

Abstract
Activation-induced deaminase (AID) is a prerequisite for immunoglobulin (Ig) class-switch recombination and somatic hypermutation, which is critical for antibody affinity maturation. IgM and IgG autoantibodies are characteristic of the systemic autoimmune disorders such as lupus. However, the relative contributions of hypermutated high-affinity IgG antibodies and germline-encoded IgM antibodies to systemic autoimmunity are not defined fully. The role of AID in autoimmunity is unclear. The current study used AID-deficient mice to investigate the role of AID in the development and pathogenesis of murine lupus. C57BL/6 mice deficient in both Fas and AID were generated. Compared to their AID-competent littermates, AID−/− lymphoproliferative (lpr) mice produced significantly elevated levels of IgM autoreactive antibodies with enhanced germinal centre (GC) response, developed more advanced splenomegaly and exhibited more severe glomerulonephritis. Thus, AID may play an important role in the negative regulation of systemic autoimmune manifestations in murine lupus. The results also indicate that hypermutated high-affinity IgG antibodies are not necessary for the development of autoimmune syndrome in lpr mice on a C57BL/6 background.
Keywords: AID, autoantibodies, autoimmunity, lupus
Introduction
High levels of autoantibodies are associated with many autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) [1–3]. SLE is considered a systemic autoimmune disease in which immune complex deposition or direct autoantibody deposition leads to complement fixation, Fc receptor (FcR) activation and subsequent tissue inflammation [2]. The majority of autoantibodies in human and murine lupus are hypermutated high-affinity immunoglobulin (Ig)G antibodies, which have usually been considered pathogenic [4–10]. The role of IgM autoantibodies in autoimmune diseases is controversial. The relative contribution of IgG and IgM antibodies to lupus-like pathogenesis remains to be defined [11–15]. Importantly, it has not been determined whether high-affinity antibodies that result from Ig gene somatic hypermutation (SHM) and subsequent clonal selection of activated B cells are critical in eliciting organ damage and systemic autoimmune syndrome in lupus.
Activation-induced cytidine deaminase (AID), a member of the cytidine deaminase family that acts as an editor of DNA and RNA, is required for Ig gene SHM and isotype class-switch recombination (CSR) [16–18]. To study the role of AID in the pathogenesis of murine lupus, we introduced the AID deficiency into the lupus-prone lymphoproliferative (lpr) mice of C57BL/6 background (B6). Our results showed that, compared to their AID-competent littermates, AID-deficient B6-lpr mice produced significantly elevated levels of IgM autoreactive antibodies, developed more severe splenomegaly and suffered from more severe glomerulonephritis. Thus, our findings indicate that AID deficiency promotes systemic autoimmune manifestations.
Materials and methods
Mice
AID-deficient mice were kindly provided by Dr T. Honjo (Kyoto University, Kyoto, Japan) and Dr David Schatz (Yale University). C57BL/6-lpr (B6-lpr) mice were purchased from the Jackson Laboratory. AID−/− mice were back-crossed to B6-lpr mice for six generations. All groups of mice (B6, AID−/−, B6-lpr and AID−/−lpr) used in the experiments were littermates. Mice of both sexes were kept in specific pathogen-free facilities. All animals were housed in autoclaved microisolators, provided with sterile bedding, food and water, and maintained on a 12-h day/night cycle. Animal experimentation was performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine.
Flow cytometry
Single-cell suspensions of spleens and lymph nodes from mice of various genotypes were depleted of erythrocytes and first incubated with purified anti-mouse CD32/CD16 (PharMingen, San Diego, CA, USA) to block antibody binding to Fcγ receptors. The cells were then stained with different antibodies to cell surface markers. The samples were acquired by a fluorescence activated cell sorter (FACS)Calibur flow cytometer (BD Bioscience, San Jose, CA, USA) and analysed using Flowjo software (TreeStar, Inc., San Carlos, CA, USA).
Serum autoantibody assays
Serum levels of anti-dsDNA antibodies were analysed by enzyme-linked immunosorbent assay (ELISA) as described previously [19]. Microplates were coated with dsDNA (Invitrogen, Carlsbad, CA, USA) and blocked with 10% fetal calf serum (FCS). Samples were added and incubated for 1 h at 37°C and washed. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgM (Southern Biotechnology Associates, Birmingham, AL, USA) was used as secondary detection antibody. HRP activity was visualized using a tetramethylbenzidine (TMB) peroxidase substrate kit (Bio-Rad Laboratories, Hercules, CA, USA), and optical densities were determined at 450 nm using an automated reader.
Urine analysis
Urine from all groups of mice was collected and levels of urine protein, leucocytes and red blood cells were determined semi-quantitatively with Multistix 8 SG (Bayer Corporation, Elkhart, IN, USA).
Histology
The procedures for freezing tissues, sectioning and immunohistochemical staining were conducted as described previously [20]. Briefly, kidneys and spleens were frozen in optical cutting temperature (OCT) compound and serial 6-µm-thick frozen sections were cut in a cryostat microtome, thaw-mounted onto poly-L-lysine-coated slides, air-dried, fixed in ice-cold acetone for 10 min and stored at −80°C. To examine immune complex deposition in glomeruli, kidney sections were incubated with fluorescein-conjugated goat anti-mouse C3 antibody (ICN Biomedicals, Inc., Irvine, CA, USA) and goat F(ab′)2 anti-mouse Ig antibody (Dako, Glostrup, Denmark) and examined under a fluorescent microscope. The numbers of positive glomeruli were counted. Then the same sections were restained with haematoxylin and eosin (H&E) to obtain total numbers of glomeruli. The percentage of glomeruli with immune complex deposition were calculated. The pathology scores of renal tissue of H&E-stained sections were graded on 0–4 scale as described [21].
Statistical analysis
Data were analysed using either a one-way analysis of variance (anova) and Newman–Keuls post-hoc test or an unpaired Student's t-test with Welch's correction (GraphPad Prism, Inc., La Jolla, CA, USA).
Results
AID deficiency promotes splenomegaly in B6-lpr mice
To study whether AID deficiency affects the development of systemic autoimmunity in lpr mice, we generated AID−/−lpr mice by back-crossing AID−/− mice to B6-lpr mice. Control animals including both wild-type B6 and AID−/− mice were their littermates. All the mice were on a B6 background. As the onset of the autoimmune syndrome in lpr mice with a B6 background is relatively delayed compared to MRL/lpr mice, all the mice were analysed at 12–14 months of age.
Compared to B6-lpr mice, AID−/−lpr mice exhibited more pronounced splenomegaly (Fig. 1). At 12 months of age, the sizes of spleens from AID−/−lpr mice were significantly larger than those of lpr mice. In addition, the total numbers of cells from the spleen of AID−/−lpr mice were significantly higher than those in lpr mice (Table 1). The total numbers of B cells in AID−/−lpr mice were increased significantly compared to those in B6-lpr mice, indicating more B cell expansion in the absence of AID. There were no significant differences in the sizes of mesenteric lymph nodes from lpr and AID−/−lpr mice (data not shown).
Fig. 1.
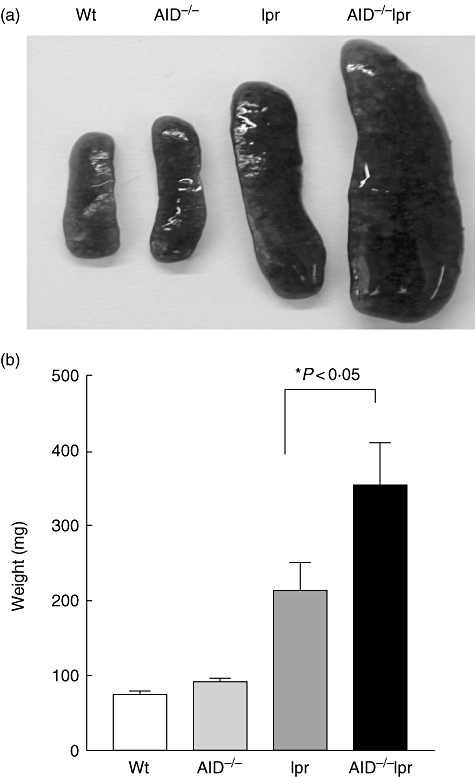
Enhanced splenomegaly in activation-induced deaminase (AID)−/− lymphoproliferative (lpr) mice. (a) Representative photographs of spleens from 12-month-old wild-type B6, AID−/−, B6-lpr and AID−/−lpr mice. (b) Weight of spleens from wild-type B6, AID−/−, B6-lpr and AID−/−lpr mice (n = 19, 26, 23, 22, respectively).
Table 1.
Cell numbers in the spleen (×106).
| Spleen | B6 | AID−/− | lpr | AID−/−lpr |
|---|---|---|---|---|
| Total | 53·7 ± 13·0 | 67·1 ± 6·2 | 96·3 ± 26·6 | 122·8 ± 46·9 |
| CD19+ B cells | 28·5 ± 4·5 | 32·1 ± 7·5 | 42·1 ± 3·5 | 61·2 ± 4·5 |
| CD3+ T cells | 19·4 ± 3·7 | 22·6 ± 2·3 | 35·8 ± 5·3 | 42·5 ± 3·8 |
| CD4+ T cells | 12·1 ± 2·0 | 13·2 ± 2·2 | 24·3 ± 3·7 | 25·2 ± 4·4 |
| CD8+ T cells | 5·9 ± 1·3 | 6·8 ± 1·5 | 5·4 ± 2·8 | 6·6 ± 1·1 |
| DN T cells* | 1·2 ± 0·5 | 2·2 ± 0·3 | 5·7 ± 1·7 | 9·5 ± 1·4 |
Double negative (DN) T cells represent CD3+CD4-CD8- T cells. Spleens were removed from 12-month old wild-type B6, AID−/−, lymphoproliferative (lpr) and activation-induced deaminase (AID)−/− mice, respectively, and numbers of total cells and lymphocytes were enumerated; n = 5–8.
Increased double negative (DN) T cells and enhanced germinal centre (GC) response in AID−/−lpr mice
It is known that a population of CD4-CD8-B220+TCRαβ+ DN T cells is greatly accumulated in both lupus patients and lpr mice, which is caused in part by fas defects, leading to abnormal lymphocyte survival with subsequent autoimmunity [22,23]. Interestingly, we found that this population was increased greatly in AID−/−lpr mice compared to B6-lpr mice (Fig. 2a and Table 1). These DN T cells were B220+ and the vast majority of them were T cell receptor (TCR)αβ+ (data not shown). The data indicate profound abnormal expansion of T cells in the absence of AID.
Fig. 2.
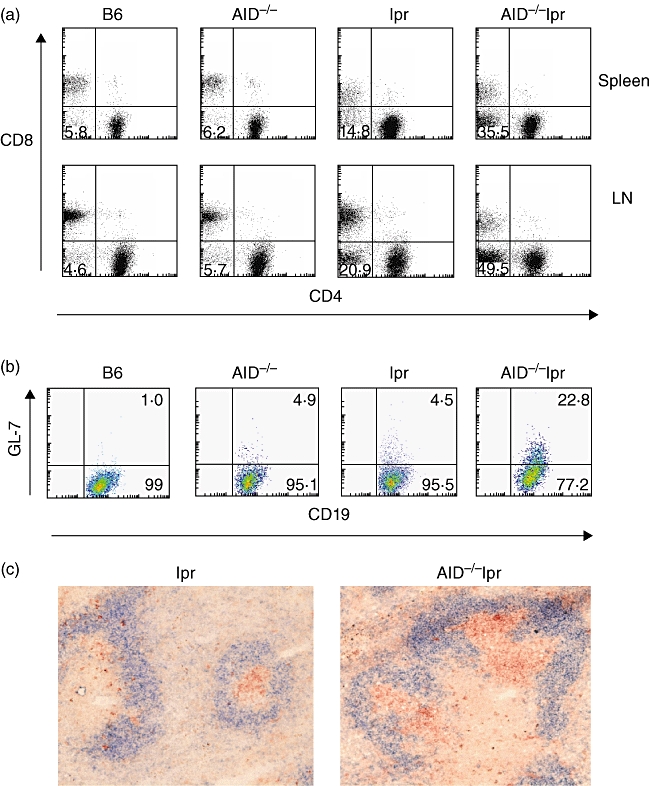
Increased double negative (DN) T cells and enhanced germinal centre (GC) response in activation-induced deaminase (AID)−/− lymphoproliferative (lpr) mice. (a) CD3+ T cells were gated and analysed further for CD4-CD8- DN T cells. The numbers shown represent the percentages of DN T cells within the T cell gate. (b) Splenic cells were gated on CD19+ B cells and analysed further for the percentages of GC B cells within the CD19+ B cell gate. The data were representative of results from eight to 10 mice (12–14 months old) in each group. (c) Representative splenic sections stained with peanut agglutinin (PNA) (red) and anti-IgD antibody (blue) to visualize GCs in the spleen of B6-lpr and AID−/−lpr mice.
Enhanced development of spontaneous GC response is a hallmark of systemic autoimmunity in lupus-prone mice. To determine whether AID deficiency promotes GC response, we analysed GC formation in AID-deficient mice and their AID-competent counterparts. At 12 months old, wild-type B6 mice had few GL-7+ GC B cells in the spleen (Fig. 2b). However, the GL-7+ B cell population was increased significantly in AID−/−lpr mice compared to that in lpr mice. The increase in GC formation in AID−/−lpr mice was confirmed by immunohistology. GCs are significantly larger in size in the spleens of AID−/−lpr mice compared to those of B6-lpr mice (Fig. 2c). The data demonstrated that activated B cells were expanded in AID−/−lpr mice.
Elevated levels of IgM autoantibodies in AID−/−lpr mice
Because AID deficiency leads to impaired Ig somatic hypermutation and isotype class-switch, antibodies other than IgM isotype are not produced in AID-deficient mice [16–18]. We analysed the serum IgM autoantibodies to dsDNA by ELISA. By 12 months of age, the levels of IgM antibodies against dsDNA were significantly higher in AID−/−lpr mice than those in lpr mice (Fig. 3a). We further determined total serum IgM levels in different groups of mice. AID−/− mice at 12 months of age had significantly elevated serum IgM levels compared to wild-type controls (Fig. 3b). The total IgM levels in AID−/−lpr mice were also high, but with no statistical significance if compared to total IgM levels in B6-lpr mice.
Fig. 3.
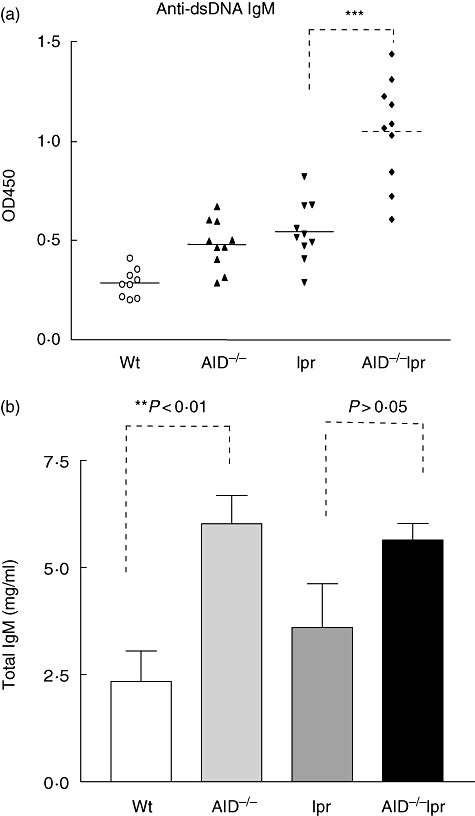
Elevated production of immunoglobulin (Ig)M autoantibodies in activation-induced deaminase (AID)−/− lymphoproliferative (lpr) mice. (a) Anti-dsDNA autoantibodies in the sera from wild-type B6, AID−/−, lpr and AID−/−lpr mice at 12 months old were measured by enzyme-linked immunosorbent assay. (b) Total serum IgM levels in 12-month-old mice were measured; 10 mice each group. **P < 0·01; ***P < 0·001 by Student's t-test.
AID−/−lpr mice develop more severe glomerulonephritis
To determine the impact of AID deficiency on glomerulonephritis, we examined the occurrence and severity of glomerulonephritis in mice of various genotypes. The results showed that compared to B6-lpr mice, AID−/−lpr mice developed more severe proliferative glomerulonephritis, demonstrated by hypercellularity, enlarged Bowman's capsules and more severe destruction of glomerular structure (Fig. 4a). The renal pathology score was significantly higher in AID−/−lpr mice compared to that in lpr mice (Fig. 4d). However, the percentage of glomeruli positive for immune complex deposition in AID−/−lpr mice was increased slightly compared to that in lpr mice, but was not statistically significant (Fig. 4b, c, e and f).
Fig. 4.
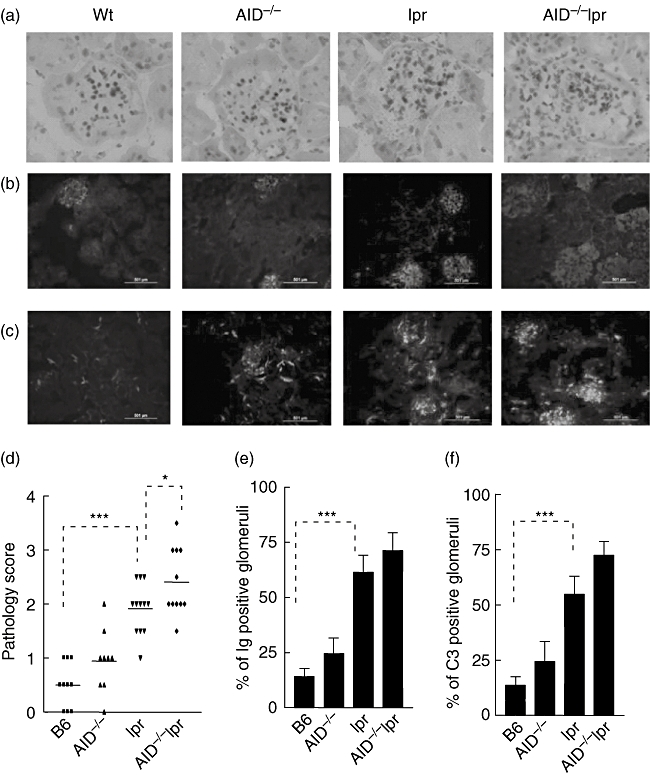
AID-deficiency leads to more severe glomerulonephritis in lpr mice with B6 background. (a) Haematoxylin and eosin (H&E) staining showed glomerulonephritis. (b,c) Immunofluorescent staining showed immunoglobulin (Ig) deposition (b) and C3 deposition (c) in the kidney. (d) The pathology scores of renal tissue were determined. (e,f) The numbers of glomeruli positive for Ig deposition (e) and C3 deposition (f) were counted and calculated as percentage of total glomeruli. Kidney sections from eight to ten mice in each group were analysed.
To assess kidney function, urine of different strains of mice was analysed for the levels of protein, leucocytes and red blood cells. Consistent with more severe destruction of glomeruli, the levels of protein, leucocytes and blood in the urine of AID−/−lpr mice were significantly higher than those in lpr littermates (Fig. 5). Thus, our data indicated that AID deficiency in B6-lpr mice leads to more severe glomerulonephritis and deteriorated kidney function.
Fig. 5.
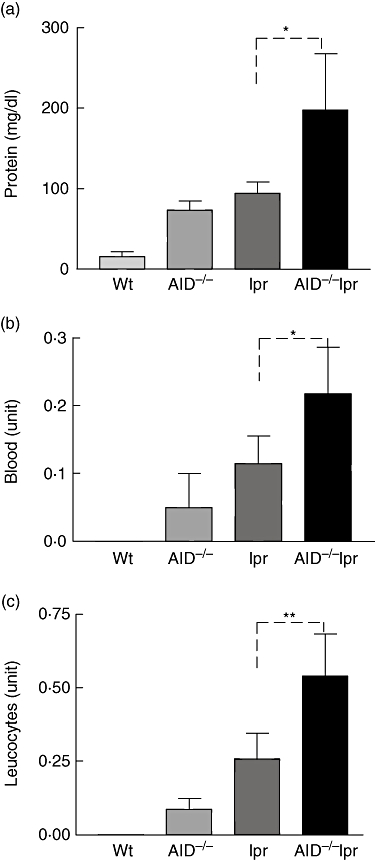
Urinalysis of protein, leucocytes and blood. Data (mean ± standard deviation) are shown for the levels of protein (a), blood (b) and leucocytes (c) in the urine of different strains [B6, n = 19; activation-induced deaminase (AID)−/−, n = 30; lymphoproliferative (lpr), n = 35; AID−/−lpr, n = 35] of mice at 12–14 months of age. *P < 0·05; **P < 0·01.
Discussion
It is clear that B cells play a critical role in the development of systemic autoimmune disease [24]. B cells can promote autoimmune pathology by several potential mechanisms including antigen presentation, cytokine production and autoantibody production. Although autoantibodies are characteristic of the systemic autoimmune disease, their involvement in the pathogenesis has long been debated. The present study revealed that the absence of AID did not alleviate the autoimmune syndrome in lpr mice on a B6 background. Interestingly, AID−/−lpr mice had more profound B cell expansion and severe disease manifestations than lpr mice. As AID deficiency results in little class-switching and somatic hypermutation, the results indicate that IgG autoantibodies are not required for the disease manifestation. AID deficiency in B6-lpr mice had significantly higher levels of IgM antoantibodies compared to B6-lpr littermates, which may contribute to the increased IC deposition in kidney and more severe kidney pathology in AID−/−lpr mice. In addition, it has been shown that AID deficiency drives B cell autoimmunity [25]. AID−/− mice develop tertiary lymphoid organs spontaneously in non-lymphoid tissues and develop severe gastritis with elevated gastric autoantigen-specific serum IgM levels [25]. Thus, the severe autoimmune syndrome in AID−/−lpr mice may also be contributed by AID deficiency. Indeed, our results also revealed that AID deficiency resulted in significantly enhanced GC formation in AID−/−lpr mice, which is consistent with previous reports showing that there is intense proliferation of GC B cells leading to giant GCs in AID-deficient patients [17,26]. Although the mechanism for the dramatically increased GC B cell population caused by AID deficiency is not clear, this property may promote expansion of autoreactive B cells in vivo. In combination with fas deficiency, AID deficiency may accelerate the expansion of autoreactive B cells. The increased GC B cell population may contribute to the severity of autoimmunity in AID−/−lpr mice. Thus, AID may play an important role in the containment of autoimmune diseases by negative regulation of autoreactive B cells.
In summary, the current study demonstrated that AID deficiency results in more severe autoimmune syndrome in the relatively less susceptible C57BL/6 background. Our results are supported by previous reports from AID-deficient patients showing that AID-deficient patients are not only prone to infections, but also to lymphoid hyperplasia, autoimmune diseases and inflammatory disorders [26]. Twenty-one per cent of AID-deficient patients developed autoimmune disorders, including polyarthritis, autoimmune hepatitis and Crohn's disease [26]. Thus, attempts to limit AID function as a therapeutic strategy for the treatment of autoimmune diseases should be cautious.
Acknowledgments
This work was supported by National Institute of Health grant AI051532 (to S. Han).
Disclosure
The authors declare that they have no conflict of interest related to the publication of this manuscript.
References
- 1.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 2.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–6. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–10. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 4.Tsao BP, Ohnishi K, Cheroutre H, et al. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J Immunol. 1992;149:350–8. [PubMed] [Google Scholar]
- 5.Ohnishi K, Ebling FM, Mitchell B, Singh RR, Hahn BH, Tsao BP. Comparison of pathogenic and non-pathogenic murine antibodies to DNA: antigen binding and structural characteristics. Int Immunol. 1994;6:817–30. doi: 10.1093/intimm/6.6.817. [DOI] [PubMed] [Google Scholar]
- 6.Ehrenstein MR, Katz DR, Griffiths MH, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995;48:705–11. doi: 10.1038/ki.1995.341. [DOI] [PubMed] [Google Scholar]
- 7.Radic MZ, Ibrahim SM, Rauch J, Camper SA, Weigert M. Constitutive secretion of transgene-encoded IgG2b autoantibodies leads to symptoms of autoimmune disease. J Immunol. 1995;155:3213–22. [PubMed] [Google Scholar]
- 8.Vaughan JH. 1992 Joseph J. Bunim Lecture. Pathogenetic concepts and origins of rheumatoid factor in rheumatoid arthritis. Arthritis Rheum. 1993;36:1–6. doi: 10.1002/art.1780360102. [DOI] [PubMed] [Google Scholar]
- 9.Korganow AS, Ji H, Mangialaio S, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–61. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 10.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 11.Katz MS, Foster MH, Madaio MP. Independently derived murine glomerular immune deposit-forming anti-DNA antibodies are encoded by near-identical VH gene sequences. J Clin Invest. 1993;91:402–8. doi: 10.1172/JCI116214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito MR, Terasaki S, Kondo E, Shiwaku H, Fukuoka Y, Nose M. Experimental lupus nephritis in severe combined immunodeficient (SCID) mice: remodelling of the glomerular lesions by bystander IgM antibodies. Clin Exp Immunol. 2000;119:340–5. doi: 10.1046/j.1365-2249.2000.01133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youd ME, Luus L, Corley RB. IgM monomers accelerate disease manifestations in autoimmune-prone Fas-deficient mice. J Autoimmun. 2004;23:333–43. doi: 10.1016/j.jaut.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Forger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus. 2004;13:36–44. doi: 10.1191/0961203304lu485oa. [DOI] [PubMed] [Google Scholar]
- 15.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. 2000;97:1184–9. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 17.Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–75. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 18.Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002;20:165–96. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 19.Zheng B, Zhang X, Guo L, Han S. IgM plays an important role in induction of collagen-induced arthritis. Clin Exp Immunol. 2007;149:579–85. doi: 10.1111/j.1365-2249.2007.03440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S, Cao S, Bheekha-Escura R, Zheng B. Germinal center reaction in the joints of mice with collagen-induced arthritis: an animal model of lymphocyte activation and differentiation in arthritis joints. PG-1438-43. Arthritis Rheum. 2001;44:1438–43. doi: 10.1002/1529-0131(200106)44:6<1438::AID-ART239>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 21.Kelley VE, Roths JB. Interaction of mutant lpr gene with background strain influences renal disease. Clin Immunol Immunopathol. 1985;37:220–9. doi: 10.1016/0090-1229(85)90153-9. [DOI] [PubMed] [Google Scholar]
- 22.Bleesing JJ, Brown MR, Novicio C, et al. A composite picture of TcR alpha/beta(+) CD4(-)CD8(-) T cells (alpha/beta-DNTCs) in humans with autoimmune lymphoproliferative syndrome. Clin Immunol. 2002;104:21–30. doi: 10.1006/clim.2002.5225. [DOI] [PubMed] [Google Scholar]
- 23.Shi X, Xie C, Kreska D, Richardson JA, Mohan C. Genetic dissection of SLE: SLE1 and FAS impact alternate pathways leading to lymphoproliferative autoimmunity. J Exp Med. 2002;196:281–92. doi: 10.1084/jem.20010955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hase K, Takahashi D, Ebisawa M, Kawano S, Itoh K, Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS ONE. 2008;3:e3033. doi: 10.1371/journal.pone.0003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quartier P, Bustamante J, Sanal O, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol. 2004;110:22–9. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]