

Abstract
Gram-negative infections can result in endotoxic shock, which is the most common cause of death in intensive care units. Most of the undesirable effects in sepsis and septic shock have been ascribed to lipopolysaccharide (LPS), a normal constituent of the bacterial wall. The response to LPS involves rapid secretion of proinflammatory cytokines [tumour necrosis factor-α, interleukin (IL)-1, IL-6, IL-8, interferon-γ] and the concomitant induction of anti-inflammatory mediators such as IL-10 and transforming growth factor-β and glucocorticoids (GC), which render the host temporarily refractory to subsequent lethal doses of LPS challenge in a process known as LPS or endotoxin tolerance. Although protective from the development of sepsis or systemic inflammation, endotoxin tolerance has also been pointed out as the principal cause of the non-specific immunosuppression described in these patients. In this report we demonstrate, using a mouse model, that while the maintenance of tolerance is dependent upon GC, the establishment of tolerance by LPS could be inhibited by dexamethasone (Dex), a synthetic GC. Conversely, we demonstrated that mifepristone (RU486), a known GC receptor antagonist, was capable of inducing a transient and reversible disruption of endotoxin tolerance, also permitting partial restoration of the humoral immune response in LPS tolerant/immunosuppressed mice. These results are encouraging for the management of immunosuppression in sepsis and/or non-infectious shock, and deserve further investigation in the future.
Keywords: dexamethasone, endotoxin tolerance, glucocorticoids, LPS, RU486
Introduction
Severe Gram-negative infections can result in endotoxic shock, which is the most common cause of death in intensive care units [1–5]. Most of the undesirable effects in sepsis and septic shock caused by Gram-negative bacteria have been ascribed to lipopolysaccharide (LPS), a normal constituent of the bacterial wall [3,6–9].
Substantial evidence suggests that the response to LPS involves not only a rapid secretion of proinflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-8 and interferon (IFN)-γ, but also the concomitant induction of potent anti-inflammatory factors secreted by monocytes/macrophages such as IL-10, transforming growth factor (TGF)-β[10–13] or glucocorticoids (GC) [10,13–15], which render the host temporarily refractory to subsequent lethal doses of LPS challenge [16–19].
This refractoriness to LPS, known as LPS or endotoxin tolerance, is characterized by a decreased production of proinflammatory cytokines in response to LPS following a first exposure to the same stimulus, and is thought to be a host adaptation to limit overwhelming inflammation that occurs during bacterial Gram-negative infection [1,15,20].
However, although protective from the development of sepsis or systemic inflammation, endotoxin tolerance has also been pointed out as the principal cause of the non-specific immunosuppression reported in these patients, which can lead to fatal blunting of immunological responses to subsequent infections in survivors of sepsis or septic shock [18,21–23].
GC and anti-inflammatory cytokines such as IL-10 and TGF-β have been considered to be the main agents responsible for support of endotoxin tolerance [18,24,25]. The importance of GC was demonstrated essentially by the fact that adrenalectomized mice did not become tolerant to LPS [15,18,26].
However, the mode of action of GC in tolerance is not understood fully. For instance, LPS injection of galactosamine-treated mice did not generate endotoxin tolerance, despite the fact that the level of corticosterone in these animals was similar to that found in LPS-treated naive mice [15]. In addition, although it is known that the hypothalamic–pituitary–adrenal axis plays an active role in endotoxin tolerance [14,27], GC treatment in high doses have been used historically in sepsis with no benefit to patients. However, more recently low doses of GC have been used to treat septic shock in patients with adrenal insufficiency [28,29]. In addition, the management of endotoxin tolerance/immunosuppression is controversial and constitutes a crucial problem in the treatment of sepsis [23,30,31].
The aim of our studies was to gain insight into the role of GC on the mechanisms of establishment and maintenance of endotoxin tolerance, as well as immunosuppression induced by the tolerance phenomenon, through the use of dexamethasone (Dex), a synthetic GC, and mifepristone (RU486), an inhibitor of GC and progesterone receptors.
For this purpose, and considering that de-activation of endotoxin tolerance and/or restoration of the immune response might potentially be beneficial in the treatment of sepsis or septic shock [23,30–33], we used LPS-induced tolerant/immunosuppressed mice as an experimental model to analyse events during early and late stages of human sepsis.
In brief, our results indicate that GC could play an important and differential role in the establishment and maintenance of endotoxin tolerance with opposing effects on these two processes. Conversely, the humoral immune response could be restored partially in tolerant/immunosuppressed animals through inhibition of endogenous GC activity by RU486. All these effects were dependent upon the time-point of exposure to GC or to RU486.
Materials and methods
Reagents
Mouse recombinant IFN-γ and rabbit anti-murine anti-TNF-α were purchased from PeproTech Inc. (Mexico, DF). Soluble TNF-α receptor (sTNFR – etanercept) was obtained from Wyeth Pharmaceuticals Inc. (Collegeville, PA, USA). Mifepristone [RU486-17-hydroxy-11-(4-dimethylaminophenyl) 17-(1-propynyl) estra-4, 9-diene-3-one], thioglycollate broth, mouse recombinant TNF-α and lipopolysaccharide (LPS) from Escherichia coli O111:B4, catalogue no. L2630 purified by phenol extraction, were obtained from Sigma-Aldrich (St Louis, MO, USA). Synthetic glucocorticoid dexamethasone (Dex) (Decadrón Shock) was obtained from Sidus S.A. (C.A. Buenos Aires, Argentina). Cytokines and reagents were prepared in sterile pyrogen-free saline. Corticosterone level was determined by a commercially available radioimmunoassay (RIA) kit from ICN Biomedicals (Costa Mesa, CA, USA). [3H]-dexamethasone ([3H]-Dex) in ethanol was from New England Nuclear (Boston, MA, USA) and had a specific activity of 35·00 Ci/mM (1254·00 GBq/mM). Sheep red blood cells (SRBC) were obtained from Alfredo Gutierrez® (C.A.). The following anti-mouse antibodies were used: phycoerythrin (PE)-conjugated rat anti-immunoglobulin (Ig)M monoclonal antibody (mAb) (BD-Pharmingen, San Diego, CA, USA) and fluorescein isothiocyanate (FITC)-conjugated goat anti-IgG polyclonal antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA).
Mice
BALB/c mice were bred in the animal facility of the Department of Experimental Medicine, Academia Nacional de Medicina, Buenos Aires. Female mice aged 12–16 weeks weighing 20–25 g were used throughout the experiments. They were maintained under a 12 h light–dark cycle at 22 ± 2°C and fed with standard diet and water ad libitum. The experiments performed herein were conducted according to the principles set forth in the Guide for the Care and Use of Laboratory Animals[34].
Endotoxin tolerance models
Classical tolerance model
Mice were tolerized by intraperitoneal (i.p.) inoculation of LPS (80 µg/kg) for 4 consecutive days. Twenty-four hours after the last injection animals were resistant to a lethal dose (LD) of LPS (2 LD50 = 8 mg/kg i.p.).
Tolerance/immunosuppression model
Because immunosuppression is a quantitative effect dependent upon the number of doses and concentration of LPS injections, a stronger immunosuppression was obtained by treatment of mice with different doses of LPS for 13 consecutive days. The inoculation regimen began with 200 µg/kg i.p. for the first 3 days, followed by 4 mg/kg i.p. for 9 days.
Lethality studies
Mice were injected i.p. with a lethal dose of LPS (2 LD50 = 200 µg) in pyrogen-free saline and followed up to 72 h. This dose induces 100% mortality between 24 and 48 h after injection. The same batch of LPS was used throughout the experiments.
Treatment with RU486 and immunization in tolerant/immunosuppressed mice
Twenty-four hours after the last dose of endotoxin, LPS tolerant/immunosuppressed mice were inoculated with RU486 (30 mg/kg i.p.) and 30 min later they were immunized with SRBC (5 × 108/mouse i.p.). Then, at 24 and 30 h after the immunization, mice were treated again with RU486. Control mice (naive) were either treated or not with RU486 and immunized using the same regimen.
Seven days after immunization the animals were bled and serum sample were collected and frozen at −20°C until to use.
Thioglycolate-elicited mouse peritoneal macrophages
Mice were injected intraperitoneally with 2 ml of 3% (wt/vol) thioglycollate broth. After 4 days they were killed and cells were harvested by peritoneal lavage with cold phosphate-buffered saline (PBS) and cultured in 48-well tissue culture plates (Costar, Cambridge, MA, USA) at a concentration of 2·5 × 105 cells/well in RPMI-1640, supplemented with 10% fetal calf serum (FCS), 1% penicillin and 1% streptomycin. After overnight incubation at 37°C in a humidified atmosphere of 5% CO2 (and 95% air), wells were washed twice with PBS to remove non-adherent cells. As judged by morphological criteria and Turk colourant staining, more than 90–95% of the adherent cells were macrophages.
TNF-α bioassay
The biological activity of TNF-α was determined using a sensitive actinomycin D-treated murine L-929 fibroblast assay, as described previously [35]. Briefly, L-929 cells were plated in 96-well plates (Costar) at 1·8×104 cells/well in 0·1 ml and allowed to grow to near confluence overnight at 37°C in 95% air, 5% CO2. Serially diluted macrophage supernatants were added to the L-929 cells. After 18 h of incubation in the presence of 10 µg/ml actinomycin D (Amersham Biosciences, Piscataway, NJ, USA), the plates were washed with PBS and viable cells were fixed and stained with violet crystal solution (0·1% in 20% methanol) for 20 min at 37°C. Then, absorbance of the blue colour extracted with 30% acetic acid was measured with a microtitre plate reader (Organon Tecnika, C.A. Buenos Aires Argentina) at 550 ηm. The activity titre of TNF-α in lytic units/ml (LU50/ml) was calculated from the reciprocal of the dilution necessary for 50% cell lysis.
Enzyme-linked immunosorbent assay (ELISA)
Plasma were collected and frozen at −20°C until use. TNF-α and IL-10 ELISA were performed on flat-bottomed polystyrene microtitre plates (OptEIA set; BD Biosciences, San Diego, CA, USA) according to the manufacturer's instructions.
Haemagglutination assay
The antibody response to SRBC was evaluated through a haemagglutination assay. Briefly, serum samples were inactivated at 56°C for 30 min and diluted in a double dilution test using PBS–bovine serum albumin (BSA) 0·2%. Then, 50 µl of each dilution was dispensed in a round-bottomed 96-well microplate and 50 µl of 0·25% SRBC in PBS–BSA was added. Finally, the plates were incubated for 24 h at room temperature and the titre was considered as the reciprocal of the last positive dilution.
Flow cytometry to determine IgG, IgM anti-SRBC
To measure mouse IgG and IgM, anti-SRBC serum samples were prepared at different dilutions in PBS–BSA 0·5%. Then, 10 µl of serum were incubated with 3 µl of 1% SRBC (PBS–BSA 0·5%) for 30 min at 4°C. The cells were washed three times and (PE) anti-IgM or (FITC) anti-IgG was added and incubated for 30 min at 4°C. Cells were washed and immunoglobulins were evaluated in a Becton Dickinson FACScan using CellQuest software (Becton Dickinson, San Jose, CA, USA). Controls of SRBC incubated with labelled antibodies in the absence of serum were also carried out.
Statistical analysis
Values are expressed as the mean ± standard error of the mean (s.e.m.) of n observations. The statistical significance of differences between TNF-α samples measured by the L-929 bioassay was determined using the non-parametric Friedman test followed by Wilcoxon's signed-rank test. ELISA and haemagglutination assays were analysed using the Mann–Whitney unpaired test. All statistical tests were interpreted in a two-tailed fashion and P < 0·05 was considered significant.
Results
Dexamethasone induces refractoriness to a lethal dose of LPS
A daily i.p. injection of LPS (80 µg/kg) in mice for 4 days induces the establishment of tolerance to LPS, a phenomenon characterized by low secretion of TNF-α in response to subsequent doses of LPS [19,36] and high levels of corticosterone in serum 3 h after the last LPS injection (tolerants: 1099·6 ng/ml ± 23·2 versus normal: 163·7 ng/ml ± 5·8; n = 5) [15,37–39]. This increase of GC in tolerant animals seems to be important in the refractoriness to LPS, as naive mice (n = 6) survived when they were pretreated with Dex 2·5 mg/kg i.p. between 0 and 3 h before a lethal dose of LPS (8 mg/kg i.p.). However, when LPS was injected 10 h after Dex, the mortality was 57·2% (n = 7) and after 24 h reached values of 92·3% (n = 13). This LPS refractoriness induced by Dex correlated with the low amount of TNF-α in mice plasma 90 min after the simultaneous injection of Dex and LPS (Dex–LPS = 183 ± 67 pg/ml versus LPS = 8431 ± 1027 pg/ml) (n = 6).
Similar results were obtained in vitro when mouse peritoneal macrophages were treated with Dex (40 µg/ml) for 30 min, and later with LPS (20 ηg/ml) for 6 h. After this period the supernatants were collected and the biological activity of TNF-α was determined using the L-929 assay. The LPS-induced secretion of TNF-α was reduced significantly by Dex to 6·7 ± 2% of control (LPS alone) (n = 6).
Taking into account the schedules used for these in vivo and in vitro experiments we investigated if the effect of Dex could be due to a mere interaction or blockade of LPS by Dex. For this purpose, LPS and [3H]-Dex were incubated at 37°C for 1 h and passed through a Sephadex G-10. The first peak eluted from the column (LPS) was devoid of radioactivity, indicating that [3H]-Dex was not bound to LPS. In addition, the capacity of this peak of LPS to induce TNF-α secretion from mouse macrophages remained intact (not shown).
Dexamethasone inhibits the establishment of LPS tolerance
Considering that GC are increased in plasma of tolerant mice and that Dex was responsible for animal protection to a lethal dose of LPS, we speculated that Dex would be also capable of inducing tolerance to LPS. However, daily injections of Dex (2·5 mg/kg) for 4 days instead of LPS did not induce a tolerant state indicating that, although important for protection, Dex is not involved in the establishment of the tolerant state (not shown).
Conversely, when we tried to tolerize animals through the simultaneous injection of LPS and Dex instead of LPS alone, the animals did not become tolerant to endotoxin, indicating that Dex prevented the establishment of LPS tolerance. This effect correlated with the increase in TNF-α and IL-10 after exposure to a lethal dose of LPS, which is in agreement with the lack of tolerance in these animals (Table 1).
Table 1.
Dexamethasone inhibits the establishment of lipopolysaccharide (LPS) tolerance.
| Control (n = 6) | LPS (n = 6) | Dex + LPS (n = 9) | |
|---|---|---|---|
| Mortality % | 100 | 0 | 100 |
| TNF-α (pg/ml) | 7211 ± 1174 | 43 ± 14 | 4303 ± 1429* |
| IL-10 (pg/ml) | 2877 ± 403 | 298 ± 89 | 3756 ± 776* |
P < 0·001 significantly different from tolerant (LPS). BALB/c mice were injected with LPS [80 µg/kg intraperitoneally (i.p.)]. The endotoxin was injected alone (LPS) or simultaneously with dexamethasone (Dex) (2·5 mg/kg; i.p.) (Dex + LPS) for 4 days. One group of naive mice was inoculated only with saline (control). After 24 h of the last LPS dose all groups received a lethal dose of LPS (8 mg/kg i.p.) and mortality was evaluated up to 72 h. The mortality of mice treated only with Dex for 4 days was 100% after the challenge with lethal dose of LPS (n = 6) (not shown). Mice treated under the same schedule (LPS and Dex + LPS) were bled 90 min after LPS lethal dose and plasma level of tumour necrosis factor (TNF)-α and interleukin (IL)-10 were determined by enzyme-linked immunosorbent assay. Results are expressed as the mean ± standard error of the mean, four to six per group.
TNF-α is not relevant for the establishment of endotoxin tolerance
Because TNF-α is one of the first cytokines induced by LPS and is capable of inducing a lethal shock similar to LPS [32], the TNF-α effect in the establishment of tolerance to LPS was studied. For this purpose, three groups of mice (n = 6/group) were injected with 25, 50 or 100 ηg of TNF-α for 4 consecutive days. After this period a lethal dose of LPS was injected. The animals from all three groups died between 48 and 72 h, while those treated with LPS instead of TNF-α survived (data not shown). Moreover, peritoneal macrophages could still be made tolerant to LPS in the presence of anti-TNF-α antibodies or soluble TNF-α receptors (Fig. 1). Taken together these results indicate that, at least in our hands, TNF-α is not a relevant cytokine for the establishment of endotoxin tolerance.
Fig. 1.
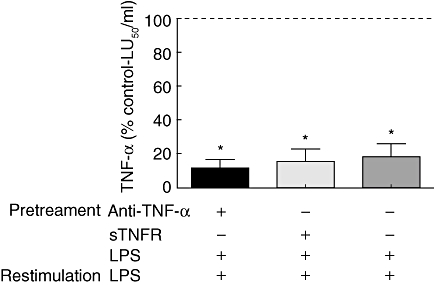
Peritoneal macrophages were pretreated with anti-tumour necrosis factor (TNF)-α, soluble TNF receptor or saline for 15 min and incubated with lipopolysaccharide (LPS) 20 ηg/ml for 20 h. Then, cells were washed and restimulated with LPS 100 ηg/ml for 6 h. Supernatants were collected and the biological activity of TNF-α was determined by L-929 assay. Results are expressed as the mean ± standard error of the mean (% control) of six independent experiments. *P < 0·05, significantly different from control. The control cells were treated only with LPS 100 ng/ml for 6 h and their TNF-α activity was considered as the 100% and is represented by a dashed line.
RU486 disrupts the maintenance but not the establishment of LPS tolerance
In order to analyse the importance of Dex in refractoriness to LPS, RU486, a well-known GC and progesterone receptor antagonist, was assayed. Thus, when RU486 (12 mg/kg s.c.) was injected 5 min before a protective dose of Dex, all animals died (n = 6) when challenged with a lethal dose of LPS, indicating that the effect of RU486 was exerted on GC and not on progesterone receptors.
We then analysed whether RU486 was able to overcome the tolerant state. Tolerant mice were treated with RU486 and the animals were injected with a lethal dose of LPS at different times. Mortality was evaluated up to 72 h post-LPS. The results shown in Table 2 indicate that RU486 abrogates endotoxin tolerance completely up to 3 h after injection, and mice then return gradually to the initial tolerance state (8 h), indicating that the effect of RU486 was limited to induce a transient and reversible effect.
Table 2.
RU486 disrupts the maintenance of lipopolysaccharide (LPS) tolerance.
| Tolerant + RU486 |
||||||||
|---|---|---|---|---|---|---|---|---|
| 0 h (n = 20) | 1 h (n = 11) | 3 h (n = 11) | 5 h (n = 10) | 8 h (n = 10) | 24 h (n = 10) | Tol (n = 11) | N (n = 10) | |
| Mortality % | 100 | 100 | 100 | 30 | 0 | 0 | 0 | 100 |
| TNF-α (pg/ml) | 2474 ± 565* | 219 ± 63** | 247 ± 67** | 205 ± 68*** | 104 ± 35n.s. | 83 ± 20n.s. | 43 ± 14 | 8431 ± 1027 |
| IL-10 (pg/ml) | 1676 ± 392** | 2507 ± 712* | 2589 ± 264* | 1963 ± 430** | 358 ± 84n.s. | 260 ± 122n.s. | 336 ± 107 | 3083 ± 456 |
P < 0·001,
P < 0·01,
P < 0·05, significantly different from tolerant (Tol). BALB/c mice were tolerized with LPS [80 µg/kg intraperitoneally (i.p.)] for 4 days. After 24 h of the last LPS dose, mice were inoculated with mifepristone (RU486) (12 mg/kg subcutaneously) and at different times indicated in the table, a lethal dose of LPS (8 mg/kg i.p.) was injected. The mortality was evaluated until 72 h after lethal LPS inoculation. One control group of tolerant mice (Tol) was inoculated with propyleneglycol (RU486 vehicle) and all animals survived a lethal dose of LPS. Control naive (N) were inoculated with saline instead of LPS tolerizing dose and challenge with a lethal dose of LPS. Mice treated under the same schedule were bled 90 min after of LPS lethal dose and plasma levels of tumour necrosis factor (TNF)-α and interleukin (IL)-10 were determined by enzyme-linked immunosorbent assay. Results are expressed as the mean ± standard error of the mean of four to seven per group; n.s.: not significant.
Disruption of the mechanism of endotoxin tolerance by RU486 correlates with the increase of TNF-α in these animals, this being another marker of tolerance de-activation. The high levels of IL-10 observed in RU486-treated tolerant mice also suggest limited importance of IL-10 in the maintenance of tolerance. Conversely, pretreatment or simultaneous injection of naive mice with RU486 and LPS did not prevent the establishment of tolerance (data not shown).
In order to compare the overcoming of LPS tolerance induced by RU486 to that obtained by IFN-γ[17,33] in the treatment of septic/immunosuppressed patients, mouse peritoneal macrophages were made tolerant with LPS and then treated with mouse IFN-γ for 18 h, washed and restimulated with LPS, and the production of TNF-α was evaluated at different times. We observed an increase in TNF-α production at 0 h and 24 h later, indicating that mouse IFN-γ, similar to human IFN-γ, induces disruption to the LPS tolerance state. However, after 72 h this effect disappears and cells return to the tolerant state (Fig. 2). This transient and reversible effect resembles those observed with RU486, although it should be taken into account that IFN-γ was studied in vitro, whereas the effects of RU486 were studied in vivo.
Fig. 2.

Peritoneal macrophages were pretreated with lipopolysaccharide (LPS) 20 ηg/ml or saline for 20 h, washed and incubated for 18 h with mouse interferon (IFN)-γ 500 U/ml. They were then restimulated at 0, 24 and 72 h with LPS 100 ηg/ml for 6 h (a–c). Supernatants were collected and biological activity of tumour necrosis factor (TNF)-α was determined by L-929 cytotoxicity assay. Results are expressed as the mean ± standard error of the mean (% control) of six independent experiments and were represented in log10 scale. ##P < 0·01, #P < 0·05, significantly different from control; **P < 0·01, significantly different from tolerant (dark bar). The control cells were treated only with LPS 100 ng/ml for 6 h; this TNF-α activity was considered to be 100% and is represented by a dashed line.
RU486 restores the humoral immune response
Taking into account that endotoxin tolerance may be one of the causes of the immunosuppression observed frequently in late sepsis [40,41], and considering that RU486 induces a transient overcoming of tolerance, finally we analysed the effect of RU486 on humoral immune response in LPS-induced tolerant/immunosuppressed mice. Although immunosuppression was observed in mice tolerized with the classical endotoxin tolerance model (four doses of LPS), stronger immunosuppression was obtained by increasing the concentration and the number of LPS doses.
The results shown in Fig. 3 indicate that RU486 can partially restore or enhance the primary humoral immune response in immunosuppressed mice. In addition, using a flow cytometry assay we observed that restoration of the primary humoral immune response involved the production of both IgM and IgG antibodies (Fig. 4). At 1 : 300 dilutions the IgM anti-SRBC of the control group appears to be similar to RU486-treated immunosuppressed mice. However at 1 : 5000 dilutions the IgM response was still detected in the control group, while it was negative in the RU486-treated immunosuppressed group (data not shown).
Fig. 3.
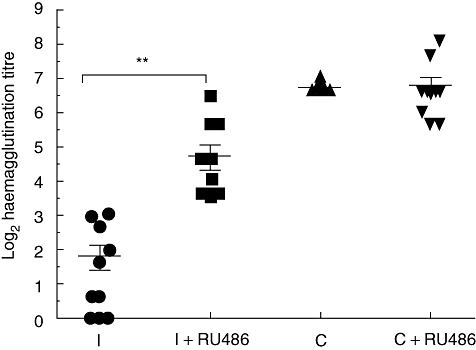
BALB/c mice were tolerized/immunosuppressed with lipopolysaccharide (LPS) [200 µg/kg intraperitoneally (i.p.) for the first 3 days, followed by 4 mg/kg i.p. for 9 days]. After 24 h, mice were treated with mifepristone (RU486) (30 mg/kg i.p.) and 30 min later were immunized with sheep red blood cells (SRBC) (5 × 108/mouse i.p.). Twenty-four and 30 h after the immunization, mice were treated again with RU486. Control mice (naive) were either treated or not with RU486 and immunized using the same schedule. Seven days after immunization the animals were bled and serum antibodies were evaluated through a haemagglutination assay. Results are expressed as mean ± standard error of the mean (% control average) and each point represents an individual mouse. The data represent three independent experiments with three to five mice per group. I = LPS tolerant/immunosuppressed mice; I + RU486 = RU486-treated LPS tolerant/immunosuppressed mice; C = control mice (naive); C + RU486 = control mice (naive) + RU486. **P < 0·01.
Fig. 4.
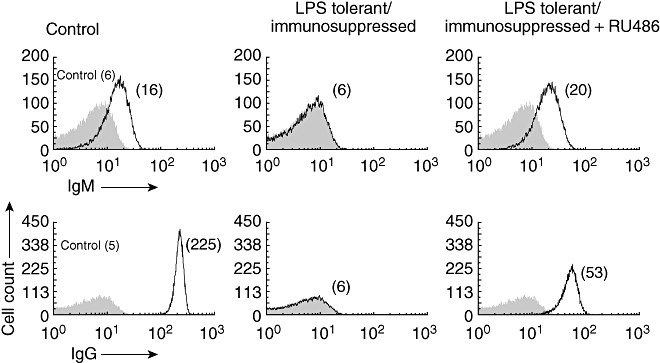
Immunoglobulin (Ig)M and IgG anti- sheep red blood cell (SRBC) levels were compared between the different groups using a 1 : 300 dilution. Data show a representative experiment of three. Number in parentheses represents the median fluorescent intensity (MFI). Control: naive mice immunized.
Discussion
Endotoxin tolerance has been considered to be one of the main causes of immunosuppression reported in patients with sepsis due to Gram-negative infections [17,23]. It has also been described that patients who succumb to septic shock after 72 h (late sepsis) show similar clinical signs of endotoxin tolerance [32,33].
These are some of the reasons why studies on the regulation of LPS tolerance have merited the attention of research groups around the world. However, despite these efforts, the complex phenomenon of endotoxin tolerance has not yet been elucidated completely. Part of this complexity could be due to the different agents, factors or mechanisms involved in LPS-induced tolerance/immunosuppression, such as chemokines induced by IL-13 and IL-4 [40], 1α-25-dihydroxyvitamin D3[42], GC [15,20], catecholamines [43,44], depletion of dendritic cells [45], IL-10 and TGF-β[25] or the decreased expression of fractalkine receptors [46]. In addition, LPS has been found to regulate as many as 1500 genes [47].
Although the relevance of GC in LPS-induced tolerance/immunosuppression has long been recognized, some of their effects are controversial and not understood completely [15,18,28].
This may be due to the different models used or, more probably, to conclusions resulting from studies directed to investigate a particular stage of endotoxin tolerance (i.e. maintenance), and later generalized inappropriately.
The aim of our study was essentially to evaluate the participation of endogenous and exogenous (Dex) GC in two relevant and different steps of endotoxin tolerance: establishment, a short period with prevalence of inflammatory cytokines, and maintenance, a longer period with predominance of anti-inflammatory agents.
Considering that endotoxin induces the increase of GC in serum through activation of the hypothalamic–pituitary–adrenal axis, we speculated that Dex would also be responsible for inducing tolerance to LPS. However, a daily injection of Dex was not capable of inducing a tolerant state. On the other hand, the simultaneous injection of LPS and Dex instead of LPS alone inhibited the induction of tolerance, suggesting that although important for the protection of animals against a lethal dose of LPS, paradoxically, Dex inhibited the establishment of endotoxin tolerance.
Because, concomitantly to this effect, Dex inhibits TNF-α production, we investigated whether TNF-α could be a necessary cytokine for the establishment of LPS tolerance. However, the negative results obtained by daily injections of TNF-α and the fact that anti-TNF-α or soluble TNF-α receptors (etanercept) did not modify the tolerance induced by LPS in vitro indicated clearly that, in our hands, TNF-α is not a cytokine responsible for the establishment of tolerance.
Our results are in agreement with those of Medvedev et al. [48], but not with other authors, who suggested that TNF-α was capable of inducing LPS tolerance [49,50]. This discrepancy could be the result of using a different animal model (rat) and/or the fact that these experiments were carried out using a non-physiological dose of TNF-α (200 µg/kg/day for 5 consecutive days) [49] or from a different species [50].
However, as GC and Dex inhibit the production of a set of proinflammatory cytokines such as TNF-α, IL-1-α, IL-1β, IL-12, IFN-γ, IL-6 and IL-8 [28,51,52], this suggests that inflammatory agent(s) other than TNF-α would be necessary for the establishment of LPS tolerance. In line with this, we have found previously that IL-1β was capable of inducing the establishment of endotoxin tolerance, an effect determined through protection against LPS, increasing the level of GC and by down-regulation of Toll-like receptor 4 (TLR-4) and up-regulation of GC receptors, both indicators of endotoxin tolerance [53].
Considering that RU486 can overcome the tolerant state, and taking into account all the previously described data, a central role for GC in the maintenance of endotoxin tolerance is suggested.
Similarly to GC, IL-10 has been recognized as an important cytokine in tolerance, although its mode of action is also controversial. In fact, some authors consider IL-10 to be a central cytokine for the establishment of tolerance [25], while others consider that IL-10 is critical for the maintenance but not for the establishment of endotoxin tolerance [54,55]. The fact that we found a low level of IL-10 in tolerant animals and high values in RU486-treated tolerant mice suggests that this cytokine is not crucial in the maintenance of tolerance. This is in line with Baykal et al. [56] and with those authors who show that IL-10 knock-out mice (IL-10–/–) can be tolerized by LPS [54]. However, we cannot discard a possible role for IL-10, as redundant mechanisms in the regulation of endotoxin tolerance could be possible, although it has been shown that this anti-inflammatory cytokine regulates GC synthesis in a negative manner through the inhibition of adrenocorticotrophic hormone (ACTH) effects [57,58].
During recent years, endotoxin tolerance has been reported as one of the causes of immunosuppression in Gram-negative infections and considered to be one of the principal causes of mortality in late sepsis [23,32]. Thus, in this situation many patients die much later with signs of opportunistic infections accompanied by down-regulation of monocyte human leucocyte antigen D-related (HLA-DR) expression or reduced ability to produce TNF-αin vitro[33]. This assumption was important in defining different treatment strategies, because most of the previous treatments using anti-inflammatory therapies were unsuccessful [57,59].
Many researchers have tried to reverse the state of immunosuppression in sepsis using IFN-γ, granulocyte colony stimulation factor (G-CSF) or granulocyte–macrophage colony stimulation factor (GM-CSF) [12,33,60].
In fact, IFN-γ administered to septic patients restored deficient HLA-DR expression, LPS-induced TNF-α production and bacterial clearance in many patients, although the effect on the immune response is not known.
In this report we have demonstrated a RU486-driven disruption of tolerance that, although using a mouse model, resembles those obtained by treatment with IFN-γ. In addition, in our case RU486 treatment was capable of restoring immunological competence in LPS tolerant/immunosuppressed mice. Considering that RU486 exerts a transient and reversible disruption of the regulation of tolerance/immunosuppression, but not a dismantling effect (Table 2), this suggests that RU486 opens a window that, although transient, is central for initiation of the humoral immune response (Figs 3 and 4).
In summary, in our mouse experimental model the establishment of tolerance by LPS could be inhibited by simultaneous injection of LPS with Dex, the maintenance of tolerance is dependent on GC, and overcoming endotoxin tolerance can be achieved by a competitive inhibitor of GC, RU486. These data and the preliminary observation that RU486 can restore the primary humoral immune response in immunosuppressed mice, are important and encouraging results that deserve further investigation in a situation where the loss of immune competence can be fatal [31].
Acknowledgments
We thank Dr Susana Fink for critical reading of the manuscript, Mr Antonio Morales for technical assistance and Dr Oscar Bottasso for his help in statistical analysis. This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (PICT-2005-38197) and Fundación Alberto J. Roemmers.
Disclosure
The authors have no conflicts of interest.
References
- 1.Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. Corticosteroids for severe sepsis and septic shock: a systematic review and meta-analysis. BMJ. 2004;329:480. doi: 10.1136/bmj.38181.482222.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parrish WR, Gallowitsch-Puerta M, Czura CJ, Tracey KJ. Experimental therapeutic strategies for severe sepsis: mediators and mechanisms. Ann NY Acad Sci. 2008;1144:210–36. doi: 10.1196/annals.1418.011. [DOI] [PubMed] [Google Scholar]
- 3.Rangel-Frausto MS. The epidemiology of bacterial sepsis. Infect Dis Clin North Am. 1999;13:299–312. doi: 10.1016/s0891-5520(05)70076-3. [DOI] [PubMed] [Google Scholar]
- 4.Shimaoka M, Park EJ. Advances in understanding sepsis. Eur J Anaesthesiol Suppl. 2008;42:146–53. doi: 10.1017/S0265021507003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zacharowski K, Zacharowski PA, Koch A, et al. Toll-like receptor 4 plays a crucial role in the immune-adrenal response to systemic inflammatory response syndrome. Proc Natl Acad Sci USA. 2006;103:6392–7. doi: 10.1073/pnas.0601527103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosshart H, Heinzelmann M. Targeting bacterial endotoxin: two sides of a coin. Ann NY Acad Sci. 2007;1096:1–17. doi: 10.1196/annals.1397.064. [DOI] [PubMed] [Google Scholar]
- 7.Ianaro A, Tersigni M, D'Acquisto F. New insight in LPS antagonist. Mini Rev Med Chem. 2009;9:306–17. doi: 10.2174/1389557510909030306. [DOI] [PubMed] [Google Scholar]
- 8.Opal SM. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int J Med Microbiol. 2007;297:365–77. doi: 10.1016/j.ijmm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Opal SM, Cross AS. Clinical trials for severe sepsis. Past failures, and future hopes. Infect Dis Clin North Am. 1999;13:285–97. doi: 10.1016/s0891-5520(05)70075-1. [DOI] [PubMed] [Google Scholar]
- 10.Carroll JA, Reuter RR, Chase CC, et al. Profile of the bovine acute-phase response following an intravenous bolus-dose lipopolysaccharide challenge. Innate Immun. 2009;15:81–9. doi: 10.1177/1753425908099170. [DOI] [PubMed] [Google Scholar]
- 11.Fock RA, Vinolo MA, Crisma AR, Nakajima K, Rogero MM, Borelli P. Protein-energy malnutrition modifies the production of interleukin-10 in response to lipopolysaccharide (LPS) in a murine model. J Nutr Sci Vitaminol (Tokyo) 2008;54:371–7. doi: 10.3177/jnsv.54.371. [DOI] [PubMed] [Google Scholar]
- 12.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–24. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Lemus E, Benicky J, Pavel J, Saavedra JM. In vivo angiotensin II AT(1) receptor blockade selectively inhibits LPS-induced innate immune response and ACTH release in rat pituitary gland. Brain Behav Immun. 2009;23:945–57. doi: 10.1016/j.bbi.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beishuizen A, Thijs LG. Endotoxin and the hypothalamo–pituitary–adrenal (HPA) axis. J Endotox Res. 2003;9:3–24. doi: 10.1179/096805103125001298. [DOI] [PubMed] [Google Scholar]
- 15.Evans GF, Zuckerman SH. Glucocorticoid-dependent and -independent mechanisms involved in lipopolysaccharide tolerance. Eur J Immunol. 1991;21:1973–9. doi: 10.1002/eji.1830210902. [DOI] [PubMed] [Google Scholar]
- 16.Cavaillon JM. The nonspecific nature of endotoxin tolerance. Trends Microbiol. 1995;3:320–4. doi: 10.1016/s0966-842x(00)88963-5. [DOI] [PubMed] [Google Scholar]
- 17.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotox Res. 2003;9:101–7. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 18.Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–7. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 19.West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 20.Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Limaye AP, Kirby KA, Rubenfeld GD, et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA. 2008;300:413–22. doi: 10.1001/jama.300.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luyt CE, Combes A, Deback C, et al. Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am J Respir Crit Care Med. 2007;175:935–42. doi: 10.1164/rccm.200609-1322OC. [DOI] [PubMed] [Google Scholar]
- 23.Morrison DC, Silverstein R, Luchi M, Shnyra A. Structure–function relationships of bacterial endotoxins. Contribution to microbial sepsis. Infect Dis Clin North Am. 1999;13:313–40. doi: 10.1016/s0891-5520(05)70077-5. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda T, Yang L, Ikenoue T, Mallard C, Hagberg H. Endotoxin-induced hypoxic–ischemic tolerance is mediated by up-regulation of corticosterone in neonatal rat. Pediatr Res. 2006;59:56–60. doi: 10.1203/01.pdr.0000191140.87314.ce. [DOI] [PubMed] [Google Scholar]
- 25.Randow F, Syrbe U, Meisel C, et al. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;181:1887–92. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuckerman SH, Qureshi N. In vivo inhibition of lipopolysaccharide-induced lethality and tumor necrosis factor synthesis by Rhodobacter sphaeroides diphosphoryl lipid A is dependent on corticosterone induction. Infect Immun. 1992;60:2581–7. doi: 10.1128/iai.60.7.2581-2587.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borges BC, Antunes-Rodrigues J, Castro M, Bittencourt JC, Elias CF, Elias LL. Expression of hypothalamic neuropeptides and the desensitization of pituitary–adrenal axis and hypophagia in the endotoxin tolerance. Horm Behav. 2007;52:508–19. doi: 10.1016/j.yhbeh.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Allary J, Annane D. Glucocorticoids and sepsis. Minerva Anestesiol. 2005;71:759–68. [PubMed] [Google Scholar]
- 29.Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009;301:2362–75. doi: 10.1001/jama.2009.815. [DOI] [PubMed] [Google Scholar]
- 30.Kox WJ, Volk T, Kox SN, Volk HD. Immunomodulatory therapies in sepsis. Intens Care Med. 2000;26(Suppl. 1):S124–8. doi: 10.1007/s001340051129. [DOI] [PubMed] [Google Scholar]
- 31.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–7. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–76. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 33.Docke WD, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–81. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 34.National Institutes of Health. Guide for the care and use of laboratory animals. Bethesda, MD: National Institutes of Health; 1985. NIH Publication no. 85-23. [Google Scholar]
- 35.Wang AM, Creasey AA, Ladner MB, et al. Molecular cloning of the complementary DNA for human tumor necrosis factor. Science. 1985;228:149–54. doi: 10.1126/science.3856324. [DOI] [PubMed] [Google Scholar]
- 36.Fitting C, Dhawan S, Cavaillon JM. Compartmentalization of tolerance to endotoxin. J Infect Dis. 2004;189:1295–303. doi: 10.1086/382657. [DOI] [PubMed] [Google Scholar]
- 37.Szabo C, Thiemermann C, Wu CC, Perretti M, Vane JR. Attenuation of the induction of nitric oxide synthase by endogenous glucocorticoids accounts for endotoxin tolerance in vivo. Proc Natl Acad Sci USA. 1994;91:271–5. doi: 10.1073/pnas.91.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegler-Heitbrock HW. Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm. 1995;45:13–26. [PubMed] [Google Scholar]
- 39.Zuckerman SH, Evans GF, Butler LD. Endotoxin tolerance: independent regulation of interleukin-1 and tumor necrosis factor expression. Infect Immun. 1991;59:2774–80. doi: 10.1128/iai.59.8.2774-2780.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamim CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. J Leukoc Biol. 2004;75:408–12. doi: 10.1189/jlb.0503214. [DOI] [PubMed] [Google Scholar]
- 41.Yadavalli GK, Auletta JJ, Gould MP, Salata RA, Lee JH, Heinzel FP. Deactivation of the innate cellular immune response following endotoxic and surgical injury. Exp Mol Pathol. 2001;71:209–21. doi: 10.1006/exmp.2001.2387. [DOI] [PubMed] [Google Scholar]
- 42.Helming L, Bose J, Ehrchen J, et al. 1alpha,25-Dihydroxyvitamin D3 is a potent suppressor of interferon gamma-mediated macrophage activation. Blood. 2005;106:4351–8. doi: 10.1182/blood-2005-03-1029. [DOI] [PubMed] [Google Scholar]
- 43.Aninat C, Seguin P, Descheemaeker PN, Morel F, Malledant Y, Guillouzo A. Catecholamines induce an inflammatory response in human hepatocytes. Crit Care Med. 2008;36:848–54. doi: 10.1097/CCM.0B013E31816532BE. [DOI] [PubMed] [Google Scholar]
- 44.Bergmann M, Gornikiewicz A, Sautner T, et al. Attenuation of catecholamine-induced immunosuppression in whole blood from patients with sepsis. Shock. 1999;12:421–7. doi: 10.1097/00024382-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Gautier EL, Huby T, Saint-Charles F, Ouzilleau B, Chapman MJ, Lesnik P. Enhanced dendritic cell survival attenuates lipopolysaccharide-induced immunosuppression and increases resistance to lethal endotoxic shock. J Immunol. 2008;180:6941–6. doi: 10.4049/jimmunol.180.10.6941. [DOI] [PubMed] [Google Scholar]
- 46.Pachot A, Cazalis MA, Venet F, et al. Decreased expression of the fractalkine receptor CX3CR1 on circulating monocytes as new feature of sepsis-induced immunosuppression. J Immunol. 2008;180:6421–9. doi: 10.4049/jimmunol.180.9.6421. [DOI] [PubMed] [Google Scholar]
- 47.Bjorkbacka H, Fitzgerald KA, Huet F, et al. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics. 2004;19:319–30. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- 48.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–74. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 49.Fraker DL, Stovroff MC, Merino MJ, Norton JA. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J Exp Med. 1988;168:95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogle CK, Guo X, Chance WT, Ogle JD. Induction of endotoxin tolerance in rat bone marrow cells by in vivo infusion of tumor necrosis factor. Crit Care Med. 1997;25:827–33. doi: 10.1097/00003246-199705000-00019. [DOI] [PubMed] [Google Scholar]
- 51.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94:557–72. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 52.Beer HD, Fassler R, Werner S. Glucocorticoid-regulated gene expression during cutaneous wound repair. Vitam Horm. 2000;59:217–39. doi: 10.1016/s0083-6729(00)59008-6. [DOI] [PubMed] [Google Scholar]
- 53.Alves-Rosa F, Vulcano M, Beigier-Bompadre M, Fernandez G, Palermo M, Isturiz MA. Interleukin-1beta induces in vivo tolerance to lipopolysaccharide in mice. Clin Exp Immunol. 2002;128:221–8. doi: 10.1046/j.1365-2249.2002.01828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berg DJ, Kuhn R, Rajewsky K, et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–47. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zingarelli B, Hake PW, Cook JA. Inducible nitric oxide synthase is not required in the development of endotoxin tolerance in mice. Shock. 2002;17:478–84. doi: 10.1097/00024382-200206000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Baykal A, Kaynaroglu V, Hascelik G, Sayek I, Sanac Y. Epinephrine and endotoxin tolerance differentially modulate serum cytokine levels to high-dose lipopolysaccharide challenge in a murine model. Surgery. 1999;125:403–10. [PubMed] [Google Scholar]
- 57.Natanson C, Hoffman WD, Suffredini AF, Eichacker PQ, Danner RL. Selected treatment strategies for septic shock based on proposed mechanisms of pathogenesis. Ann Intern Med. 1994;120:771–83. doi: 10.7326/0003-4819-120-9-199405010-00009. [DOI] [PubMed] [Google Scholar]
- 58.Koldzic-Zivanovic N, Tu H, Juelich TL, et al. Regulation of adrenal glucocorticoid synthesis by interleukin-10: a preponderance of IL-10 receptor in the adrenal zona fasciculata. Brain Behav Immun. 2006;20:460–8. doi: 10.1016/j.bbi.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Deans KJ, Haley M, Natanson C, Eichacker PQ, Minneci PC. Novel therapies for sepsis: a review. J Trauma. 2005;58:867–74. doi: 10.1097/01.ta.0000158244.69179.94. [DOI] [PubMed] [Google Scholar]
- 60.Meisel C, Schefold JC, Pschowski R, et al. GM-CSF to reverse sepsis-associated immunosuppression: a double-blind randomized placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180:640–8. doi: 10.1164/rccm.200903-0363OC. [DOI] [PubMed] [Google Scholar]