects on tissue cells and several immune cells. IL-17 mobilizes neutrophils, partly through increasing their local survival and partly through granulopoiesis and CXC chemokine induction. Various chemokines and cytokines are induced by IL-17A and F, including tumour necrosis factor (TNF)-α, IL-1β, IL-8, IL-6, growth regulated-α (GRO-α), monocyte chemoattractant protein (MCP)-1 and gingival crevicular fluid (GCF), as well as intercellular adhesion molecule (ICAM)-1 by monocytes, airway epithelial cells, keratinocytes, vein endothelial cells and fibroblasts. TNF-α and IFN-γ may enhance the expression of IL-17-induced chemokines and cytokines [12,15].
Accumulating data shows that Th17 cells are important in host protection against various bacterial and fungal species [16–21]. Indeed, protection against Streptococcus pneumoniae[22] is impaired in IL-17-deficient mice. Moreover, it has been demonstrated by various groups that antigen-presenting cells (APCs) primed with bacteria or bacterial products promote Th17 cells in the human system [23,24].
IL-17-secreting cells and Th17-associated cytokines have been implicated in a number of human autoimmune and inflammatory diseases. There is compelling evidence that IL-17 plays an important role in the pathogenesis of rheumatoid arthritis. IL-17 levels were enhanced in the synovium of RA patients [25]. Th17 effector functions in RA were identified as receptor activator nuclear factor kappa-B ligand (RANKL) expression on the surface of Th17 cell-induced osteoclastogenesis, promoting cartilage and bone destruction/resorption independently of TNF and IL-1 [26]. Moreover, in psoriasis, T cells obtained from psoriatic skin lesions showed a predominantly Th17 phenotype [27]. Similar findings emerged from biopsies of lesions of Crohn's disease [28]. IL-17 and IL-6 were among the most highly expressed genes in multiple sclerosis (MS) lesions [29] and high levels of IL-17 were detected in the serum and cerebrospinal fluid of MS patients [30]. Although the relationship between IL-17 and pathogenesis of a number of diseases is still unclear, there have been publications on the overexpression of Th17 pathway genes in chronic inflammatory diseases such as systemic lupus erythomatosus [31], asthma [32] and human tumours [33], as well as allograft rejection [34] and infections; for example, Helicobacter pylori-associated gastritis [35]. However, it is unclear whether IL-17 plays a direct role as a causative agent in the pathogenesis of a number of those diseases or, rather, is implicated only as a consequence of feedback mechanisms and amplification of inflammatory responses. The function and regulation of human Th17 cells in human health and disease is covered in more detail by Crome et al. in this series [36].
Development of murine Th17 cells
Recently, it has become clear that the cytokines IL-6 and transforming growth factor (TGF)-β can induce naive murine T cells to develop into Th17 cells, which are characterized by the expression of the transcription factor RORγt (Fig. 2) [7,37–39]. In contrast, the signature cytokines of Th1 and Th2 cells, IFN-γ and IL-4 were demonstrated to inhibit the development of Th17 cells [6]. Moreover, there is considerable evidence of the involvement of Th17 in immunopathogenesis from work on experimental mice models such as experimental autoimmune encephalomyelitis (EAE) [6] and collagen-induced arthritis (CIA) [40,41].
Fig. 2.
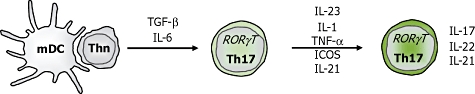
Development of murine interleukin (IL)-17 producing CD4+ T cells.
Initially, it was reported that the IL-12 family member IL-23, a heterodimer consisting of the IL-12/23 p40 and the unique IL-23p19 subunit, was an important factor for the development of Th17 cells [42,43]. However, naive murine T cells do not express the IL-23 receptor and do not differentiate into Th17 cells in the presence of IL-23 in vitro. Instead, IL-23 is essential for the maintenance and/or expansion of murine Th17 cells. The proinflammatory cytokines IL-1β and TNF-α have been shown to amplify the development of Th17 cells [44,45]. IL-1 may enhance the TGF-β/IL-6-induced IL-17 production on its own or synergistically with IL-23. However, none of these cytokines can substitute for TGF-β or IL-6 to promote murine Th17. More recently, other molecules have also been shown to influence the development of murine Th17 cells. In this respect, inducible co-stimulatory molecule–programmed death 1 (ICOS–PD1) interaction enhances the production of IL-17 [46]. Another cytokine that may also be produced by Th17 cells, IL-21, seems to act as an autocrine regulatory factor supporting the Th17 development, but also has various other effects throughout the immune system, such as regulation of B cell responses [47,48].
Development of Th17 cells in human memory versus naive CD4 T cells
Given that IL-17 is associated with the pathology of a number of human inflammatory and autoimmune diseases, it is essential to understand how this cytokine is controlled in human T cells and to define the conditions under which human naive T cells may become Th17 cells. One of the initial studies in human T cells reported that T cell receptor (TCR) cross-linking leads to IL-17 production [49]. Another study indicated that IL-23 induced IL-17 expression [50]. However, the problem with these early studies was that they did not separate effects on memory T cells versus naive T cells. It was only more recently that memory and naive T cells were isolated to assess the various conditions necessary in each population for promoting the production of IL-17 (Fig. 3).
Fig. 3.
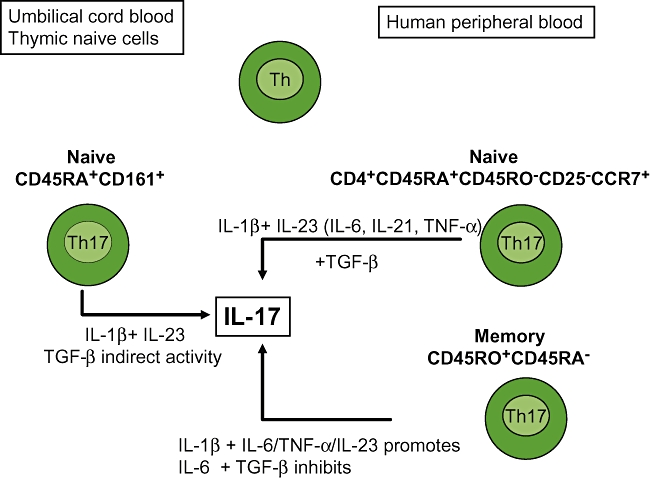
Production of interleukin (IL)-17 from naive versus memory T cells skewed towards T helper type 17 (Th17) conditions. Different skewing conditions are used for different cell types.
Somewhat in contrast to the murine system, the optimal conditions for the development of a robust Th17 population secreting large amounts of IL-17 from human naive T cells remain elusive. This may be analogous to the situation with generating primary human Th2 cells secreting IL-4 in vitro, and appears to be similarly donor-dependent. It is, however, much easier to demonstrate IL-17 secretion from human memory T cells. It was proposed that human memory CD4+CD45RO+CD45RA- T cells up-regulated the RORC2 transcription factor (known as RORγt in mice) and IL-17 in response to TCR occupancy. In one study human Th17 differentiation did not occur in response to TGF-β and IL-6, even though these cytokines promoted RORC2 expression. In memory T cells, IL-23 up-regulated its own receptor and promoted RORC2 and IL-17 expression [51]. Subsequent studies then attempted to characterize the cytokines and in vitro conditions that regulate IL-17-secreting memory T cells. IL-17-secreting memory T cells can be identified by the combined expression of CCR6 and CCR4 [52]. Another study identified the CD4+CD45RO+CCR7-CCR6+ effector memory T cell population as the main IL-17-secreting T cells [53]. IL-1β, IL-23 and, to a lesser extent, IL-6 promoted production of IL-17 with an additive effect seen when IL-1β was added together with IL-23 or IL-6. IL-23 with IL-6 enhanced IL-17 production but the effect was similar to that seen with IL-23 alone. However, TGF-β inhibited IL-17 when added alone or together with either IL-1β, IL-23 or IL-6, an effect that was replicated in other studies [23,24,53]. The regulatory effects of these cytokines were seen both at the protein level and at mRNA transcription. A recent study reported that prostaglandin E2 (PGE2), an inflammatory mediator abundant in chronically inflamed tissues, acted directly on memory T cells by promoting IL-17 production and simultaneously inhibiting IFN-γ production [54]. PGE2 stimulated the EP2 and EP4 receptors expressed on T cells, affecting the activity of transcription factors and leading to an increase in RORC2 and a decrease in T-bet mRNA.
A number of studies have attempted to understand the mechanism underlying human Th17-mediated immunity against bacteria through the production of IL-17 in memory T cells. One study showed that IL-17 could be induced in memory CD4 T cells by human dendritic cells upon sensing of NOD2 ligand MDP, a peptidoglycan derivative expressed by many bacterial species [24]. Other data showed that DCs promoted IL-17 production exclusively in memory T cells through IL-23 and IL-1β[42,43]. In addition, we have shown previously that secretion of IL-17 by memory cells was enhanced by T cell receptor ligation in the context of Toll-like receptor (TLR)-activated monocytes [23]. Furthermore, we have shown recently that monocytes from inflamed joints of rheumatoid arthritis patients can specifically induce the secretion of IL-17 by CD4+ T cells, indicating that these findings have relevance to human autoimmune disease in vivo[55].
The issue of inducing substantial and reliable IL-17 secretion from human naive T cells is much more difficult than in mice. A study by Acosta Rodriguez and colleagues demonstrated that IL-17 could be induced from cell sorted CD45RA+CD25–CCR7+ naive human T cells in the presence of IL-1β and IL-6 [56]. Another study found that the differentiation factors for human Th17 cells from magnetic bead-selected CD4+CD45RA+CD45RO- naive T cells were IL-1β and IL-23 [57]. Recently, Boniface showed that PGE2 together with IL-1β and IL-23 promoted the development of Th17 cells [58]. Chen and colleagues showed a much lower secretion of IL-17 from naive T cells versus memory T cells by enzyme-linked immunosorbent assay (ELISA) [51]. These studies suggested that TGF-β might not be necessary for the differentiation of human Th17 cells in vitro, with the implication that the conditions required for human and murine Th17 differentiation were different from each other. However, as these groups used different techniques of isolating the naive T cell population, such as fluorescence activated cell sorting (FACS), utilizing varied cell parameters, versus magnetic affinity cell sorting (MACS) bead selection, there was doubt as to whether the cells skewed subsequently were properly ‘naive’, as there was a potential for a small contaminating effector population that might explain the apparent disparity of these results.
The role of TGF-β in the induction of Th17 cells from naive human T cells has been somewhat controversial. The Th17 differentiation performed in the above studies took place in RPMI-1640 supplemented with bovine or human serum as the culture medium. There was a suggestion that contaminating platelets and serum contained TGF-β, which could mask the induction of IL-17 production. More recently, three independent research groups tackled these problems by using either serum-free X-vivo 15 culture medium or umbilical cord blood, and concluded that TGF-β was an essential requirement for the differentiation of Th17 from naive human T cells. Manel and colleagues showed that TGF-β, IL-1β and IL-23 were important cytokines required for human Th17 polarization in serum-free conditions. They also indicated that TGF-β induced RORC2 expression but paradoxically blocked its transcriptional activity, thereby preventing IL-17 expression [59]. A cocktail of the proinflammatory cytokines IL-1β and IL-6, IL-21 or IL-23 counteracted that inhibition and contributed to RORC expression, promoting IL-17 induction. It appeared that TGF-β had a dose-dependent effect on the induction of IL-17 in naive human T cells. TGF-β alone at 1 ng/ml, even under the effect of RORC2 overexpression from a lentiviral vector, did not induce IL-17. However, TGF-β at 10 ng/ml, in synergy with IL-1β and IL-23, promoted RORC and IL-17 expression. A study carried out by Yang and colleagues further confirmed the dose-dependent effect of TGF-β because concentrations of TGF-β ranging from 0·1 to 10 ng/ml, in this case with IL-21, enhanced IL-17 induction and RORC expression, whereas 50 ng/ml TGF-β suppressed IL-17 differentiation [60]. Volpe and colleagues also showed that TGF-β, particularly at concentrations of 1–10 ng/ml, regulated IL-17 production positively in a dose-dependent manner in the presence of specific inflammatory cytokines such as IL-1β, IL-6, IL-23 and TNF (of which the latter could be excluded without any significant negative effect) [61]. They also indicated that although absolute IL-17 production was higher in the absence versus the presence of serum, the cytokine requirements remained identical in those two types of culture mediums and concluded that exogenous TGF-β played an important role independently of the experimental system. These data, showing that that TGF-β could induce the development of Th17 cells from naive CD4 T cells, suggested that at a molecular level the requirements for human and mouse Th17 differentiation were similar and that mouse models could still be useful to clarify Th17-mediated immune mechanisms in humans. The role of TGF-β on the expression of IL-17 from human memory cells seems to be different from its effect on naive cells. At doses that others have shown necessary for IL-17 production in naive T cells, IL-17 production from memory T cells is actually inhibited [23,24,53]. This may have relevance to human disease mechanisms in vivo, as it is likely that memory/effector T cells will be the cell population responsible for the majority of IL-17 production at inflammatory sites.
CD161 has been identified as a novel surface marker for human IL-17-secreting cells [62,63]. CD161 is the human homologue of murine NK1·1, which is expressed on almost all natural killer (NK) cells and also by a subset of T cells called NK T cells [64,65]. One study showed that human Th17 cells originated exclusively from a CD161+CD4+ T cell precursor using umbilical cord blood (UCB) or single-positive CD4+CD8+ thymocytes in the presence of IL-1β and IL-23. Naive CD161+CD4+ T cells were found to express both RORC2 and IL-23 receptor constitutively even before culture, indicating that RORC2 expression did not depend upon TGF-β that had been added exogenously or had been present in the culture serum [62,66,67]. In agreement with other studies, there was an inability to induce Th17 from purified CD45RA+CD45RO- naive circulating T cells of adult volunteers, and this was presumed to be due to the fact that that population did not possess a CD161+ fraction. It is unclear why CD161+ UCB or thymic naive cells can induce Th17 differentiation, while a purified CD45RA+CD45RO- naive population cannot. An explanation could be that Th17 cell precursors are more abundant in UCB than adult blood, possibly from the fact that they migrate very early into tissues, where they can differentiate into IL-17-secreting cells later in life.
A recent paper by the same group claimed that TGF-β did not have a direct critical role on the differentiation of human IL-17-secreting T cells, but rather acted indirectly to favour IL-17 induction by suppressing the expression of T-bet and inhibiting selectively the expansion of IFN-γ-secreting T cells [67]. This would result in a greater degree of positive selection of an already existing RORC2-expressing T cell population and relative expansion of an IL-17 positive population induced by IL-1β and IL-23. They suggested that the effect of TGF-β on the development of Th17 and Th1 cells could be due to the lower susceptibility of Th17 versus Th1 and Th2 cells to its suppressive activity, as shown by the relatively weaker inhibitory effect of TGF-β on the proliferation of human Th17 clones/IL-17-secreting circulating CD4+ T cells in comparison to the strong inhibition on that of Th1 and Th2 clones and IFN-γ-secreting T cells. A further study by Gerosa and colleagues agreed with the premise that the requirement for TGF-β by naive T cells to produce IL-17 might be due partially to its inhibiting effect on IFN-γ production [68]. Their particular study looked at naive CD4+ T cells co-cultured with supernatants from monocyte-derived dendritic cells that had been activated with zymosan or β-glucan. They clarified further that although IL-1β, IL-23, IL-6 and TGF-β were all present in the supernatants collected from their APC system, IL-1β was the essential cytokine needed, along with one or more of the other cytokines, to induce IL-17 production.
It has emerged recently that Th17 differentiation is enhanced by stimulation of the aryl hydrocarbon receptor (AhR), a ligand-dependent transcription factor that responds to a wide range of ligands [69]. Exposure of mouse CD4+ T cells to the tryptophan metabolite and AhR ligand, 6-formylin-dolo [3,2-b] carbazole (FICZ), under Th17 polarizing conditions enhances Th17 development and exacerbates autoimmune pathology in EAE. The same group has also shown that human Th17 cells express AhR and that AhR ligands support Th17 differentiation in human CD4+ T cells [70]. The disparities between different levels of Th17 polarization found by other groups was suggested to be due to differences in the generation of endogenous AhR ligands in different culture media, with RPMI-1640 having less activity than IMDM. IMDM contains 3–5 times higher amounts of aromatic amino acids, such as tryptophan, tyrosine and phenylalanine than RPMI-1640, hence giving rise to different levels of endogenous AhR ligands in the two media. It was shown further that human Th17 cells from bulk CD4+ T cells from peripheral blood was improved markedly in IMDM compared with RPMI-1640 and reduced strongly in the presence of an AhR antagonist. A summary of the different in vitro culture conditions used by different groups is shown in Table 1, which indicates the large number of protocols currently in use.
Table 1.
Summary of the diverse skewing conditions used for human interleukin (IL)-17 differentiation in naive and memory CD4+ T cells by various research groups.
| Group | Cells | Separation | TCR activity | Cyt Sk. (ng/ml) | Culture conditions | Outcome |
|---|---|---|---|---|---|---|
| Wilson [55]: Malefyt | CD4+CD45RO- | CD45RO Bead dep | CD2/3/28 beads | IL-1β + IL-23 (50) | Yssel's medium + Hu antibody serum | TGF-β inhibits IL-17 induction |
| Acosta-Rodriguez [54]: Sallusto | CD45RA+CCR7+CD25- | Cell sort | CD3/28 beads/allogeneic monocyte | IL-1β (10) + IL-6 (50) | RPMI | TGF-β inhibits IL-17 induction |
| Chen [49]: O-Shea | PBMCs Naive: RA+RO- (90%) | CD45RO- CD45RA+ MACS: 90% | pbCD3 + sCD28 | TGFB IL-6 IL-23 (10) | Unspecified | Low levels of IL-17 secreted by naive versus memory cells |
| Evans [23]: Taams, Lord | CD25-CD45RO- CD45RA+/CD25- CD45RA-CD45RO+ | MACS | pbCD3/28 | Anti-IFN-γ antibody (10 µg/ml) | RPMI | IL-17 not induced in naive cells. In memory cells: 1.TGF-β inhibits IL-17 production 2.Anti-IFN-γ produces optimal IL-17 |
| Van Beelen [24] | CD45RA+CD45RO- CCR7+ | MACS and FACS | 1.Bacteria-primed dendritic cells 2.pbCD3/28 (1 µg/ml) | IL-1β (10), IL-23 (10) | IMDM + 10%FCS, gentamycin | Promote IL-17 production from memory and not naive cells Best condition for producing IL-17 from memory cells: IL-23 + IL-1β from an APC-free system or from dendritic cells |
| Liu & Rohowsky-Kochan [51] | Memory CD45RO+CCR7+CD45RO+CCR7- | Neg. MACS Neg. MACS | pbCD3 (5 µg/ml) sCD28 | IL-1β (10) + IL-6 (40) + IL-23 (50) | RPMI 10% supplemented with anti-CD28 (2 µg/ml) | IL-17 produced from CD45RO+CCR7-CCR6+ |
| Manel [57]: Littman | Buffy coat: (1. Naive; 2. Memory) 1.CD3+CD4+RO-CD25-2.CD3+CD5+CD25-RO+UCB: CD3+CD4+CD25- HLA-DR-CD45RA+ | MACS for CD4 FACS for naive and memory | Home-made T cell expander 1 : 1 1 µg/ml CD3 and CD28 | IL-1β (10) IL-23 (10) TGF-β (10) | X-vivo 15 supplemented with pen/strep RPMI fully supplemented/RPMI w/o FCS | TGF-β (1) alone inhibits RORγt-induced IL-17 production. TGF-β (10) with proinflammatory (IL-1β/IL-23/IL-6) is required for RORγt and IL-17 induction |
| Volpe [59]: Soumelis | 1.CD3+CD4+CD45RA+CD 45RO- (buffy coat) 2.CD4+CD45RA 3.Human cord blood | 1.MACS (96%) 2.Cell sorted (99%) | Dynabeads CD3/28 | IL-1β + IL-6 + TNF (all 10) + IL-23 (100) + TGF-β (1) | Yssel's medium + serum X-vivo 15 | IL-23+TGF-β essential TNF not essential IL-1β + IL-6 increase No effect of serum on IL-17 production Exogeneous TGF-β promotes IL-17 differentiation |
| Gerosa [66]: Trinchieri | Naive: CD4+RO- | StemCell Tech Kit (98%) | pbCD3+CD28 | IL-1β (10) IL-23 (1·5–15) IL-6, TGF-β | RPMI + 10%FBS + L-Glu | IL-1β essential for IL-17 induction Used supernatants from T cell/monocyte-derived DCs stimulated with zymosan and β-glucan |
| Yang [58]: Hafler | Naive: CD4+CD25- CD62L+CCR7+CD45RA Central memory: CD4+ | Cell sorted | pbCD3 (1·5 µg/ml) + sCD28 (1 µg/ml) | IL-6 (25) TGF-β not acidified (5) IL-1β (12·5) IL-21 (25), IL-23 (25) | X-vivo 15 supplemented with 1 µg/ml soluble CD28 | IL-17 induction in naive: TGF-β + IL-21 Effector: IL-1β and IL-1β + IL-6 Effect of TGF-β: enhances from 0·1 to 10, inhibits 50 |
| Cosmi [60]: Annunziato | CD4+CD161+UCB | MACS | CD3+CD28 (5 µg/ml) | IL-1β 10 + IL-23 20 (IL-6 2); IL-12 2; TGF-β 5; IL-21 50 | RPMI + goodies + 10% FBS | Unable to differentiate Th17 from naive CD4 PB (no CD161+ cells) Enrichment of CD161 in UCB and thymocytes promotes IL-17 |
| Veldhoen*[67]: Stockinger | Bulk CD4 | MACS | pbCD3 (1 µg/ml) pbCD28 (2·5 µg/ml) | TGF-β(0·5), IL-6 (30) IL-1β (10), IL-23 (10) | IMDM or RPMI + 10% serum replacement factor (Invitrogen) | Increased IL-17 in IMDM and RPMI Supplemented with AhR agonist |
| Santarlasci [65]: Annunziato | UCB CD161+ cells | MACS | CD3/CD28 (5 µg/ml) | IL-1β (10), IL-23 (20) TGF-β (0·1–5) | RPMI + FCS + 2-mercaptoethanol X-vivo 15 | TGF-β does not inhibit Th17 proliferation TGF-β favours Th17 development by inhibiting T-bet (Th1) |
More mouse than human data. APC: antigen presenting cells; Cyt Sk.: cytokine skewing; Neg: negative; pb: plate bound; s: soluble; IFN: interferon; TGF: transforming growth factor; TCR: T cell receptor; DC: dendritic cell; HLA-DR: human leucocyte antigen D-related; PBMC: peripheral blood mononuclear cells; FCS: fetal calf serum; FBS: fetal bovine serum; MACS: magnetic affinity cell sorting; RORγt: retinoic acid-related organ receptor gamma-t; UCB: umbilical cord blood; IMDM: Iscove's modified Eagle's medium; AhR: aryl hydrocarbon receptor.
Plasticity of Th17 cells
CD4+ T cell lineages are relatively plastic, with the possibility of one committed lineage shifting to another [5,71]. Both IFN-γ, the Th1 signature cytokine, and IL-4, the Th2 signature cytokine, inhibit IL-17 induction in mice and humans [5,23]. In mice, the loss of the transcription factor T-bet results in enhanced IL-17 induction in CD4+ T cells [3,5,6]. We found that blocking IFN-γ signalling in a lipopolysaccharide (LPS)-activated monocyte system as well as in an APC-free system increased the number of IL-17-secreting memory cells, suggesting that T cell polarity was relatively flexible in human CD4+ T cells [23]. It could be inferred that there was a distinct possibility of a switch in lineage commitment from previously committed effector T cells. This hypothesis was supported by the presence of the single positive IL-17 population (IFN-γ-/IL-17+) as well as a substantial percentage of IFN-γ+/IL-17+ double-positive cells in humans observed by a number of research groups [23,51,52,72]. Those two cell types were found to express both RORC2 and T-bet and the Th17 cells could be shifted to Th1 by the addition of IL-12, which indicated that Th1 and Th17 cells shared a common pathway [72]. It will be interesting to determine whether those two populations represent different subsets of helper T cells derived from a common precursor or different stages of lineage polarization. Moreover, growth factor independent-1 (Gfi-1) induced by IL-4 promotes Th2 differentiation and at the same time inhibits Th17 differentiation in murine models [73,74]. It is therefore apparent that there is a degree of flexibility between Th1, Th2 and Th17 phenotypes in vivo, although it is uncertain whether this plasticity works effectively in all directions.
The Th17/regulatory T cell (Treg) differentiation pathways also point towards such plasticity [71,75]. Both Th17 effector cells and regulatory T cells play a critical role in autoimmune inflammation, despite the fact that their mechanisms of action are opposed to one other. Tregs, defined by high expression of forkhead box P3 (FoxP3) in murine and human systems, are strong immunomodulators of T cell activation that suppress proliferation and cytokine production by effector T cells [37,76,77]. The reciprocal relationship between Th17 and Tregs is indicated by their mutual requirement for TGF-β[37,39,78]. Murine studies showed that TGF-β induced FoxP3 Tregs from naive T cells, but the addition of IL-6 inhibited FoxP3 development and instead promoted Th17 differentiation [37]. There are several lines of evidence to support the reciprocal relationship between Th17 and Tregs. This was further illustrated by the fact that IL-2, a growth factor for Tregs, could also inhibit the induction of Th17 cells [79]. This study demonstrated that mice lacking IL-2 signalling pathway components (STAT5−/−) possessed lower levels of Tregs and increased levels of Th17 in the peripheral repertoire. Retinoic acid, a vitamin D metabolite, could stimulate the generation of Treg cells by promoting TGF-β signalling and FoxP3 promoter activity while inhibiting Th17 differentiation by blocking IL-6 signalling [80]. Focusing on the human system, the phenomenon of loss in Treg function and induction of Th17 cells has been demonstrated in the pathogenesis of human autoimmune diseases such as multiple sclerosis [81], rheumatoid arthritis [82], Crohn's disease [72] and psoriasis [83]. Beriou and colleagues showed recently that human FoxP3 Treg cells could be induced to produce IL-17 in an inflammatory medium, pointing towards a mechanism of immune regulation where inflammation could drive a subset of Tregs to adopt a Th17 phenotype that allowed for the rapid shut-down of suppression and induction of proinflammatory responses [84]. IL-17 production by FoxP3+/IL-17+ cells was induced by proinflammatory cytokines (IL-1β/IL-6) and inhibited by TGF-β. These data were confirmed by the group led by Koenen, who demonstrated IL-17 production in Tregs stimulated with allogenic antigen-presenting cells [85]. Both ex vivo and in vitro, expression of FoxP3 was maintained with IL-17 production leading to co-expression of FoxP3 and IL-17, a finding that was also seen at the transcriptional level [84,86].
Looking at the transcriptional mechanism of Th17 cells and Tregs in mice, Zhou and colleagues showed recently that TGF-β alone was sufficient to induce both RORγt and FoxP3 [86]. A low dose of TGF-β along with IL-6/IL-21 stimulated Th17 induction; a higher dose of TGF-β inhibited IL-23R expression with a corresponding elevation of FoxP3 expression, inducing the production of Treg cells. In fact, the study demonstrated that as FoxP3 expression increased, it could interact directly with RORγt, leading to inhibition of its transcriptional activity. A recent study, this time in human cells, agreed that T cells could co-express RORγt and FoxP3 [87]. Moreover, a particular FoxP3 sequence was demonstrated to associate with RORα, another Th17-specific transcription factor, indicating that FoxP3 suppressed Th17 development through inhibition of both RORγt and RORα[88]. Another study found that the Runt-related transcription factor 1 (Runx1) bound to RORγt and synergized to induce Th17 differentiation [89]. Runx1 also interacted with FoxP3 to inhibit Th17 differentiation. The plasticity of the Th17 and Treg developmental pathways may indicate that Th17 and induced Tregs have a common T cell precursor that can differentiate into one or the other, depending upon the cytokines present and the activity of a number of transcription factors. The potential of Tregs to become IL-17-producing cells and the underlying mechanisms are discussed further by Afzali et al. in this series [90].
It is important to understand this reciprocal relationship in autoimmune diseases such as rheumatoid arthritis, psoriasis or multiple sclerosis. Although many of the results on the Th17/Tregs relationship come from murine studies, the periods of flares and quiescence in human autoimmune diseases also argue for a similar reciprocal relationship between those two T lineages in humans. It has been proposed that during acute flares of inflammation immune responses become dysregulated, and T cell phenotypes have the tendency to lean towards proinflammatory lineages (Th17 or Th1) and away from anti-inflammatory phenotypes (Treg) depending upon the cytokine milieu and the local DC population [91]. During recovery from flares or periods of quiescence, the balance would be redressed and in this instance the anti-inflammatory phenotype would have a dominant effect over proinflammatory effector populations.
Conclusion
It is clear that many similarities exist between human and mouse Th17 cells, and that further understanding of disease mechanisms in vivo will require accurate dissection in preclinical models. Nevertheless, it is important to recognize that there may be some differences between human and mouse disease in the context of T cell polarity, due perhaps to the effect of genetic modifiers when looking at outbred human populations. The next few years will provide exciting opportunities to increase both our understanding of fundamental mechanisms in T cell biology and also how we can translate these discoveries into better diagnosis and therapy of human disease.
Acknowledgments
This work was supported by the Medical Research Council (GML), and BBSRC. G. M. L. acknowledges financial support from the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.
Disclosure
None.
References
- 1.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–93. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 2.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 3.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–42. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 4.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 5.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 6.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 8.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao Z, Fanslow WC, Seldin MF, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–21. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 10.Yao Z, Spriggs MK, Derry JM, et al. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- 11.Gaffen SL. An overview of IL-17 function and signaling. Cytokine. 2008;43:402–7. doi: 10.1016/j.cyto.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–76. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Fort MM, Cheung J, Yen D, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–95. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 14.Liang SC, Long AJ, Bennett F, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–9. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- 15.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 16.Chung DR, Kasper DL, Panzo RJ, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–63. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- 17.Happel KI, Dubin PJ, Zheng M, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–31. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 19.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–15. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 20.LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–8. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 21.Ye P, Garvey PB, Zhang P, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–40. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 22.Malley R, Srivastava A, Lipsitch M, et al. Antibody-independent, interleukin-17A-mediated, cross-serotype immunity to pneumococci in mice immunized intranasally with the cell wall polysaccharide. Infect Immun. 2006;74:2187–95. doi: 10.1128/IAI.74.4.2187-2195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans HG, Suddason T, Jackson I, Taams LS, Lord GM. Optimal induction of T helper 17 cells in humans requires T cell receptor ligation in the context of Toll-like receptor-activated monocytes. Proc Natl Acad Sci USA. 2007;104:17034–9. doi: 10.1073/pnas.0708426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–9. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miranda-Carus ME, Benito-Miguel M, Balsa A, et al. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis Rheum. 2006;54:1151–64. doi: 10.1002/art.21731. [DOI] [PubMed] [Google Scholar]
- 27.Pene J, Chevalier S, Preisser L, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180:7423–30. doi: 10.4049/jimmunol.180.11.7423. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. 2008;57:1682–9. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 29.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 30.Matusevicius D, Kivisakk P, He B, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 31.Wong CK, Ho CY, Li EK, Lam CW. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus. 2000;9:589–93. doi: 10.1191/096120300678828703. [DOI] [PubMed] [Google Scholar]
- 32.Molet S, Hamid Q, Davoine F, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. 2001;108:430–8. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 33.Kato T, Furumoto H, Ogura T, et al. Expression of IL-17 mRNA in ovarian cancer. Biochem Biophys Res Commun. 2001;282:735–8. doi: 10.1006/bbrc.2001.4618. [DOI] [PubMed] [Google Scholar]
- 34.Loong CC, Hsieh HG, Lui WY, Chen A, Lin CY. Evidence for the early involvement of interleukin 17 in human and experimental renal allograft rejection. J Pathol. 2002;197:322–32. doi: 10.1002/path.1117. [DOI] [PubMed] [Google Scholar]
- 35.Luzza F, Parrello T, Monteleone G, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–7. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 36.Crome SQW, Wang AY, Levings MK. Function and regulation of human T helper 17 cells in health and disease. Clin Exp Immunol. 2009 doi: 10.1111/j.1365-2249.2009.04037.x. doi: 10.1111/j.1365-2249.2009.04037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 38.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 39.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Induction of cartilage damage by overexpression of T cell interleukin-17A in experimental arthritis in mice deficient in interleukin-1. Arthritis Rheum. 2005;52:975–83. doi: 10.1002/art.20885. [DOI] [PubMed] [Google Scholar]
- 41.Koenders MI, van den Berg WB. Are T helper 17 cells really pathogenic in autoimmunity? Clin Exp Immunol. 2009 doi: 10.1111/j.1365-2249.2009.04039.x. doi: 10.1111/j.1365-2249.2009.04039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 43.Hoeve MA, Savage ND, de Boer T, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–70. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 44.Kidoya H, Umemura M, Kawabe T, et al. Fas ligand induces cell-autonomous IL-23 production in dendritic cells, a mechanism for Fas ligand-induced IL-17 production. J Immunol. 2005;175:8024–31. doi: 10.4049/jimmunol.175.12.8024. [DOI] [PubMed] [Google Scholar]
- 45.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–91. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauquet AT, Jin H, Paterson AM, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–75. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ettinger R, Sims GP, Fairhurst AM, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175:7867–79. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 48.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–74. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 49.Liu XK, Lin X, Gaffen SL. Crucial role for nuclear factor of activated T cells in T cell receptor-mediated regulation of human interleukin-17. J Biol Chem. 2004;279:52762–71. doi: 10.1074/jbc.M405764200. [DOI] [PubMed] [Google Scholar]
- 50.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–46. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Rohowsky-Kochan C. Regulation of IL-17 in human CCR6+ effector memory T cells. J Immunol. 2008;180:7948–57. doi: 10.4049/jimmunol.180.12.7948. [DOI] [PubMed] [Google Scholar]
- 54.Napolitani G, Acosta-Rodriguez EV, Lanzavecchia A, Sallusto F. Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-gamma production by memory CD4+ T cells. Eur J Immunol. 2009;39:1301–12. doi: 10.1002/eji.200838969. [DOI] [PubMed] [Google Scholar]
- 55.Evans HG, Gullick NJ, Kelly S, et al. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci USA. 2009;106:6232–7. doi: 10.1073/pnas.0808144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 57.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 58.Boniface K, Bak-Jensen KS, Li Y, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–48. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–2. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–7. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 62.Cosmi L, De Palma R, Santarlasci V, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903–16. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romagnani S, Maggi E, Liotta F, Cosmi L, Annunziato F. Properties and origin of human Th17 cells. Mol Immunol. 2009 doi: 10.1016/j.molimm.2008.12.019. in press. [DOI] [PubMed] [Google Scholar]
- 64.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 65.Godfrey DI, Berzins SP. Control points in NKT-cell development. Nat Rev Immunol. 2007;7:505–18. doi: 10.1038/nri2116. [DOI] [PubMed] [Google Scholar]
- 66.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int Immunol. 2008;20:1361–8. doi: 10.1093/intimm/dxn106. [DOI] [PubMed] [Google Scholar]
- 67.Santarlasci V, Maggi L, Capone M, et al. TGF-beta indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur J Immunol. 2009;39:207–15. doi: 10.1002/eji.200838748. [DOI] [PubMed] [Google Scholar]
- 68.Gerosa F, Baldani-Guerra B, Lyakh LA, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. 2008;205:1447–61. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veldhoen M, Hirota K, Westendorf AM, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 70.Veldhoen M, Hirota K, Christensen J, O'Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–9. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–55. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu J, Davidson TS, Wei G, et al. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J Exp Med. 2009;206:329–41. doi: 10.1084/jem.20081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu J, Guo L, Min B, et al. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity. 2002;16:733–44. doi: 10.1016/s1074-7613(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 75.Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009;21:281–5. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–53. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 77.O'Connor RA, Taams LS, Anderton SM. CD4+ T helper cells: functional plasticity and differential sensitivity to regulatory T cell-mediated regulation. Clin Exp Immunol. 2009 doi: 10.1111/j.1365-2249.2009.04040.x. doi: 10.1111/j.1365-2249.2009.04040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–7. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 80.Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 81.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200:277–85. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sugiyama H, Gyulai R, Toichi E, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–73. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beriou G, Costantino CM, Ashley CW, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood. 2009;113:4240–9. doi: 10.1182/blood-2008-10-183251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112:2340–52. doi: 10.1182/blood-2008-01-133967. [DOI] [PubMed] [Google Scholar]
- 86.Zhou L, Lopes JE, Chong MM, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA. 2009;106:4793–8. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180:4785–92. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- 89.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Induction of interleukin-17 production by regulatory T cells. Clin Exp Immunol. 2009 doi: 10.1111/j.1365-2249.2009.04038.x. doi: 10.1111/j.1365-2249.2009.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]