

Abstract
Studies have demonstrated that B cells play important roles in systemic sclerosis (SSc), especially through the CD19/CD22 autoimmune loop. CD22 is a B cell-specific inhibitory receptor that dampens B cell antigen receptor (BCR) signalling via tyrosine phosphorylation-dependent mechanism. In this study, we examined the presence and functional property of circulating autoantibodies reacting with CD22 in systemic sclerosis. Serum samples from 10 tight skin (TSK/+) mice and 50 SSc patients were assessed for anti-CD22 autoantibodies by enzyme-linked immunosorbent assays using recombinant mouse or human CD22. The association between anti-CD22 antibodies and clinical features was also investigated in SSc patients. Furthermore, the influence of SSc serum including anti-CD22 autoantibodies for CD22 tyrosine phosphorylation was examined by Western blotting using phosphotyrosine-specific antibodies reacting with four major tyrosine motifs of CD22 cytoplasmic domain. Anti-CD22 autoantibodies were positive in 80% of TSK/+ mice and in 22% of SSc patients. Patients positive for anti-CD22 antibodies showed significantly higher modified Rodnan skin thickness score compared with patients negative for anti-CD22 antibodies. Furthermore, anti-CD22 antibodies from patients' sera were capable of reducing phosphorylation of all four CD22 tyrosine motifs, while sera negative for anti-CD22 antibodies did not affect CD22 phosphorylation. Thus, a subset of SSc patients possessed autoantibodies reacting with a major inhibitory B cell response regulator, CD22. Because these antibodies can interfere CD22-mediated suppression onto B cell activation in vitro, SSc B cells produce functional autoantibodies that can enhance their own activation. This unique regulation may contribute to the autoimmune aspect of SSc.
Keywords: autoantibody, B cell activation, CD22, systemic sclerosis
Introduction
Systemic sclerosis (SSc) is a connective tissue disorder presenting fibrosis and vascular changes with an autoimmune background [1–3]. Autoimmunity in SSc is characterized by the presence of anti-nuclear antibodies, which is positive in more than 90% of patients [4]. Furthermore, the specificities of autoantibodies correlate closely with the clinical manifestations: anti-DNA topoisomerase I (topo I) antibodies and anti-RNA polymerase antibodies are associated with diffuse cutaneous SSc (dcSSc), while anti-centromere antibodies are found in limited form [1,5]. None the less, while it is likely that autoantibody production is linked closely to the development of SSc, the pathogenic relationship between systemic autoimmunity and the clinical manifestations of SSc remains unclear. Recently, several studies have revealed that autoantibodies may play pathogenic roles in SSc [6]: studies have shown that anti-topo I antibodies can bind directly to the cell surface of fibroblasts, which can induce monocyte adhesion and activation [7,8]. Furthermore, agonistic autoantibodies that recognize and stimulate platelet-derived growth factor (PDGF) receptor have been reported [9], although other studies failed to reproduce the finding [10,11].
Recent studies have demonstrated that B cells play critical roles in the manifestation of connective tissue diseases by a variety of functions other than well-established autoantibody-mediated mechanisms [12–16]. In addition to autoantibody production, SSc patients exhibit polyclonal B cell hyperactivity and hyper-γ-globulinaemia, suggesting that abnormal B cell activation exists in SSc [17]. Several studies have revealed the presence of B cell infiltration in the skin and the lung of patients with SSc [18,19]. However, the underlying mechanisms inducing B cell hyperactivation in SSc remains unclear. B cell functions are regulated by the B cell antigen receptor (BCR) and specialized cell surface co-receptors, or ‘response regulators’, which inform B cells of their microenvironment [20,21]. Among these response regulators, CD22 is a B cell-specific inhibitory receptor that contains immunoreceptor tyrosine-based inhibitory motifs [22–25]. The lack of CD22 in mouse leads to abnormal B cell activation and autoantibody production such as anti-DNA antibodies [26]. CD22 dampens B cell signalling via the tyrosine phosphorylation-dependent mechanism. Tyrosine residues within CD22 are phosphorylated upon BCR ligation [22,27], and then recruit signalling molecules such as SHP-1 and Grb2 [28,29]. Studies have demonstrated that B cell signalling mediated by the CD19/CD22 autoimmune loop is disrupted in patients with SSc as well as other autoimmune diseases [30,31]. We have reported that phosphorylation of CD22 was decreased in B cells from the tight skin (TSK/+) mouse, a genetic animal model for SSc [32]. The phenotype of B cells from hemizygous TSK/+ mice resembles that observed in CD22-deficient mice [32]. Indeed, B cells from TSK/+ mice exhibit an exaggerated [Ca2+]i response after BCR ligation due to CD22 dysfunction. These studies suggest that disruption of CD22 function may contribute to the autoimmune aspect of SSc.
In this study, we assessed the presence of autoantibodies against CD22 in the sera from TSK/+ mice and SSc patients. Furthermore, anti-CD22 antibodies decreased CD22 phosphorylation induced by BCR ligation. Thus, that anti-CD22 antibodies affect functionally signalling events from BCR raises the possibility of autocrine B cell activation that SSc B cells produce functional autoantibodies, which then regulate B cell activation.
Materials and methods
Mice and cells
Heterozygous TSK/+ mice on a C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). TSK/+ mice deficient for CD22 were generated as described previously [32]. To verify the TSK genotype, polymerase chain reaction (PCR) amplification of a partially duplicated fibrillin 1 gene was carried out using genomic DNA from each mouse [33]. C57BL/6 mice were used as controls. All mice were housed in a specific pathogen-free barrier facility. The Committee on Animal Experimentation of Kanazawa University Graduate School of Medical Science approved all studies and procedures. Human B lymphoblastoid cell lines (BJAB cells) were cultured at 37°C in RPMI-1640 medium supplemented with 5% fetal calf serum (FCS).
Patients and sera
Serum samples were obtained from 50 Japanese patients with SSc (40 females and 10 males). All patients fulfilled the criteria proposed by the American College of Rheumatology (ACR) [34]. These patients were grouped according to the classification system proposed by LeRoy et al. [35]: 20 patients (six females and 14 males) had dcSSc and 30 patients (26 females and four males) had limited cutaneous SSc (lcSSc). Detailed data were available in 43 patients, and the age of patients with SSc [mean ± standard deviation (s.e.)] was 49 ± 15. The disease duration of patients with dcSSc and lcSSc was 2·3 ± 2·2 and 8·9 ± 10 years, respectively. Raynaud's phenomenon being excluded, the disease duration of all patients was less than 5 years. None of the SSc patients were treated with corticosteroids, d-penicillamine or immunosuppressive therapy. As a disease control, 20 patients with systemic lupus erythematosus (SLE), who fulfilled the ACR criteria [36], were also included. These patients had active SLE as determined by the SLE Disease Activity Index [37]. Age- and sex-matched 17 Japanese healthy individuals were used as normal controls. All serum samples were stored at −80°C prior to use. Melon™ Gel IgG Spin Purification Kits (Pierce, Rockford, IL, USA) were used for immunoglobulin (Ig)G purification from serum samples.
Complete medical histories, physical examinations and laboratory tests were conducted for 43 of 50 patients. Skin score was measured using the scoring technique of the modified Rodnan total skin thickness score (modified Rodnan TSS) [38]. Organ system involvement was defined as described previously [1]. The protocol was approved by Kanazawa University Graduate School of Medical Science and Kanazawa University Hospital, and informed consent was obtained from all patients.
Enzyme-linked immunosorbent assay (ELISA) and Western blot analysis for anti-CD22 antibodies
Mouse or human recombinant CD22/Fc chimera protein (R&D Systems, Minneapolis, MN, USA) was used as antigen. In this recombinant protein, extracellular domain of mouse or human CD22 was fused to the C-terminal of Fc region of human IgG1. Serum anti-CD22 antibody levels were determined by ELISA. Briefly, 96-well plates [enzyme immunoassay/radioimmunoassay (EIA/RIA) plate; Coster, Cambridge, MA, USA] were coated with mouse or human recombinant CD22/Fc chimera protein (5 mg/ml) at 4°C overnight. The wells were blocked with 2% bovine serum albumin and 1% gelatin in Tris-buffered saline for 1 h at 37°C. Serum samples were preabsorbed on wells coated with human IgG1 Fc (R&D Systems) for 1 h at 20°C. Then, the serum samples were diluted 1:100 and added to duplicated wells for 90-min incubation at 20°C. After washing four times, the bound antibodies were detected with alkaline phosphatase-conjugated goat anti-mouse or anti-human IgG antibodies reacting with F(ab)′ fragments (Cappel, Durham, NC, USA), using p-nitrophenyl phosphate (Sigma-Aldrich, St Louis, MO, USA) as substrate. The optical density (OD) of the wells was determined subsequently.
Human recombinant CD22 (0·5 µg/lane) was subjected to electrophoresis on 7·5% sodium dodecyl sulphate-polyacrylamide slab gels. The proteins were electrotransferred from the gels to nitrocellulose sheets for immunoblotting analysis. The nitrocellulose sheets were cut into strips and incubated overnight with serum samples diluted 1:50 at 4°C. The strips were then incubated for 1·5 h with alkaline phosphatase-conjugated goat anti-human IgG antibody (Cappel), and colour was developed using an amplified alkaline phosphatase assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Four SSc patients positive for CD22 autoantibodies by ELISA and four healthy individuals were evaluated.
B cell activation and Western blot analysis
B cell activation and Western blot analysis were performed as described previously [39]. BJAB cells were incubated in RPMI-1640 medium containing 20% serum samples for 10 min at 37°C. Serum samples were from SSc and SLE patients who showed high anti-CD22 antibody levels or healthy individuals. Subsequently, BJAB cells were stimulated with F(ab′)2 anti-human IgM antibody (20 g/ml; Cappel), before cells were lysed in buffer containing 1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl (pH 8·0), 1 mM Na orthovanadate, 2 mM ethylenediamine tetraacetic acid (EDTA), 50 mM nafamostat mesylate (NaF) and protease inhibitors. The lysates were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and electrotransferred to nitrocellulose membranes. The membranes were incubated with phosphospecific polyclonal antibodies to four CD22 tyrosine motifs (Y762, Y807, Y822 and Y842) [27] or anti-phospho-CD19 (Tyr531) antibody (Cell Signaling Technology, Beverly, MA, USA), followed by incubation with horseradish peroxidase (HRP)-conjugated anti-rabbit antibodies (Pierce). The membrane was developed using a chemiluminescent substrate (SuperSignal; Pierce). To verify equivalent amounts of protein in each lane, the blots were probed with anti-extracellular-regulated kinase 2 (ERK2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). ERK2 expression levels were confirmed not to change during the assay. Band intensity was quantified using Quantity One software (Bio-Rad). To preabsorb anti-CD22 antibodies in the sera, samples were incubated with protein A-coated agarose beads conjugated with CD22/Fc proteins for 1 h.
Statistical analysis
Statistical analysis was performed using Mann–Whitney U-test for comparison of antibody levels, Fisher's exact probability test for comparison of frequencies and Bonferroni's test for multiple comparisons. Spearman's rank correlation coefficient was used to examine the relationship between two continuous variables. A P-value less than 0·05 was considered significant.
Results
TSK/+ mice produce anti-CD22 autoantibodies
TSK/+ mice exhibit skin fibrosis and autoimmunity, and have a partial resemblance to human SSc patients. Previously, we have reported that B cells isolated freshly from TSK/+ mice mounted impaired CD22 phosphorylation and exaggerated [Ca2+]i response and upon BCR stimulation [32], suggesting that TSK/+ B cells are activated in vivo by soluble factors, such as circulating autoantibodies. Based on this, we first assessed whether TSK/+ mice had anutoantibodies to CD22. The presence and levels of anti-CD22 autoantibodies in 10 serum samples from TSK/+ mice were assessed by ELISA using recombinant mouse CD22 as an antigen (Fig. 1b). TSK/+ mice exhibited significantly higher mean anti-CD22 antibody levels than those in wild-type C57BL/6 mice as normal controls (1·126 versus 0·129 OD, P < 0·005). Eight of 10 serum samples from TSK/+ mice exceeded the mean ± 2 s.d. of the samples from wild-type mice. Thus, TSK/+ mice produce autoantibodies reacting with CD22.
Fig. 1.

Anti-CD22 antibody levels in serum samples from tight skin (TSK/+) mice and C57BL/6 mice. Antibody levels were determined by an enzyme-linked immunosorbent assay using a recombinant mouse CD22 protein as an antigen. The short bar indicates the mean value in each group. A broken line indicates the cut-off value (mean ± 2 standard deviations of the C57BL/6 mice). *P < 0·05.
Anti-CD22 autoantibodies in human SSc patients
Next, we assessed the presence of anti-CD22 autoantibodies in human SSc patients. The presence and levels of anti-CD22 autoantibodies in serum samples from SSc patients, SLE patients and healthy individuals were assessed by ELISA (Fig. 2). Serum anti-CD22 antibody levels were significantly higher in SSc patients compared with healthy individuals (0·756 versus 0·52 OD, P < 0·05, Fig. 2). Furthermore, serum anti-CD22 antibody levels were significantly higher in SSc patients than in SLE patients (0·756 versus 0·563 OD, P < 0·05, Fig. 2). Anti-CD22 antibody levels tended to be high in dcSSc patients compared with lcSSc patients, although there was no statistical difference. When an absorbance value higher than the mean ± 2 s.d. (0·982) of the healthy individuals' samples was considered positive, anti-CD22 autoantibodies were positive in 22% of SSc patients (11 of 50, Fig. 2). By contrast, anti-CD22 antibodies were negative in all healthy individuals. While SLE patients did not exhibit a significant elevation of serum anti-CD22 antibodies, four patients were positive for anti-CD22 antibodies.
Fig. 2.
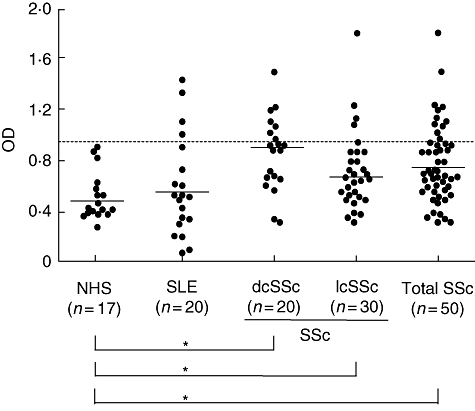
Anti-CD22 antibody levels in serum samples from patients with systemic sclerosis (SSc), those with systemic lupus erythematosus (SLE) and normal human subjects (NHS). SSc patients included 20 with diffuse cutaneous SSc and 30 with limited cutaneous SSc. Antibody levels were determined by an enzyme-linked immunosorbent assay using recombinant human CD22. The short bars indicate the mean values in each group. A broken line indicates the cut-off value (mean ± 2 standard deviations of the control samples). *P < 0·05.
The presence of anti-CD22 antibodies was confirmed by immunoblotting analysis using human recombinant CD22. Serum samples from SSc patients positive for anti-CD22 antibodies by ELISA exhibited reactivity with CD22 (∼127 kDa) by immunoblotting (lanes 1 and 2; Fig. 3). By contrast, no serum samples from healthy individuals reacted with CD22 (lane 3). Thus, the presence of anti-CD22 autoantibodies in patients with SSc was confirmed by immunoblotting analysis.
Fig. 3.
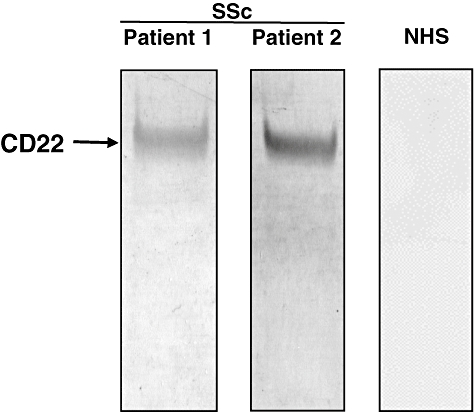
Representative immunoblotting of recombinant CD22 with sera from patients with SSc positive for anti-CD22 autoantibody by Western blotting. Lanes 1 and 2, serum samples from patients with systemic sclerosis (SSc) positive for anti-CD22 antibodies by enzyme-linked immunosorbent assay; lane 3, a serum sample from a normal human subject (NHS).
Frequency of anti-CD22 positivity and clinical correlation in SSc
We assessed whether patients positive for anti-CD22 antibodies have common clinical features when compared with those negative for anti-CD22 antibodies. SSc patients positive for anti-CD22 antibodies had higher modified Rodnan TSS than those negative for anti-CD22 antibodies (16 ± 11 versus 8·4 ± 8·6, P < 0·05, Table 1). There was no association with disease duration. Thus, anti-CD22 autoantibody levels were increased in SSc patients, especially in dcSSc patients. Patients with anti-CD22 antibodies showed higher values of SP-D, a marker for interstitial lung disease, than those without anti-CD22 antibodies [Table 1; SP-D, 137 (44–502) ng/dl versus 73·2 (29·2–311) ng/dl, P < 0·05]. When dcSSc patients were divided into those positive and negative for anti-CD22 antibodies, the presence of anti-CD22 antibodies showed a tendency to be associated with lower values of %vital capacity (VC), although the difference was not statistically significant (78·2% versus 99·2%, P = 0·06). Oesophageal involvement was also observed at higher positivity in patients positive than in those negative for anti-CD22 antibodies, although it was also not significant (P = 0·06). There was no other association with the presence or absence of disease duration or organ involvement, including heart, kidney, joint and muscle. Anti-CD22 autoantibody levels were not associated significantly with other anti-nuclear antibody specificities, including antibodies against topo I, centromere and U1RNP, although anti-topo I antibodies were found more frequently in anti-CD22-positive patients (Table 1; 55% versus 35%). When SSc patients positive for anti-topo I antibodies were divided into those positive and negative for anti-CD22 antibodies, patients positive for anti-CD22 antibodies showed higher titres of anti-topo I antibodies by approximately twofold than those negative (151·3 ± 60·2 IU/l versus 78·7 ± 43·2 IU/l, P < 0·05). Cell-surface CD22 expression on peripheral B cells from SSc patients was also assessed. Patients positive for anti-CD22 antibodies exhibited slightly lower expression of CD22 than those negative, although the difference was not significant (92·3 ± 8·4% of patients negative; P = 0·11).
Table 1.
Clinical and laboratory characteristics of patients with systemic sclerosis (SSc) positive for anti-CD22 autoantibodies
| Anti-CD22 positive (n = 11) | Anti-CD22 negative (n = 32) | |
|---|---|---|
| Sex (female/male) | 9/2 | 26/6 |
| Clinical features | ||
| Median (range) TSS | 13 (1–39)* | 5 (1–40) |
| Pitting scar | 56 | 42 |
| Nailfold bleeding | 78 | 85 |
| Raynaud's phenomenon | 77 | 87 |
| Organ involvement | ||
| Pulmonary fibrosis | 64 | 70 |
| Decreased %VC | 36 | 13 |
| Decreased %DLco | 64 | 59 |
| Heart | 0 | 21 |
| Kidney | 11 | 0 |
| Joint | 33 | 29 |
| Oesophagus | 89 | 47 |
| Muscle | 11 | 29 |
| Laboratory data | ||
| Positive anti-topoisomerase I antibody | 55 | 35 |
| Positive anti-centromere antibody | 33 | 35 |
| Positive anti-U1RNP antibody | 0 | 5·8 |
| Elevated CRP level | 0 | 13 |
| Elevated ESR | 36 | 25 |
| IgG, median (range) mg/dl | 1420 (1199–2790) | 1385 (846–2541) |
| SP-A, median (range) ng/dl | 46·7 (19·1–56·9) | 36·8 (8·3–158) |
| SP-D, median (range) ng/dl | 137 (44–502)* | 73·2 (29·2–311) |
| KL-6, median (range) U/ml | 370 (201–1156) | 269 (138–2629) |
P < 0·05. Unless noted otherwise, values are percentages. TSS, modified Rodnan total skin thickness score; VC, vital capacity; Dlco, diffusing capacity for carbon monoxide; SP-A, pulmonary surfactant protein A; SP-D, pulmonary surfactant protein D; KL-6, sialyated carbohydrate antigen KL-6; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein.
Autoantibodies to CD22 in sera from SSc patients diminish tyrosine phosphorylation of CD22
As shown above, a proportion of SSc patients produce autoantibodies to B cell surface molecule CD22, while whether these autoantibodies can affect B cell functions was unclear. Because CD22 functions are regulated by tyrosine phosphorylation on the cytoplasmic domain of CD22 [27], we examined whether anti-CD22 autoantibodies can affect their tyrosine phosphorylation using a human B cell line, BJAB cells.
CD22 has six tyrosine residues, which are phosphorylated by Lyn upon BCR ligation. We have previously generated polyclonal antibodies that recognize phosphorylation of four tyrosine residues that are demonstrated to have critical functions [27]. BJAB cells were preincubated with sera from SSc patients with anti-CD22 antibodies or from healthy individuals before they were stimulated with F(ab′)2 fragments of anti-human IgM antibody. Preincubation with serum samples from patients or controls did not alter cell surface CD22 expression significantly (data not shown). Stimulation using F(ab′)2 anti-human IgM antibody was confirmed to induce Lyn kinase activation as well as intracellular [Ca2+] increase (data not shown). Anti-IgM stimulation induced intense phosphorylation on all four tyrosine residues of CD22, as reported previously (Fig. 4a, centre lane) [27]. However, preincubation with serum samples positive for anti-CD22 antibodies reduced phosphorylation of the four tyrosines significantly when BJAB cells were stimulated identically otherwise (Fig. 4a, right lane and 4b; P < 0·05). This reduction in CD22 phosphorylation was not observed when BJAB cells were preincubated with serum samples from controls (Fig. 4b). The same assay was performed using serum samples of SSc patients from whom anti-CD22 antibodies were removed using recombinant CD22 protein. Removal of anti-CD22 antibodies in the patients' sera restored normal CD22 phosphorylation in all four tyrosines (Fig. 4b), while preincubation with control Fc did not affect CD22 phosphorylation (data not shown). That the antibodies were responsible for decreasing CD22 tyrosine phosphorylation was also confirmed by purified IgG fraction from the patients' sera, which showed identical results to whole serum samples (Fig. 4b). Therefore, anti-CD22 autoantibodies had the ability to reduce BCR-induced tyrosine phosphorylation of CD22.
Fig. 4.
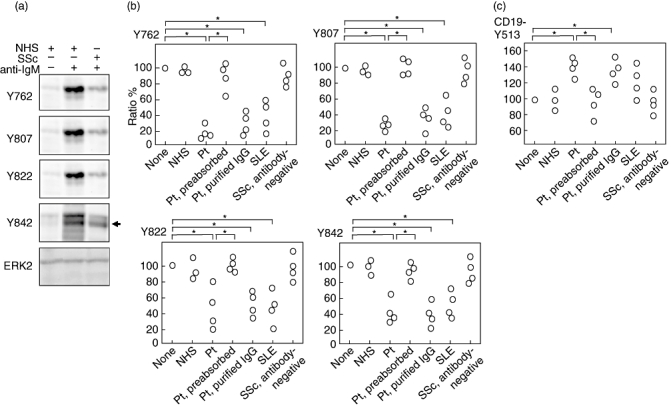
The effect of anti-CD22 autoantibodies on B cell antigen receptor (BCR)-induced tyrosine phosphorylation of CD22. (a) B lymphoblastoid cell lines (BJAB) cells were preincubated with sera from systemic sclerosis (SSc) patients positive for anti-CD22 antibodies (right lane) or from healthy individuals (left and centre lanes) before they were stimulated with F(ab′)2 fragments of anti-human immunoglobulin (Ig)M antibody for 0 min (left lane) or 3 min (centre and right lanes). Lysed cells were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent Western blotting with antibodies to phosphotyrosines of CD22. Anti-extracellular-regulated kinase 2 (ERK2) blotting was performed for a loading control. (b) The intensity of each CD22 tyrosine phosphorylation in BJAB cells was quantified as a ratio to the phosphorylation level induced by anti-IgM ligation. Phosphorylation levels induced by anti-IgM ligation without serum preincubation were defined as 100% (none). As described in (a), prior to stimulation, BJAB cells were preincubated with serum samples from normal healthy subjects (NHS), those from SSc patients positive for anti-CD22 antibodies (Pt), the same anti-CD22-positive sera from which anti-CD22 antibodies were removed using recombinant CD22 protein-conjugated beads (Pt, preabsorbed), purified IgG from the same anti-CD22-positive sera (Pt, purified IgG), serum samples from systemic lupus erythematosus (SLE) patients positive for anti-CD22 antibodies (SLE) or SSc patients negative for anti-CD22 antibodies (SSc, antibody-negative). *P < 0·05. (c) The effect of anti-CD22 autoantibodies on BCR-induced tyrosine phosphorylation of CD19. BJAB cells were processed as described in (b), followed by Western blotting by anti-phospho-CD19 antibody. The intensity of each CD19 tyrosine phosphorylation in BJAB cells was quantified as a ratio to the phosphorylation level induced by anti-IgM ligation. Phosphorylation levels induced by anti-IgM ligation without serum preincubation were defined as 100% (none). *P < 0·05.
Several SLE patients were also positive for anti-CD22 antibodies (Fig. 2). While preincubation with serum samples from SLE patients positive for anti-CD22 antibodies reduced phosphorylation of the four tyrosines (Fig. 4b, P < 0·05), sera from SSc patients negative for anti-CD22 antibodies did not affect normal CD22 phosphorylation (Fig. 4b). Therefore, the existence of serum anti-CD22 antibodies that were capable for reducing CD22 phosphorylation was not specific for SSc patients.
To clarify the consequence of decreased CD22 phosphorylation, tyrosine phosphorylation status of CD19 was assessed. Preincubation with serum samples positive for anti-CD22 antibodies increased tyrosine phosphorylation of CD19 significantly (Fig. 4c, P < 0·05). Cell-surface CD19 expression was not altered (data not shown). Therefore, anti-CD22 autoantibodies had the ability to not only reduce BCR-induced tyrosine phosphorylation of CD22, but also to enhance CD19 tyrosine phosphorylation induced by BCR cross-linking.
Discussion
The current study revealed that anti-CD22 autoantibodies were present in the sera from SSc patients (Figs 2 and 3). Anti-CD22 antibodies from SSc patients' sera were capable of reducing BCR-mediated phosphorylation of all four major CD22 tyrosine motifs (Fig. 4), suggesting that anti-CD22 antibodies can modulate the B cell response. Thus, a subset of SSc patients possessed autoantibodies reacting to a major inhibitory B cell response regulator, CD22, and these antibodies can interfere with the CD22-mediated suppression onto B cell activation in vitro, suggesting that autoantibodies produced by B cells can modulate the functions of B cells themselves.
While anti-CD22 antibodies were detected at high frequency in TSK/+ mice (80%, Fig. 1), human SSc patients possessed anti-CD22 antibodies at a much lower frequency (22%). Therefore, B cell hyperactivation in SSc patients is not explained solely by the presence of anti-CD22 antibodies. With regard to clinical correlation, modified Rodnan TSS was significantly higher in SSc patients positive than those negative for anti-CD22 autoantibodies. Thus, anti-CD22 antibodies may be associated with an accelerating fibrotic process. None the less, while anti-CD22 antibodies were not detected in healthy subjects, anti-CD22 antibodies were also detected in comparable frequency in sera from patients with SLE (Fig. 2). Anti-CD22 autoantibodies from SLE patients' sera also had the ability to reduce CD22 phosphorylation at the same levels observed in those from SSc patients' sera (Fig. 4b). This suggests that these autoantibodies are not specific to SSc and may occur in other autoimmune disorders. Therefore, anti-CD22 autoantibodies may drive B cell activation in autoimmune diseases and that, in SSc, augmented B cell activation possibly contributes co-operatively to the fibrotic process with other multiple fibrosing factors that are specific for SSc and are not dysregulated in other autoimmune diseases. Thus, anti-CD22 autoantibodies may not have the ability to induce sclerosis solely, but may serve as an exacerbating factor in a proportion of SSc patients. The mechanisms of how CD22 become a target in SSc are also unclear. As CD22 is a highly glycosylated surface protein, unglycosylated forms expressed during the inflammatory process may serve as an antigenic molecule. Alternatively, unknown cross-reactivity with microorganisms may trigger the induction. Collectively, this novel mechanism of autoantibody-mediated B cell activation may contribute to B cell-driven autoimmune mechanism in SSc and other connective tissue diseases.
Although the mechanism of how anti-CD22 antibodies decrease CD22 phosphorylation remains unsolved, there are several possibilities. CD22 expression appears on the cell surface of IgM+IgD+ mature B cells and disappears with differentiation to plasma cells. CD22 is a potent intercellular adhesion molecule recognized as a member of the ‘sialoadhesin’ or ‘SIgLec’ family [40,41]. Through binding with sialic acids, CD22 is associated constitutively with the BCR by way of cis interactions [42]. Anti-CD22 antibodies may disturb the association between BCR and CD22. Alternatively, as antibody binding to the CD22 ligand-recognition site is reported to lead to prompt internalization of the molecule [43], anti-CD22 ligation may lead to the internalization of surface CD22 into the cytoplasm, which results in inhibitory feedback onto BCR from CD22. Decreased CD22 phosphorylation by anti-CD22 antibody ligation is likely to result in B cell activation, as it also increased tyrosine phosphorylation of CD19 (Fig. 4c). CD19 is also a B cell-specific cell-surface molecule that critically regulates B cell activation thresholds [31]. Thus, the dysregulated CD19/CD22 loop may lead to amplification of autoimmunity.
Whether anti-CD22 autoantibodies have an agonistic or antagonistic role in autoimmunity is important. Because anti-CD22 antibodies diminish BCR-induced tyrosine phosphorylation of CD22, anti-CD22 antibodies can be considered theoretically to serve as an accelerator of BCR signalling. However, this is not simple, because CD22 itself has both positive and negative effects on B cell function. CD22 regulates B cell activation negatively, while CD22 regulates B cell survival positively [23,44]. Moreover, by contrast, CD22 can also regulate B cell survival negatively in the presence of CD40 signalling [45]. Thus, CD22 function is determined by the circumstance (such as the presence of other signals) as well as the nature of B cell itself (such as the developmental stage and naive/memory). Furthermore, B cells can also be classified into effector and regulatory subsets [16]. B cells can play not only a pathogenic role but also a protective role. Therefore, the effect of anti-CD22 antibodies also depends upon the B cell role in the disease. This issue is especially important because CD22 is a promising therapeutic target by monoclonal antibody therapy. Epratuzumab, an anti-CD22 humanized monoclonal antibody, is currently undergoing clinical trials in SLE and Sjögren's syndrome [46–48], where its efficacy has been proved. In mice, anti-CD22 antibody treatment is effective particularly for marginal zone B cell population [43]. Because marginal zone B cells are important both for accelerating and protecting roles for autoimmunity [14,49], the relevance of anti-CD22 autoantibodies in vivo is intriguing and needs further clarification.
We have reported herein that functional autoantibodies to CD22 are present in sera from SSc patients. Several studies have demonstrated that the CD19/CD22 autoimmune loop plays a critical role in SSc and autoimmune diseases [31]. CD19 deficiency in TSK/+ mice results in remarkable reduction of hypodermal thickening [50]. B cells from TSK/+ mice exhibited chronically activated phenotypes, such as decreased IgM expression and increased I-A expression [32]. In particular, TSK/+ B cells showed lower phosphorylation of CD22 and augmented phosphorylation of CD19, hence mounting an exaggerated [Ca2+]i response upon BCR stimulation [32]. Thus, anti-CD22 autoantibodies can interfere with the suppressive mechanism onto B cell activation provided by the CD19/CD22 regulatory loop. This suggests the hypothesis that in autocrine B cell activation, B cells produce functional autoantibodies with enhanced activation of B cells themselves, resulting in amplifying the immune response. This potential mechanism of autoantibody-mediated B cell activation may be involved in the pathogenesis of SSc. Recently, B cells have been considered to be an attractive therapeutic target in SSc [17,51]. We have reported that B cell depletion therapy by anti-mouse CD22 antibody resulted in reduction of hypodermal fibrosis in TSK/+ mice by approximately 50% [52]. In human SSc patients, B cell depletion therapy by rituximab has been reported not to affect TSS in a study [53], while two other studies reported efficacy in reducing TSS [54,55]. Therefore, while it is still controversial whether B cell depletion therapy is useful for SSc, modulating B cell activation that may contribute to the fibrotic process regulated co-operatively by other multiple factors can be a candidate for therapeutic strategies for SSc.
Acknowledgments
We thank Ms M. Matsubara for technical assistance. This work is supported by a research on intractable diseases from the Ministry of Health, Labour and Welfare of Japan.
Disclosure
We have nothing to disclose.
References
- 1.Steen VD, Powell DL, Medsger TA., Jr Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 1988;31:196–203. doi: 10.1002/art.1780310207. [DOI] [PubMed] [Google Scholar]
- 2.Yazawa N, Fujimoto M, Tamaki K. Recent advances on pathogenesis and therapies in systemic sclerosis. Clin Rev Allergy Immunol. 2007;33:107–12. doi: 10.1007/s12016-007-8009-2. [DOI] [PubMed] [Google Scholar]
- 3.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–67. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okano Y. Antinuclear antibody in systemic sclerosis (scleroderma) Rheum Dis Clin North Am. 1996;22:709–35. doi: 10.1016/s0889-857x(05)70297-0. [DOI] [PubMed] [Google Scholar]
- 5.Reimer G, Steen VD, Penning CA, Medsger TA, Jr, Tan EM. Correlates between autoantibodies to nucleolar antigens and clinical features in patients with systemic sclerosis (scleroderma) Arthritis Rheum. 1988;31:525–32. doi: 10.1002/art.1780310409. [DOI] [PubMed] [Google Scholar]
- 6.Gabrielli A, Svegliati S, Moroncini G, Avvedimento EV. Pathogenic autoantibodies in systemic sclerosis. Curr Opin Immunol. 2007;19:640. doi: 10.1016/j.coi.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Henault J, Tremblay M, Clement I, Raymond Y, Senecal JL. Direct binding of anti-DNA topoisomerase I autoantibodies to the cell surface of fibroblasts in patients with systemic sclerosis. Arthritis Rheum. 2004;50:3265–74. doi: 10.1002/art.20515. [DOI] [PubMed] [Google Scholar]
- 8.Henault J, Robitaille G, Senecal JL, Raymond Y. DNA topoisomerase I binding to fibroblasts induces monocyte adhesion and activation in the presence of anti-topoisomerase I autoantibodies from systemic sclerosis patients. Arthritis Rheum. 2006;54:963–73. doi: 10.1002/art.21646. [DOI] [PubMed] [Google Scholar]
- 9.Baroni SS, Santillo M, Bevilacqua F, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 2006;354:2667–76. doi: 10.1056/NEJMoa052955. [DOI] [PubMed] [Google Scholar]
- 10.Classen JF, Henrohn D, Rorsman F, et al. Lack of evidence of stimulatory autoantibodies to platelet-derived growth factor receptor in patients with systemic sclerosis. Arthritis Rheum. 2009;60:1137–44. doi: 10.1002/art.24381. [DOI] [PubMed] [Google Scholar]
- 11.Loizos N, Lariccia L, Weiner J, et al. Lack of detection of agonist activity by antibodies to platelet-derived growth factor receptor alpha in a subset of normal and systemic sclerosis patient sera. Arthritis Rheum. 2009;60:1145–51. doi: 10.1002/art.24365. [DOI] [PubMed] [Google Scholar]
- 12.Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–99. doi: 10.1111/j.1600-065X.2008.00646.x. [DOI] [PubMed] [Google Scholar]
- 13.St Clair EW, Tedder TF. New prospects for autoimmune disease therapy: B cells on deathwatch. Arthritis Rheum. 2006;54:1–9. doi: 10.1002/art.21525. [DOI] [PubMed] [Google Scholar]
- 14.Viau M, Zouali M. B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. 2005;114:17–26. doi: 10.1016/j.clim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. 2006;24:467–96. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 16.Lund FE. Cytokine-producing B lymphocytes – key regulators of immunity. Curr Opin Immunol. 2008;20:332–8. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto M, Sato S. B lymphocytes and systemic sclerosis. Curr Opin Rheumatol. 2005;17:746–51. doi: 10.1097/01.bor.0000179945.73518.28. [DOI] [PubMed] [Google Scholar]
- 18.Lafyatis R, O'Hara C, Feghali-Bostwick CA, Matteson E. B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum. 2007;56:3167–8. doi: 10.1002/art.22847. [DOI] [PubMed] [Google Scholar]
- 19.Whitfield ML, Finlay DR, Murray JI, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci USA. 2003;100:12319–24. doi: 10.1073/pnas.1635114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tedder TF. Introduction: response-regulators of B lymphocyte signaling thresholds provide a context for antigen receptor signal transduction. Semin Immunol. 1998;10:259–65. doi: 10.1006/smim.1998.0118. [DOI] [PubMed] [Google Scholar]
- 21.Tsubata T. Co-receptors B lymphocytes. Curr Opin Immunol. 1999;11:249–55. doi: 10.1016/s0952-7915(99)80041-7. [DOI] [PubMed] [Google Scholar]
- 22.Schulte RJ, Campbell MA, Fischer WH, Sefton BM. Tyrosine phosphorylation of CD22 during B cell activation. Science. 1992;258:1001–4. doi: 10.1126/science.1279802. [DOI] [PubMed] [Google Scholar]
- 23.Tedder TF, Poe JC, Haas KM. CD22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005;88:1–50. doi: 10.1016/S0065-2776(05)88001-0. [DOI] [PubMed] [Google Scholar]
- 24.Nitschke L, Carsetti R, Ocker B, Kohler G, Lamers MC. CD22 is a negative regulator of B-cell receptor signalling. Curr Biol. 1997;7:133–43. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 25.Walker JA, Smith KG. CD22: an inhibitory enigma. Immunology. 2008;123:314–25. doi: 10.1111/j.1365-2567.2007.02752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. 1999;189:1307–13. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimoto M, Kuwano Y, Watanabe R, et al. B cell antigen receptor and CD40 differentially regulate CD22 tyrosine phosphorylation. J Immunol. 2006;176:873–9. doi: 10.4049/jimmunol.176.2.873. [DOI] [PubMed] [Google Scholar]
- 28.Nitschke L, Tsubata T. Molecular interactions regulate BCR signal inhibition by CD22 and CD72. Trends Immunol. 2004;25:543–50. doi: 10.1016/j.it.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Doody GM, Justement LB, Delibrias CC, et al. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269:242–4. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 30.Fujimoto M, Bradney AP, Poe JC, Steeber DA, Tedder TF. Modulation of B lymphocyte antigen receptor signal transduction by a CD19/CD22 regulatory loop. Immunity. 1999;11:191–200. doi: 10.1016/s1074-7613(00)80094-1. [DOI] [PubMed] [Google Scholar]
- 31.Fujimoto M, Sato S. B cell signaling and autoimmune diseases: CD19/CD22 loop as a B cell signaling device to regulate the balance of autoimmunity. J Dermatol Sci. 2007;46:1–9. doi: 10.1016/j.jdermsci.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Asano N, Fujimoto M, Yazawa N, et al. B Lymphocyte signaling established by the CD19/CD22 loop regulates autoimmunity in the tight-skin mouse. Am J Pathol. 2004;165:641–50. doi: 10.1016/S0002-9440(10)63328-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGaha T, Saito S, Phelps RG, et al. Lack of skin fibrosis in tight skin (TSK) mice with targeted mutation in the interleukin-4R alpha and transforming growth factor-beta genes. J Invest Dermatol. 2001;116:136–43. doi: 10.1046/j.1523-1747.2001.00217.x. [DOI] [PubMed] [Google Scholar]
- 34.American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 35.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 36.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 37.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 38.Clements PJ, Lachenbruch PA, Seibold JR, et al. Skin thickness score in systemic sclerosis: an assessment of interobserver variability in 3 independent studies. J Rheumatol. 1993;20:1892–6. [PubMed] [Google Scholar]
- 39.Fujimoto M, Fujimoto Y, Poe JC, et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity. 2000;13:47–57. doi: 10.1016/s1074-7613(00)00007-8. [DOI] [PubMed] [Google Scholar]
- 40.Sgroi D, Varki A, Braesch-Andersen S, Stamenkovic I. CD22, a B cell-specific immunoglobulin superfamily member, is a sialic acid-binding lectin. J Biol Chem. 1993;268:7011–18. [PubMed] [Google Scholar]
- 41.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–66. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 42.Razi N, Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci USA. 1998;95:7469–74. doi: 10.1073/pnas.95.13.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haas KM, Sen S, Sanford IG, Miller AS, Poe JC, Tedder TF. CD22 ligand binding regulates normal and malignant B lymphocyte survival in vivo. J Immunol. 2006;177:3063–73. doi: 10.4049/jimmunol.177.5.3063. [DOI] [PubMed] [Google Scholar]
- 44.Sato S, Miller AS, Inaoki M, et al. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity. 1996;5:551–62. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 45.Poe JC, Haas KM, Uchida J, Lee Y, Fujimoto M, Tedder TF. Severely impaired B lymphocyte proliferation, survival, and induction of the c-Myc:Cullin 1 ubiquitin ligase pathway resulting from CD22 deficiency on the C57BL/6 genetic background. J Immunol. 2004;172:2100–10. doi: 10.4049/jimmunol.172.4.2100. [DOI] [PubMed] [Google Scholar]
- 46.Steinfeld SD, Youinou P. Epratuzumab (humanised anti-CD22 antibody) in autoimmune diseases. Exp Opin Biol Ther. 2006;6:943–9. doi: 10.1517/14712598.6.9.943. [DOI] [PubMed] [Google Scholar]
- 47.Steinfeld SD, Tant L, Burmester GR, et al. Epratuzumab (humanised anti-CD22 antibody) in primary Sjogren's syndrome: an open-label phase I/II study. Arthritis Res Ther. 2006;8 doi: 10.1186/ar2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding C, Foote S, Jones G. B-cell-targeted therapy for systemic lupus erythematosus: an update. BioDrugs. 2008;22:239–49. doi: 10.2165/00063030-200822040-00003. [DOI] [PubMed] [Google Scholar]
- 49.Lopes-Carvalho T, Kearney JF. Development and selection of marginal zone B cells. Immunol Rev. 2004;197:192–205. doi: 10.1111/j.0105-2896.2004.0112.x. [DOI] [PubMed] [Google Scholar]
- 50.Saito E, Fujimoto M, Hasegawa M, et al. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J Clin Invest. 2002;109:1453–62. doi: 10.1172/JCI15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kraaij MD, van Laar JM. The role of B cells in systemic sclerosis. Biologics. 2008;2:389–95. [PMC free article] [PubMed] [Google Scholar]
- 52.Hasegawa M, Hamaguchi Y, Yanaba K, et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol. 2006;169:954–66. doi: 10.2353/ajpath.2006.060205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lafyatis R, Kissin E, York M, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009;60:578–83. doi: 10.1002/art.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daoussis D, Liossis SN, Tsamandas AC, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatol (Oxf) 2009 doi: 10.1093/rheumatology/kep093. doi: 10.1093/rheumatology/kep093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith VP, Van Praet JT, Vandooren BR, et al. Rituximab in diffuse cutaneous systemic sclerosis: an open-label clinical and histopathological study. Ann Rheum Dis. 2008 doi: 10.1136/ard.2008.095463. doi: 10.1136/ard.2008.095463. [DOI] [PubMed] [Google Scholar]