

Abstract
Several negative regulatory mechanisms control Toll-like receptor (TLR)-mediated inflammatory responses and restore immune system balance, including the zinc-finger protein A20, a negative regulator of TLR signalling that inhibits nuclear factor kappa B (NF-κB) activity. In the present study, we investigated TLR-5-mediated A20 expression and its role in intestinal epithelial cells (IECs) during inflammation. HCT-15 and HT-29 cells were stimulated with flagellin, then the expressions of A20, interleukin-1 receptor-associated kinase (IRAK-M) and Tollip were evaluated using RNase protection assay. Furthermore, experimental colitis was induced in tlr4-deficient CH3/HeJ mice by administration of dextran sodium sulphate (DSS), then flagellin was injected anally, and the colonic expression of A20 was examined by real-time polymerase chain reaction (PCR) and immunohistochemistry. To confirm flagellin-induced expression of A20, we employed an organ culture system. The role of A20 in flagellin-induced tolerance induction was evaluated in vitro, using a gene knock-down method targeting A20. A20 expression increased rapidly and peaked at 1 h after flagellin stimulation in cultured IECs, then declined gradually to the basal level. In vivo, anal injection of flagellin induced epithelial expression of A20 in injured colonic tissue, whereas flagellin did not cause a significant increase in A20 expression in non-injured normal tissue, which was also confirmed in vitro using the organ culture system. Gene knock-down using A20 siRNA did not influence tolerance induced by restimulation with flagellin. A20 is an early response negative regulator of TLR-5 signalling in IECs that functions during intestinal inflammation. Our results provide new insights into the negative feedback regulation of TLR-5 signalling that maintains the innate immune system in the gut.
Keywords: A20, intestinal inflammation, negative regulator, NF-κB, TLR
Introduction
The innate immune system recognizes conserved pathogen-associated molecular patterns (PAMPs) via a limited number of pattern recognition receptors (PRRs) [1,2]. The family of Toll-like receptors (TLRs) is one of the important PRR groups whose members play essential roles in innate immunity in various organs [3–5]. The monolayer of epithelial cells exposed to the gut luminal environment is endowed with the capacity for first-line defence against microbial pathogens [6–9]. TLR signalling in epithelial cells induces innate immune responses immediately, which preserves the gut under physiological and pathological conditions [10–13].
TLR-mediated nuclear factor kappa B (NF-κB) activation induces inflammatory responses, and inappropriate inflammation leads to local tissue damage or development of systemic diseases [14–17]. To maintain a balance between activation and inhibition of the innate immune system, a variety of negative regulatory mechanisms control TLR-mediated cellular signalling [18,19]. These range from extracellular decoy receptors to membrane-bound suppressors, which are intracellular negative regulators that down-regulate the transcription and translation of TLR-induced genes, and degradation of these proteins [20–24]. Some of these are present constitutively to control TLR activation at a physiological level, whereas others are up-regulated by TLR signalling during inflammation to attenuate TLR response in a negative feedback loop [25–27].
The zinc-finger protein A20, a ubiquitin-modifying enzyme, is an inducible and broadly expressed cytoplasmic protein that inhibits tumour necrosis factor (TNF)-α-induced NF-κB activity [28]. A20-deficient mice develop severe inflammation and are hyper-responsive to lipopolysaccharide (LPS) [29–31]. Because overexpression of A20 inhibits TLR-induced NF-κB activation and attenuates production of inflammatory cytokines in vitro, it has been suggested that A20 is one of the intracellular negative regulators for downstream signalling of TNF receptor-associated factor (TRAF) 6 and NF-κB translocation in TLR-signalling pathways [32]. However, although TLR-dependent activation of NF-κB may play a vital role in maintaining epithelial homeostasis, as well as regulating infection and inflammation in the gut, little is known regarding the expression of A20 in intestinal epithelial cells (IECs) during inflammation.
In the present study, we investigated TLR-mediated A20 expression and its role in IECs, and compared the results to those of other TLR negative regulators, including interleukin (IL)-1-receptor-associated kinase (IRAK)-M and Toll-interacting protein (Tollip) [33,34]. Both in vitro and in vivo findings showed that A20 is rapidly induced by stimulation with TLR ligands, with flagellin a particularly potent stimulator of A20 expression in IECs. These are the first known results to show that A20 is an early responsive negative regulator of TLR-5 signalling in IECs, which may contribute to the first line of defence during inflammation.
Materials and methods
Reagents
The reagents and antibodies used in our experiments are as follows: purified flagellin from Salmonella typhimurium (Invivogen, California, CA, USA), purified LPS Escherichia coli LPS (Sigma, St Louis, MO, USA), Lipofectamine 2000 (Invitrogen, California, CA, USA), pNF-kB-Luc (Stratagene, California, CA, USA), pRL-TK (Promega, Madison, WI, USA), human IL-8 enzyme immune assay (EIA) (Biosource, California, CA, USA), mouse CXCL2 EIA (R&D Systems, Minneapolis, MN, USA), dextran sodium sulphate (DSS; 5 kDa; Wako, Osaka, Japan), anti-A20 antibody (Santa Cruz, California, CA, USA), phycoerythrin (PE)-conjugated anti-E-cadherin antibody (R&D Systems), anti-β-actin antibody (Sigma) and [32]P CTP (Amersham Biosciences, Buckinghamshire, UK).
Cell culture
Two human colorectal cancer cell lines, HCT-15 and HT-29, and a mouse colonic cancer cell line, Colon-26, were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in RPMI-1640 media (Invitrogen), supplemented with 10% fetal bovine serum (FBS) (ICN Biomedicals, Aurora, OH, USA) and penicillin–streptomycin–amphotericin B (Invitrogen), and maintained at 37°C in 5% CO2 in a humidified incubator.
Primary cultures of mouse colonic epithelial cells
To evaluate flagellin-induced CXCL2 and A20 expressions in colonic epithelial cells a primary culture system was established, as described previously [35]. Briefly, the distal colonic parts dissected from BALB/c or C3H/HeJ mice were opened and washed three times in Hanks's balanced salt solution (HBSS) (Gibco, BRL, California, CA, USA) containing 5% FBS in a sterile condition. Five-millimetre tissue segments were incubated for 90 min at 37°C in 10 ml of RPMI-1640 containing antibiotics and 10 mg of Dispase (Gibco) in 50-ml centrifuge tubes with gentle rotation. Each digested sample was then passed through a nylon mesh sieve to remove mucus and undigested tissue fragments. Isolated colonic epithelial cells were cultured in RPMI-1640 medium, and supplemented with 10% FBS and antibiotics as noted above.
Enzyme immune assay (EIA)
Human IL-8 and mouse CXCL2 contents from HCT-15, HT-29, Colon-26 and primary cell culture supernatants treated with flagellin or LPS were measured using IL-8 and CXCL2 EIA kits, following the manufacturer's protocols. Briefly, appropriate sample amounts were transferred by pipette into the appropriate wells of anti-human IL-8- or mouse CXCL2-coated microtitre strips, followed by addition of a second biotinylated monoclonal antibody, then incubation was performed at room temperature for 90 min. After removing the excess second antibody by washing, the samples were incubated with streptavidin–peroxidase, after which a substrate solution was added to produce colour that was directly proportional to the concentration of human IL-8 or mouse CXCL2 present in the sample. Quantitative results were obtained from a standard curve produced from the experimental findings.
Transient transfection and luciferase assay
HCT-15 and Colon-26 cells were grown in 24-well plates (5 × 104 cells/well). After reaching 50% confluence, they were transfected transiently with pNF-κB-Luc (200 ng/well) and pRL-TK-Renilla-Luc (20 ng/well) using Lipofectamine 2000 reagent (2·5 µl/well). At 24 h after transfection, the medium was replaced with fresh medium and the cells were stimulated with flagellin or LPS for various periods, after which the cell lysates were used for measurement of luciferase activity with a dual luciferase reporter assay system kit (Promega).
RNA extraction and real time-polymerase chain reaction (PCR)
Total RNA was extracted from each sample using Isogen (Nippon Gene, Toyama, Japan), then equal amounts of RNA were reverse-transcribed into cDNA using a quantitative PCR (qPCR) cDNA kit (Stratagene). All primers (Table 1) used were flanked by intron–exon junctions using the NCBI blast tool and Primer3 software. Quantitative real-time PCR was performed using an ABI PRISM 7700 sequence detection system with SYBR green PCR master mix (Applied Biosystems, California, CA, USA), according to the manufacturer's instructions. The levels of A20, Tollip, IRAK-M and TNF-α mRNA were normalized to that of β-actin using sequence detector software (Applied Biosystems).
Table 1.
Primer sequences.
| Gene (Accession no.) | Sequences (5′–3′) |
|---|---|
| (Human-specific) | |
| For RT–PCR | |
| TLR-4 (NM_138554) | |
| Forward: | TGAGCAGTCGTGCTGGTATC |
| Reverse: | CAGGGCTTTTCTGAGTCGTC |
| TLR-5 (NM_003268) | |
| Forward: | TCAAACCCCTTCAGAGAATCCC |
| Reverse: | TTGGAGTTGAGGCTTAGTCCCC |
| β-actin (NM_001101) | |
| Forward: | CAAGAGATGGCCACGGCTGCT |
| Reverse: | TCCTTCTGCATCCTGTCGGCA |
| For RPA probe making | |
| A20 (NM_006290) | |
| Forward: | ATGCACCGATACACACTGGA |
| Reverse: | GCGTGTGTCTGTTTCCTTGA |
| IRAK-M (NM_007199) | |
| Forward: | CTCGGAATTTCTCTGCCAAG |
| Reverse: | AACCTCAGACTGGCTGCATT |
| Tollip (NM_019009) | |
| Forward: | CTCGGAATTTCTCTGCCAAG |
| Reverse: | AACCTCAGACTGGCTGCATT |
| GAPDH (NM_002046) | |
| Forward: | AGGTCGGAGTCAACGGATTT |
| Reverse: | TTCAGCTCAGGGARGACCTT |
| For real-time PCR | |
| A20 | |
| Forward: | CTGCCCAGGAATGCTACAGATAC |
| Reverse: | GTGGAACAGCTCGGATTTCAG |
| IRAK-M | |
| Forward: | AGCTGCGGGATCTCCTTAGAG |
| Reverse: | ACCGGCCTGCCAAACAG |
| Tollip | |
| Forward: | CAGGCGTGGACTCTTTCTATCTC |
| Reverse: | GACTCCGGGATGGTGATGTG |
| GAPDH | |
| Forward: | CCACATCGCTCAGACACCAT |
| Reverse: | TGACCAGGCGCCCAATA |
| (Mouse-specific) | |
| A20 (NM_009397) | |
| Forward: | CTCAGAACCAGAGATTCCATGAAG |
| Reverse: | CCTGTGTAGTTCGAGGCATGTC |
| IRAK-M (NM_028679) | |
| Forward: | GCCAAAGCCATCCAATACTTG |
| Reverse: | TGGGTTGGAGCTGGTCATC |
| Tollip (NM_023764) | |
| Forward: | TTGGCTATGTGCCCATTGC |
| Reverse: | AGCTTTGAGGTCCTCTTCATTACAG |
| TNF-α (NM_013693) | |
| Forward: | AGACCCTCACACTCAGATCATCTTC |
| Reverse: | TCCTCCACTTGGTGGTTTGC |
| β-actin (NM_007393) | |
| Forward: | CGTGAAAAGATGACCCAGATCA |
| Reverse: | CACAGCCTGGATGGCTACGTA |
TLR, Toll-like receptor; GAPDH, glyceraldehye-3-phosphate dehydrogenase; TNF, tumour necrosis factor; IRAK, interleukin-1 receptor-associated kinase; RT–PCR, reverse transcription–polymerase chain reaction; RPA: RNase protection assay; Tollip, Toll-interacting protein.
RNase protection assay
The time–course changes of A20, IRAK-M and Tollip expressions at the mRNA level were detected using an RNase protection assay (RPA) following the RPA III kit (Ambion, Austin, TX, USA) instructions. Probes for RPA were prepared by reverse transcription (RT)–PCR from mRNA samples using gene specific primers (Table 1), with the amplicons corresponding to A20 (282 b), IRAK-M (311 b), Tollip (413 b) and glyceraldehye-3-phosphate dehydrogenase (GAPDH) (661 b) used as probes. The resulting amplicons were inserted into a pCRII TA cloning vector (Invitrogen), which carried an SP6 promoter. After confirming the sequences (Big Dye; Applied Biosystems), only reverse-orientated constructs were used for RNA polymerase-mediated in vitro transcription to generate anti-sense [32]P-labelled respective riboprobes using the protocol included with the SP6 promoter specific MAXIscript kit (Ambion). Once probe-making was conducted, approximately 25 µg of total RNA was allowed to hybridize with [32]P-labelled anti-sense riboprobes. After RNase digestion, the protected fragments were resolved by polyacrylamide gel electrophoresis (PAGE) using 5% Tris borate ethylenediamine tetraacetic acid (TBE) urea precast gels (Invitrogen). Following PAGE, the gels were transferred to filter paper and vacuum-dried, then exposed signals were detected with a bioimage analyser system (BAS 2000 II; Fujix, Tokyo, Japan). All signals were standardized to the intensity of GAPDH.
A20 expression in colonic tissues from BALB/c mice with DSS-induced inflammation
Because our in vitro results showed clearly that TLR ligands induce A20 expression in colonic epithelial cells, we examined A20 expression further in colonic tissues during DSS-induced inflammation. Seven-week-old male-specific pathogen-free BALB/c mice (Charles River, Tokyo, Japan) were housed according to our institutional guidelines with the approval of the ethics committee of Shimane Medical University. To produce a DSS colitis model, mice were fed 2·5% DSS in drinking water. The mice were euthanized with an overdose of diethyl ether at different time-points during DSS administration, then the colons were dissected out and rinsed in cold PBS, and used for the detection of A20 expression by real-time PCR.
Effect of intraperitoneal injection of TLR-5 blocking peptide on colonic A20 expression in BALB/c mice with DSS-induced inflammation
To evaluate whether endogenous luminal flagellin contributes to A20 expression in colonic tissues during DSS-induced colitis, a TLR-5 blocking peptide was used in an exogenous manner. We injected intraperitoneally 100 µl of a 10–100-fold molar excess amount of the TLR-5 blocking peptide (10 µg/mouse) into BALB/c mice at the onset of DSS-induced inflammation. At 5 or 7 days after beginning DSS administration, the colonic tissues were dissected and A20 expression was examined by real-time PCR.
Effect of anal injection of flagellin on colonic A20 expression in CH3/HeJ mice with DSS-induced inflammation
TLR-5 is localized mainly in the basolateral membrane of IECs, and in normal conditions flagellin cannot induce TLR-5-dependent signalling. Therefore, we used the DSS-induced experimental colitis model to evaluate the effect of flagellin on A20 expression in IECs, as described previously [36]. To avoid the effect of LPS on A20 expression, the tlr4-deficient mouse strain CH3/HeJ was used [37]. Seven-week-old male specific pathogen-free tlr4-deficient CH3/HeJ mice (Nihon Clea, Tokyo, Japan) were studied after receiving approval from the ethics committee for animal experimentation of Shimane University. To produce DSS-induced colonic injuries, mice were fed 2·5% DSS in drinking water for 3 days, while the control group received only normal drinking water throughout the experimental period. Following DSS administration, flagellin (500 ng/mouse) was injected anally, then distal colonic parts were dissected for real-time PCR and immunohistochemistry examinations.
Immunohistochemistry
Six-mm-thick frozen sections of distal colons from CH3/HeJ mice were fixed in cold acetone for 10 min. After washing, endogenous peroxidase activity was blocked with 3% H2O2 in water for 10 min at room temperature, followed by incubation with normal blocking serum for 30 min. Subsequently, the sections were incubated for 2 h at room temperature with anti-A20 primary antibody at a 1:100 dilution and then staining was processed using a commercial immunoperoxidase staining kit (Vectastain Elite ABC Kit; Vector Laboratories, California, CA, USA), according to the manufacturer's instructions. Sections were counterstained with haematoxylin and viewed under a phase contrast light microscope.
Organ cultures of mice distal colon specimens
To confirm TLR-mediated expression of A20 in the mice colons, we initially employed an organ culture system using colonic tissues dissected from C3H/HeJ mice. The distal colon part (0·5 × 0·5 cm) was cut open longitudinally and washed three times in PBS at a sterile condition, then cultured in RPMI-1640 media with antibiotics. The cultured tissues were stimulated with flagellin (100 ng/ml) for 1 h, after which the gene expressions of A20 and TNF-α were examined using real-time PCR.
Flow cytometry
Epithelial cells (2 × 105 cells/well) isolated from C3H/HeJ mice were cultured in 24-well plates with or without flagellin (100 ng/ml) for 8 h, then A20 expression was examined by flow cytometry, in which cell surface staining for E-cadherin as a specific maker of epithelial cells and permeabilized cell staining for A20 were evaluated. Cell permeabilization was performed using the IntraPrep Permeabilization Reagent (Beckman Coulter, Tokyo, Japan). After staining, cells were analysed using an EPICS XL (Beckman Coulter), in which 10 000 events (E-cadherin-positive cells) were counted for each condition and analysed using EXPO32™ software. Isotype controls were utilized for all the samples.
In vitro tolerance induction and RNA interference
Initially, we evaluated if prestimulation with flagellin induces tolerance in HCT-15 cells. Cells were cultured in 24-well plates (5 × 104 cells/well) with or without flagellin for 16 h, then restimulated with flagellin for 6 h. Tolerance induction was evaluated by measurement of IL-8 contents in the supernatants of culture media. Next, to elucidate the role of A20 in tolerance induction, HCT-15 cells were cultured and then custom siRNAs (Qiagen, California, CA, USA) targeting A20 were transfected (33 nM/well), according to the manufacturer's protocol. After assessing the efficiency of the transfected siRNAs by real-time PCR, tolerance experiments were performed with the A20 siRNA transfected HCT-15 cells, using the same technique as above.
Statistical analysis
All data are expressed as the mean ± standard error of the mean (s.e.m.). Values were analysed using Student's t-test and Spearman's rank correlation with Stat-View version 4·0 software (Abacus Concepts, Inc., NJ, USA). For comparisons of multiple values, analysis of variance (anova) was used. P-values less than 0·05 were considered significant.
Results
Flagellin and LPS stimulate production of IL-8 in cultured intestinal epithelial cells
The gene expressions of TLR-4 and TLR-5 in human cultured epithelial cells (HCT-15, HT-29) were confirmed by RT–PCR (Fig. 1a). Both flagellin and LPS induced IL-8 production significantly by the HCT-15 cells in dose- and time-dependent manners (Fig. 1b and c), which was related to the transcriptional activity of NF-κB (Fig. 1d). Furthermore, IL-8 and NF-κB levels in response to flagellin were markedly higher than in response to LPS. On the other hand, HT-29 cells were not significantly responsive to the stimulation with LPS (Fig. 1c).
Fig. 1.
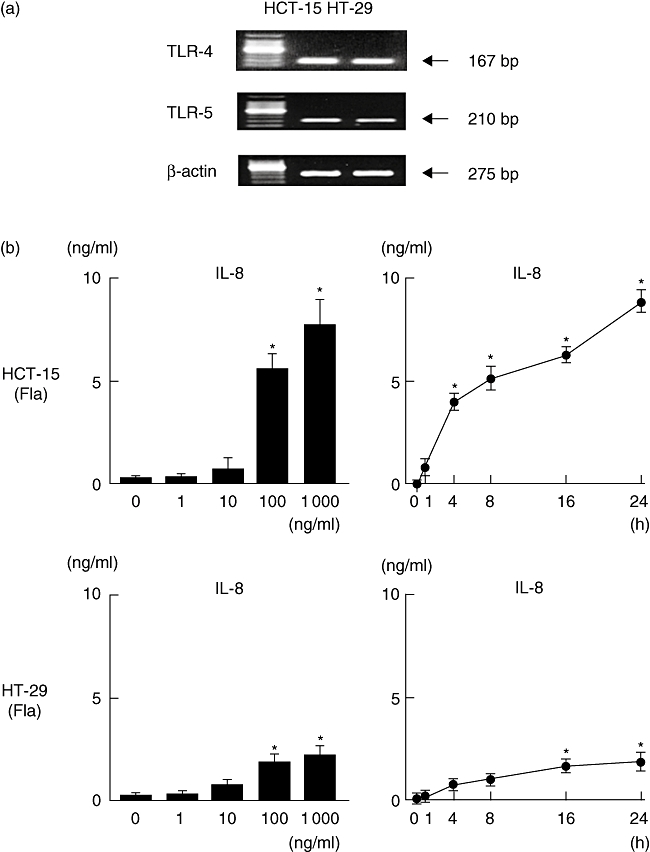
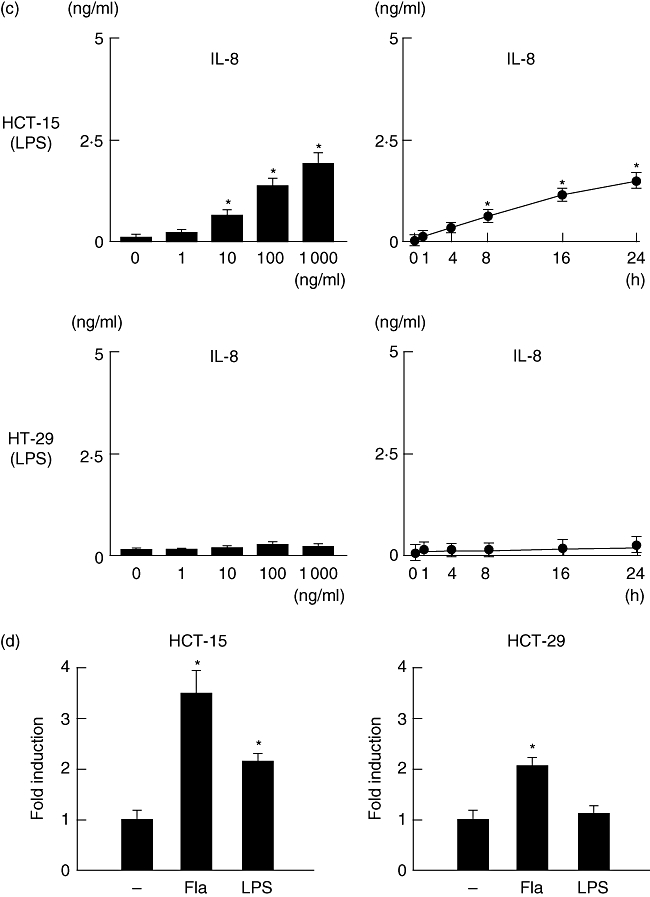
Toll-like receptor (TLR) ligand-induced interleukin (IL)-8 production and nuclear factor kappa B (NF-κB) activity in HCT-15 and HT-29 cells. (a) The expressions of TLR-4 and TLR-5 in HCT-15 and HT-29 cells were examined by reverse transcription–polymerase chain reaction (RT–PCR). (b,c) Dose- and time-dependent production of IL-8 in flagellin (Fla)- and lipopolysaccharide (LPS)-treated cells. For the dose-dependent experiments, cells were incubated with flagellin for 24 h. For the time-dependent experiments, 100 ng/ml of flagellin or LPS was used. IL-8 content in culture supernatants was measured by enzyme immune assay (EIA). (d) TLR ligand-induced NF-κB activity (flagellin; 100 ng/ml, LPS; 100 ng/ml). The transcriptional activity of NF-κB was evaluated by a luciferase assay. Error bars indicate the standard error of mean values obtained from four independent experiments. *P < 0·01 versus non-stimulated cells.
A20 expression rapidly increased after stimulation with flagellin and LPS in cultured intestinal epithelial cells
Because flagellin and LPS up-regulated IL-8 production in cultured epithelial cells, we evaluated the changes in expression by flagellin and LPS of the intracellular TLR negative regulators A20, IRAK-M and Tollip at various time-points. The expression of A20 was increased rapidly and peaked at 1 h after flagellin stimulation, although it declined gradually with the time–course change. Conversely, the expressions of IRAK-M and Tollip increased gradually, peaked after 8 h, and remained elevated for up to 16 h after stimulation with flagellin (Fig. 2a and b). The experiment using LPS as a ligand showed similar time–course changes of negative regulator expression in HCT-15 cells; however, the expression levels were relatively lower than those seen in flagellin-treated cells (Fig. 2c and d).
Fig. 2.
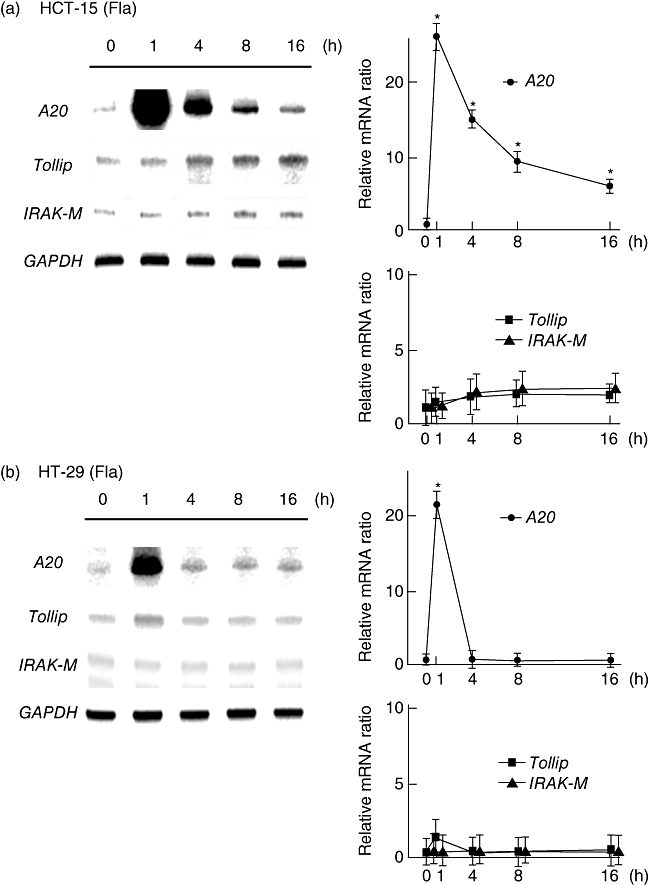
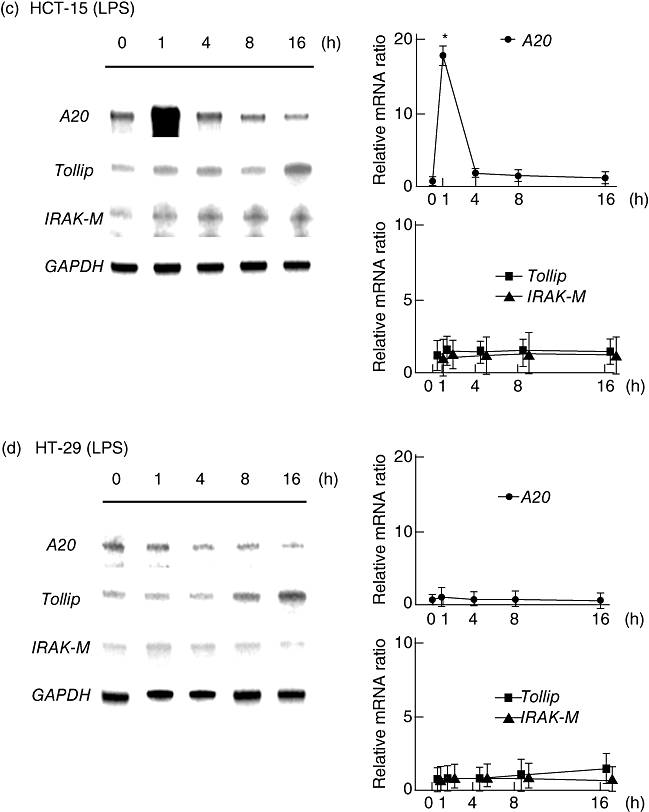
Time-dependent mRNA expression of negative regulators in HCT-15 and HT-29 cells after stimulation with Toll-like receptor (TLR) ligands. Total RNA was extracted from each sample and the expressions of negative regulators were examined by RNase protection assays. For semi-quantification of mRNA, all signals were standardized to the intensity of glyceraldehye-3-phosphate dehydrogenase (GAPDH). (a,b) Flagellin (Fla; 100 ng/ml). (c,d) Lipopolysaccharide (LPS) (100 ng/ml). Error bars indicate the standard error of mean values obtained from four independent experiments. *P < 0·01 versus non-stimulated cells.
Flagellin stimulates expression of CXCL2 and A20 in mouse cultured intestinal epithelial cells
Before carrying out an in vivo examination using the present experimental colitis model, we evaluated the flagellin-induced expressions of CXCL2 and A20 in mouse primary cultured epithelial cells (BALB/c) and Colon-26 cells. Flagellin induced CXCL2 production significantly by both cell lines in a time-dependent manner (Fig. 3a and b), which was confirmed by the transcriptional activity of NF-κB (Fig. 3c, Colon-26 cells). The expression of A20 was increased rapidly and peaked at 1 h after flagellin stimulation, and the time–course change of A20 expression was similar to that in the previous experiments with human cell lines (Fig. 3a and b).
Fig. 3.
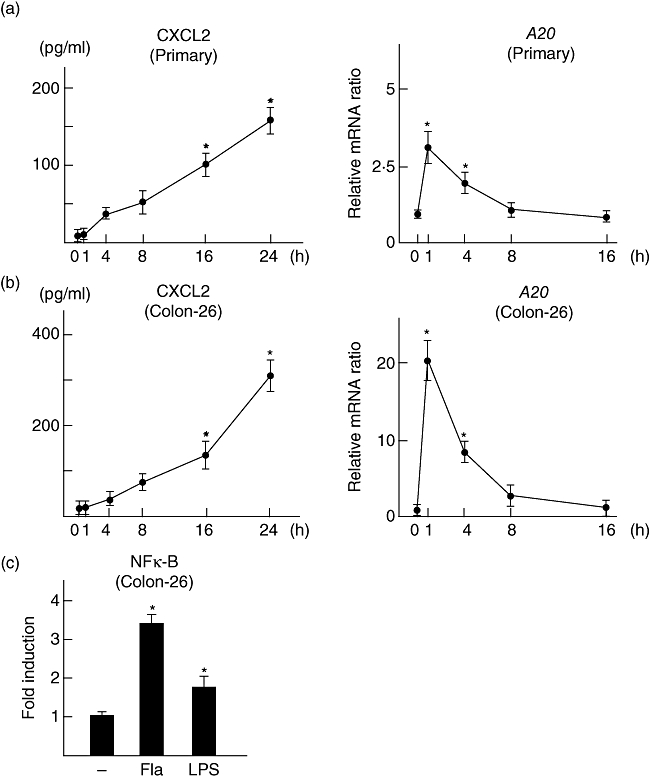
Flagellin-induced expression of CXCL2 and A20 in mouse epithelial cells. (a) Primary cultured cells isolated from BALB/c mice. (b) Colon-26 cells. For time-dependent experiments, 100 ng/ml of flagellin was used. CXCL2 contents in culture supernatants and A20 expression in cultured cells were examined by enzyme immune assay (EIA) and reverse transcription–polymerase chain reaction (RT–PCR), respectively. (c) Flagellin-induced nuclear factor kappa B (NF-κB) activity (flagellin; 100 ng/ml). The transcriptional activity of NF-κB was evaluated by a luciferase assay. Error bars indicate the standard error of mean values obtained from four independent experiments. *P < 0·01 versus non-stimulated cells.
Up-regulation of colonic A20 expression via TLR-5-mediated pathway in BALB/c mice with DSS-induced inflammation
Because flagellin induced significant A20 expression in colonic epithelial cells, we examined A20 expression further in colonic tissues during DSS-induced inflammation (BALB/c mice). As shown in Fig. 4a, A20 expression was increased significantly from days 5–9 after beginning DSS administration (Fig. 4a). Furthermore, intraperitoneal injection of the TLR-5 blocking peptide inhibited A20 expression significantly on days 5 and 7 (Fig. 4b). These findings suggest that TLR-5-mediated signalling is a potent pathway for colonic A20 expression during inflammation.
Fig. 4.
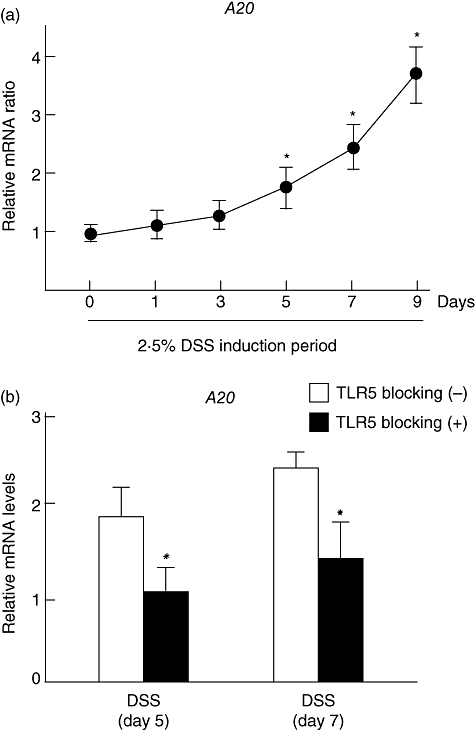
A20 expression in colonic tissues in BALB/c mice with DSS-induced inflammation. (a) Time–course changes of A20 expression in inflamed colonic tissues during dextran sodium sulphate (DSS) drinking period. Mice were given 2·5% DSS in drinking water. At each experimental point, the colons were dissected for detection of A20 mRNA by real-time polymerase chain reaction (PCR). Error bars indicate the standard error of mean values obtained independently from five mice. *P < 0·01 versus day 0. (b) Effect of Toll-like receptor (TLR)-5 blocking peptide on colonic A20 expression during DSS-induced inflammation. The TLR-5 blocking peptide (10 µg/mouse) was injected intraperitoneally into BALB/c mice at the onset of DSS induction. At 5 or 7 days after beginning DSS administration, the colonic tissues were dissected and A20 expression was examined by real-time PCR. Error bars indicate the standard error of mean values obtained independently from five mice. *P < 0·01 versus without TLR-5 blocking peptide.
Intraperitoneal injection of flagellin rapidly induces colonic A20 expression in CH3/HeJ mice pretreated with DSS
To clarify further the role of TLR-5 in colonic A20 expression, we evaluated whether intra-anal injection of flagellin induces A20 expression in CH3/HeJ mice colons. Initially, colonic injury was induced in mice by oral administration of 2·5% DSS for 3 days. Following DSS administration, flagellin was injected anally, then the expressions of A20 and TNF-α were evaluated by real-time PCR. Those were increased significantly in DSS-induced injured colonic tissues at 1 h after anal injection of flagellin, whereas there was no increase observed in non-injured colonic tissues (Fig. 5a). In addition, immunohistochemistry findings clearly showed an abundant expression of A20 in colonic epithelial cells at 8 h after injection of flagellin (Fig. 5b, i–iii). Although infiltrating mononuclear cells also expressed A20, the number of those cells was relatively low compared to that of epithelial cells (Fig. 5b, iv). These results suggest that luminal flagellin induces acute inflammation in colons pretreated with DSS and that A20 is expressed rapidly in response to flagellin stimulation in colonic epithelial cells.
Fig. 5.
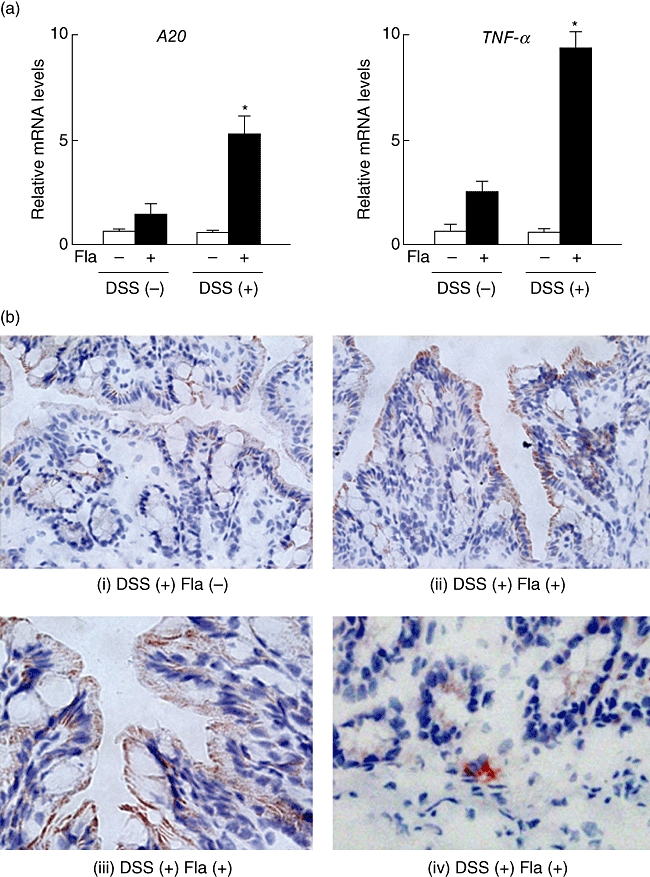
Flagellin-induced expressions of A20 and tumour necrosis factor (TNF)-α in CH3/HeJ mice colons with or without DSS colitis. Experimental colitis models were established by administering a 2·5% DSS solution as drinking water for 3 days, while the control group received only normal drinking water. Following DSS administration, flagellin (Fla; 500 ng/mouse) was injected anally, then distal colonic parts were dissected from the mice. (a) Results of real-time PCR showing gene expressions of A20 and TNF-α in colonic tissues with or without DSS-induced colonic injury at 1 h after anal injection of flagellin. Error bars indicate the standard error of mean values obtained independently from six mice. *P < 0·01 versus DSS (−) flagellin (+). (b) Representative immunohistochemistry results showing the expression of A20 in colonic epithelial cells (i; Fla −, ii, iii; Fla +) and infiltrating mononuclear cells (iv; Fla +) obtained from DSS-treated mice at 8 h after injection of flagellin.
Flagellin induces A20 expression in colonic tissues and epithelial cells isolated from C3H/HeJ mice
To assess flagellin-induced A20 expression in mouse colons, we conducted additional in vitro experiments. Initially, colonic tissues isolated from C3H/HeJ mice colons with or without DSS-treatment were cultured and treated with flagellin. A20 levels rapidly increased and peaked at 1 h after flagellin stimulation in injured colonic tissues caused by DSS administration, whereas there was no significant increase of A20 expression in non-injured normal tissue (Fig. 6a). The time–course changes of flagellin-induced TNF-α expression showed a pattern similar to that of A20 (Fig. 6a). Next, to confirm A20 expression in colonic epithelial cells, we isolated epithelial cells from CH3/HeJ mice colons and cultured them with flagellin for 8 h. The results of flow cytometry showed a significant expression of A20 in those cells (Fig. 6b).
Fig. 6.
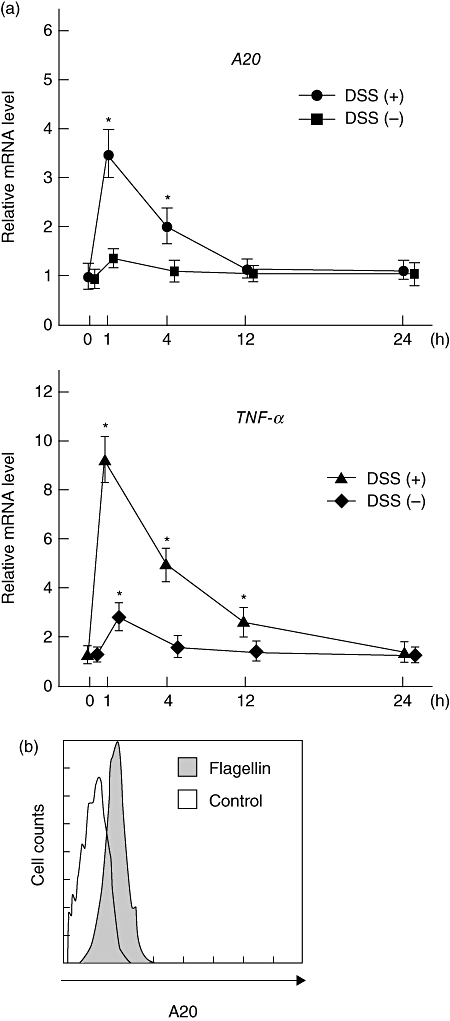
(a) Expressions of A20 and tumour necrosis factor (TNF)-α in cultured colonic tissues after stimulation with flagellin. Distal colon parts were isolated from CH3/HeJ mice with or without dextran sodium sulphate (DSS) administration, then stimulated with flagellin (100 ng/ml) for 1 h, after which the gene expressions of A20 and TNF-α were investigated using real-time polymerase chain reaction (PCR). Error bars indicate the standard error of mean values obtained independently from six mice. *P < 0·01 versus DSS (−) and flagellin (+). (b) Representative results of flow cytometry showing the expression of A20 in mice colonic epithelial cells. Colonic epithelial cells were isolated from CH3/HeJ mice as described in Materials and methods section, then stimulated with flagellin (100 ng/ml) for 8 h. The fluorescence level of cytoplasmic A20 in E-cadherin-positive cells was evaluated using flow cytometry.
A20 does not influence tolerance induced by restimulation with flagellin
First, we evaluated if prestimulation with flagellin induces tolerance in HCT-15 cells. Cells were cultured with or without flagellin for 16 h, then simulated with flagellin for 8 h. The production of IL-8 did not increase and A20 expression was not up-regulated in the prestimulated cells (Fig. 7a). Next, to clarify the role of A20 on tolerance induction by flagellin, we used a gene knock-down method with siRNA targeting A20. The siRNA used in this study showed high gene knock-down efficacy, as flagellin-induced A20 gene expression was decreased by 90%, while flagellin-induced IL-8 production in siRNA-treated cells was significantly higher than that in the control (Fig. 7b). To evaluate the role of A20 in flagellin-induced tolerance induction, cells were transfected with A20 siRNA or control siRNA, then cultured with or without flagellin for 16 h, after which they were simulated with flagellin for 8 h. Although IL-8 production in A20 siRNA-treated cells was higher than that in the control siRNA-treated cells, gene knock-down of A20 did not influence tolerance induction by prestimulation with flagellin (Fig. 7c).
Fig. 7.
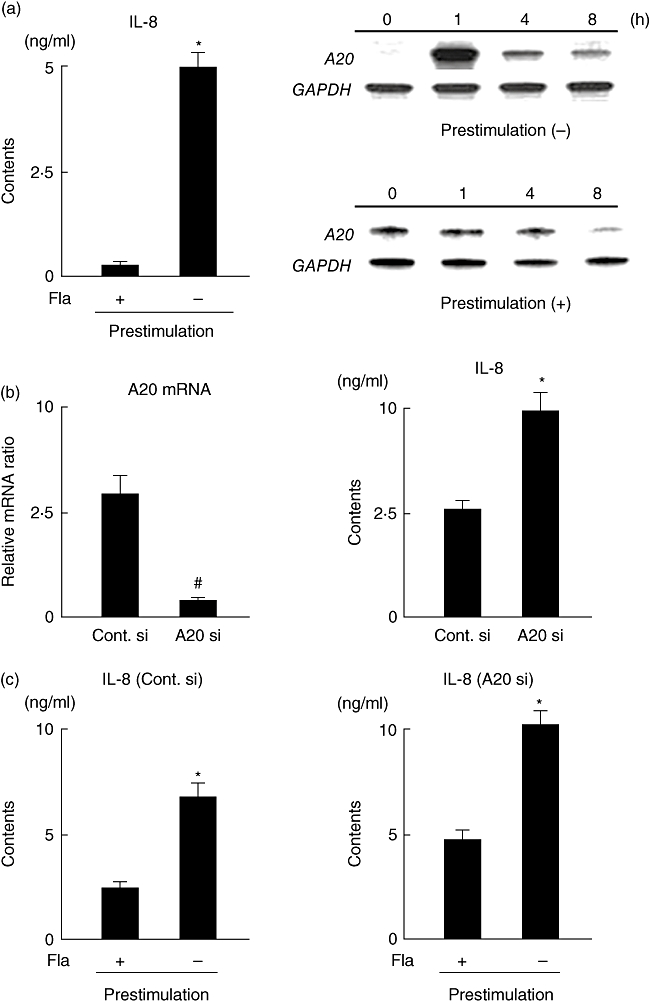
(a) Flagellin-induced tolerance induction and A20 expression in HCT-15 cells. Cells were incubated with or without flagellin (100 ng/ml) for 16 h, then restimulated with flagellin for 6 h. Tolerance induction was evaluated by measurement of interleukin (IL)-8 contents in the supernatants of culture media. A20 expression in tolerant and non-tolerant cells was examined using RNase protection assays. Error bars indicate the standard error of mean values obtained independently from six mice. *P < 0·01 versus Prestimulation (+). (b) Efficiency of A20 siRNA. HCT-15 cells were transfected with control and A20 siRNAs. A20 mRNA expression at 1 h after stimulation with flagellin (100 ng/ml) and IL-8 contents in culture supernatants at 8 h after stimulation with flagellin (100 ng/ml) were evaluated by real-time polymerase chain reaction (PCR) and enzyme immune assay (EIA), respectively. Error bars indicate the standard error of mean values obtained independently from six mice. *P < 0·01, #P < 0·05 versus control siRNA. (c) Effect of A20 on flagellin-induced tolerance induction. Cells transfected with control or A20 siRNA were incubated with or without flagellin (100 ng/ml) for 16 h, then restimulated with flagellin for 6 h. Tolerance induction was evaluated by measurement of IL-8 contents in the supernatants of culture media. Error bars indicate the standard error of mean values obtained independently from six mice. *P < 0·01 versus prestimulation (+).
Discussion
In the present study we focused upon A20, an inducible cytoplasmic negative regulator involved with TLR signalling, and evaluated its expression in IECs during intestinal inflammation. Our results revealed that flagellin-mediated TLR-5 signalling induces abundant expression of A20 rapidly in IECs, which was also confirmed in vivo in a DSS-induced acute colitis model. In addition, the present results demonstrated that A20 expression does not influence tolerance induced by restimulation with flagellin.
As a monolayer of intestinal epithelial cells is exposed to the gut luminal environment and various microbial pathogens present there, it is reasonable to speculate that epithelial cell functions are regulated by TLR signalling, and that several different negative regulators of TLR signalling may contribute to restore intestinal immune balance under physiological and pathological conditions. Recently, Otte et al. showed that TLR ligands such as LPS and lipoteichoic acid (LTA) up-regulate Tollip expression in cultured colonic epithelial cells; however, they found no expression of other inducible cytoplasmic negative regulators [38]. In the present study, the expressions of A20, IRAK-M and Tollip were increased in ligand-stimulated IECs, which were related clearly to the levels of cellular NF-κB activity and IL-8 production. These findings indicate that negative regulators may play an important role to regulate excess and uncontrolled intestinal inflammatory responses induced by TLR signalling. Our results also demonstrated that the gene expression of A20 is increased rapidly and peaks at 1 h after ligand stimulation, which preceded the up-regulation of IRAK-M and Tollip. A20 has been reported to be a rapidly inducible gene following stimulation with TNF-α and TLR ligands, and A20 dysfunction leads to aggravation of inflammation in various experimental models [39]. However, those previous studies focused mainly upon macrophages to elucidate TLR-mediated A20 expression. In contrast, our results show for the first time that A20 is an early response negative regulator of TLR signalling in IECs and that it may serve as a bridge to other negative regulators to control gut innate immunity.
Flagellin is a specific microbial ligand of TLR-5, which is a pathogenic factor that induces several inflammatory disorders, including acute infectious enteritis and inflammatory bowel disease (IBD) [40–42]. As in vitro studies have shown that flagellin is a potent stimulator of A20 expression in IECs, we focused upon flagellin and evaluated whether it induces A20 expression in mouse colons. In the present experimental colitis model mice, A20 expression was increased significantly from days 5–9 after beginning administration of DSS, which was inhibited clearly by intraperitoneal injection of the TLR-5 blocking peptide. These findings suggest that IECs sense luminal (endogenous) flagellin via TLR-5-mediated signalling and induce A20 expression as a first-line defence against excess innate immune responses.
Based on those findings, we examined further the effect of exogenous flagellin injected anally on colonic A20 expression. It has been reported that TLR-5 is expressed mainly in the basolateral membrane of IECs and flagellin in the gut lumen cannot stimulate TLR-5 directly in normal conditions [35,43]. Therefore, we induced colonic injury in mice initially by oral administration of DSS for 3 days. For this experiment, we used the tlr4-deficient mouse strain CH3/HeJ to avoid the effect of LPS on A20 expression [36]. Anal injection of flagellin induced expression of A20 rapidly as well as TNF-α in DSS-treated injured colonic tissues, which was also confirmed in vitro using an organ culture system. In addition, immunohistochemistry results clearly revealed flagellin-induced abundant expression of A20 in epithelial cells. These findings suggest that certain initial intestinal injuries caused by various luminal stimuli enable contact with flagellin and TLR-5 in IECs, after which A20 expression is induced during the early phase of TLR-5-mediated inflammation, which may contribute to the first line of defence in the gut. Although we also observed infiltrating mononuclear cells expressing A20 in immunohistochemical sections, the number of those cells was relatively low compared to that of epithelial cells. The less significant sign of colonic injury and A20 expression may be explained by the low dose and short time–course treatment of DSS, which produced mild rather than severe colonic injury.
Although A20 was characterized originally as having an auto-regulatory negative feedback approach to modulate NF-κB activation upon TNF-α stimulation, a range of other stimuli, including IL-1, LPS, CD40 and latent membrane protein (LMP)-1, also play important roles in A20 induction for gate-keeping functions against NF-κB and activator protein-1 (AP-1) in different cell types [44]. Interestingly, A20 was shown recently to inhibit NF-κB activation in response to stimulation of the intracellular NOD2 (nucleotide-binding oligomerization domain 2) receptor by muramyl dipeptide, a pathway well known in susceptibility to Crohn's disease [45]. Considering those, we assessed the effect of flagellin on A20 expression using in vitro IECs as well as in vivo with an acute model of colitis, based on the finding that the flagellin-dependent TLR-5 pathway is involved in generation of a NF-κB-mediated innate immune response. Apart from TLRs, a variety of other signalling events become active during colitis as well as in in vitro flagellin-treated cell culture conditions. Several cascades, including Janus kinases (JAK)/signal transducer and activator of transcription (STAT), mitogen-activated protein (MAP) kinases, extracellular signal-related kinase (ERK) and c-Jun N-terminal kinase (JNK), are activated strongly in patients with inflammatory bowel disease and animal models of colitis [46–48]. Therefore, A20 induction via these pathways following exposure to a variety of stimuli (TLR ligands, proinflammatory cytokines, growth factors, oxidative agents) may implicate its function as a negative regulator in a wide range of activated signalling events during colitis. The majority of these pathways lead to activation of the downstream transcription factor NF-κB while, conversely, A20 transcription is induced rapidly by a large number of stimuli following triggering of the binding of NF-κB to two specific NF-κB-binding sites in the A20 promoter [49]. Since its discovery many years ago, the mechanism of A20 activity has remained largely unexplained. Dixit and co-workers [50] found that A20 interferes with TNF-induced NF-κB activation by acting as a dual ubiquitin-editing enzyme. In addition, overexpression of A20 results in de-ubiquitination of various pathways induced by downstream mediators, indicating that it serves a negative regulatory role for controlling inflammation [44]. Based on these observations, in addition to evaluating the role of A20 in the flagellin-induced TLR-5 pathway, emphasis should also be given to other pathways by focusing upon the mechanisms of the effects of A20 on those pathways.
LPS tolerance, demonstrated by hyporesponsiveness to a second stimulation with LPS after a preceding LPS treatment, is an essential phenomenon for regulation of host innate immune response [51]. Prior exposure to other TLR ligands, including LTA, flagellin and cytosine–guanine (CPG) DNA, also induces tolerance or cross-tolerance in various immune and epithelial cells [52–54]. Induction of LPS tolerance has been reported to be associated with decreased cell surface expression of TLR-4/MD-2 complex and dysregulation of IRAK-1 activity in LPS-tolerant cells [55]. In addition, recent studies have demonstrated that intracellular negative regulators of TLR signalling such as IRAK-M and Tollip correlate with tolerance induction [38,56]. However, the detailed mechanisms involved with TLR-related tolerance induction remains unknown. We speculated that A20 functions in flagellin-induced tolerance in IECs, and conducted the present study. Although prior treatment with flagellin decreased IL-8 production by IECs significantly, A20 gene knock-down did not influence tolerance induced by prestimulation with flagellin. Recently, Boone et al. evaluated the role of A20 in LPS tolerance using bone marrow-derived macrophages isolated from A20 deficient mice and concluded that A20 is not required for induction of LPS tolerance, which is supported by the present results [30]. In addition, our in vitro findings showed that the expression of A20 was induced rapidly and abundantly after flagellin stimulation, then soon returned to the basal level. However, there were higher levels of A20 expression after 16 h in the flagellin-stimulated HCT-15 cells compared to the expressions of Tollip and IRAK-M (Fig. 2a). We also observed higher amounts of A20 being produced even during the late periods of flagellin and DSS treatments (Fig. 4a). Such diverse and prolonged A20 expression may lead to important changes in the intensity and character of immune responses by modulating NF-κB and AP-1. At the end of DSS treatment, in addition to control of inflammatory consequences, other events including tissue regeneration, angiogenesis and cell proliferation are also major concerns. Therefore, elucidating A20 roles other than immune regulation at the late phase of DSS-induced colitis may generate novel insight to understand amelioration of intestinal inflammation.
Previous reports have noted that A20 plays important roles in various pathophysiological phenomena, such as cellular proliferation, migration and apoptosis [57–59]. Another study also demonstrated that A20 is an antigen presentation attenuator and its inhibition abrogates regulatory T cell-mediated suppression [60]. Thus, A20 may be a potential therapeutic tool for treatment of several immune and inflammatory diseases. In the present study, we focused upon TLR-mediated A20 expression in IECs; however, its function remains poorly understood and additional investigations are necessary to elucidate the precise role of A20 in intestinal diseases.
In summary, we examined the expression of A20 in IECs stimulated by TLR ligands and found that A20 is an early response negative regulator of TLR-5 signalling in IECs that functions in the early phase of intestinal inflammation. Our results provide new insights into the negative feedback regulation of TLR signalling that maintains the innate immune system in the gut.
Disclosure
The authors declare that they have no conflict of interest related to the publication of this manuscript.
References
- 1.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 4.Underhill DM, Ozinsky A. Toll-like receptors: key mediators of microbe detection. Curr Opin Immunol. 2002;14:103–10. doi: 10.1016/s0952-7915(01)00304-1. [DOI] [PubMed] [Google Scholar]
- 5.Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 6.Monteleone I, Vavassori P, Biancone L, Monteleone G, Pallone F. Immunoregulation in the gut: success and failures in human disease. Gut. 2002;50:III60–4. doi: 10.1136/gut.50.suppl_3.iii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cario E, Rosenberg IM, Brandwein SL, Beck PL, Reinecker HC, Podolsky DK. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. 2000;164:966–72. doi: 10.4049/jimmunol.164.2.966. [DOI] [PubMed] [Google Scholar]
- 8.Cario E. Bacterial interactions with cells of the intestinal mucosa: toll-like receptors and NOD2. Gut. 2005;54:1182–93. doi: 10.1136/gut.2004.062794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hausmann M, Kiessling S, Mestermann S, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 10.Ortega-Cava CF, Ishihara S, Rumi MA, et al. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol. 2003;170:3977–85. doi: 10.4049/jimmunol.170.8.3977. [DOI] [PubMed] [Google Scholar]
- 11.Ishihara S, Rumi MA, Kadowaki Y, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol. 2004;173:1406–16. doi: 10.4049/jimmunol.173.2.1406. [DOI] [PubMed] [Google Scholar]
- 12.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–60. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 14.Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–94. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 15.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 16.O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003;24:286–90. doi: 10.1016/s1471-4906(03)00115-7. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–66. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 18.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 19.Shibolet O, Podolsky DK. TLRs in the gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1469–73. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- 20.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Iwami KI, Matsuguchi T, Masuda A, Kikuchi T, Musikacharoen T, Yoshikai Y. Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J Immunol. 2000;165:6682–6. doi: 10.4049/jimmunol.165.12.6682. [DOI] [PubMed] [Google Scholar]
- 22.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–9. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 23.Divanovic S, Trompette A, Atabani SF, et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–8. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wald D, Qin J, Zhao Z, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–7. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 25.Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–8. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinjyo I, Hanada T, Inagaki-Ohara K, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–91. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 27.Ishihara S, Rumi MA, Ortega-Cava CF, et al. Therapeutic targeting of toll-like receptors in gastrointestinal inflammation. Curr Pharm Des. 2006;12:4215–28. doi: 10.2174/138161206778743448. [DOI] [PubMed] [Google Scholar]
- 28.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60:1143–51. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 29.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 31.O'Reilly SM, Moynagh PN. Regulation of Toll-like receptor 4 signalling by A20 zinc finger protein. Biochem Biophys Res Commun. 2003;303:586–93. doi: 10.1016/s0006-291x(03)00389-9. [DOI] [PubMed] [Google Scholar]
- 32.Gon Y, Asai Y, Hashimoto S, et al. A20 inhibits toll-like receptor 2- and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am J Respir Cell Mol Biol. 2004;31:330–6. doi: 10.1165/rcmb.2003-0438OC. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–65. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 35.Rhee SH, Im E, Riegler M, Kokkotou E, O'Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci USA. 2005;102:13610–15. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 37.Baumgart DC, Olivier WA, Reya T, Peritt D, Rombeau JL, Carding SR. Mechanisms of intestinal epithelial cell injury and colitis in interleukin 2 (IL2)-deficient mice. Cell Immunol. 1998;187:52–66. doi: 10.1006/cimm.1998.1307. [DOI] [PubMed] [Google Scholar]
- 38.Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–70. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Turer EE, Tavares RM, Mortier E, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med. 2008;205:451–64. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vijay-Kumar M, Aitken JD, Gewirtz AT. Toll like receptor-5: protecting the gut from enteric microbes. Semin Immunopathol. 2008;30:11–21. doi: 10.1007/s00281-007-0100-5. [DOI] [PubMed] [Google Scholar]
- 41.Uematsu S, Akira S. Immune responses of TLR5(+) lamina propria dendritic cells in enterobacterial infection. J Gastroenterol. 2009;44:803–11. doi: 10.1007/s00535-009-0094-y. [DOI] [PubMed] [Google Scholar]
- 42.Mizoguchi A, Mizoguchi E. Inflammatory bowel disease, past, present and future: lessons from animal models. J Gastroenterol. 2008;43:1–17. doi: 10.1007/s00535-007-2111-3. [DOI] [PubMed] [Google Scholar]
- 43.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–5. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 44.Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. 2009;284:8217–21. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hitotsumatsu O, Ahmad RC, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–90. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitsuyama K, Matsumoto S, Rose-John S, et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263–9. doi: 10.1136/gut.2005.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki A, Hanada T, Mitsuyama K, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–81. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hommes DW, Peppelenbosch MP, van Deventer SJ. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut. 2003;52:144–51. doi: 10.1136/gut.52.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–6. [PubMed] [Google Scholar]
- 50.Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 51.Nomura F, Akashi S, Sakao Y, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164:3476–9. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 52.Cario E, Podolsky DK. Intestinal epithelial TOLLerance versus inTOLLerance of commensals. Mol Immunol. 2005;42:887–93. doi: 10.1016/j.molimm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116:203–12. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Aubel RA, Keestra AM, Krooshoop DJ, van Eden W, van Putten JP. Ligand-induced differential cross-regulation of Toll-like receptors 2, 4 and 5 in intestinal epithelial cells. Mol Immunol. 2007;44:3702–14. doi: 10.1016/j.molimm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Abreu MT, Arnold ET, Thomas LS, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem. 2002;277:20431–7. doi: 10.1074/jbc.M110333200. [DOI] [PubMed] [Google Scholar]
- 56.Nakayama K, Okugawa S, Yanagimoto S, et al. Involvement of IRAK-M in peptidoglycan-induced tolerance in macrophages. J Biol Chem. 2004;279:6629–34. doi: 10.1074/jbc.M308620200. [DOI] [PubMed] [Google Scholar]
- 57.Ortis F, Pirot P, Naamane N, et al. Induction of nuclear factor-kappaB and its downstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role in pancreatic beta cells. Diabetologia. 2008;51:1213–25. doi: 10.1007/s00125-008-0999-7. [DOI] [PubMed] [Google Scholar]
- 58.Liuwantara D, Elliot M, Smith MW, et al. Nuclear factor-kappaB regulates beta-cell death: a critical role for A20 in beta-cell protection. Diabetes. 2006;55:2491–501. doi: 10.2337/db06-0142. [DOI] [PubMed] [Google Scholar]
- 59.Li HL, Zhuo ML, Wang D, et al. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. 2007;115:1885–94. doi: 10.1161/CIRCULATIONAHA.106.656835. [DOI] [PubMed] [Google Scholar]
- 60.Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008;14:258–65. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]