

Abstract
Twenty-four novel 2,5-diaryl-1,3,4-oxadiazoline analogs of combretastatin A-4 (CA-4, 1) were designed, synthesized and evaluated for biological activities. The compounds represent two structural classes; the Type I class has three methoxy groups on the A ring and the Type II class has a single methoxy group on the A ring. Biological evaluations demonstrate that multiple structural features control the biological potency. Four of the compounds, 2-(3’-bromophenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9l), 2-(2’,5’-dimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10h), 2-(3’,4’,5’-trimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10i) and 2-(3’,5’-dimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10j), have potent antiproliferative activities against multiple cancer cell lines. Mechanistic studies indicate that they retain the microtubule disrupting effects of compound 1 including microtubule loss, the formation of aberrant mitotic spindles, and mitotic arrest. Compound 10i inhibits purified tubulin polymerization and circumvents drug resistance mediated by P-glycoprotein and βIII tubulin expression. The oxadiazoline analog 10i is a promising lead candidate worthy of further investigation.
Introduction
A worldwide effort led by the US National Cancer Institute to identify anticancer agents from plants led to the identification of the combretastatins from the African bush willow Combretum caffrum.1, 2 Combretastatin A-4 (CA-4, 1, Figure 1) is the most potent of the natural combretastatins and early work showed it inhibits tubulin polymerization and the proliferation of murine and human cancer cells3, 4 Early reports of the antitumor activity of the more water soluble prodrug CA-4P (2) against murine P388 and B16 tumors were not encouraging.5 However, detailed mechanistic studies showed that compound 1 has specific effects on tumor vasculature, causing rapid vascular shutdown leading to central tumor necrosis.6, 7 The effects of prodrug 2 on tumor vasculature have been verified in human clinical trials.8, 9 While compound 2 causes central hemorrhagic necrosis in animal tumor models, a rim of viable tumor cells remains, suggesting that the optimal use of 2 would be in combination with agents that target the tumor periphery. Compound 2 is currently being evaluated in Phase II clinical trials for activity in anaplastic thyroid carcinoma, non-small cell lung cancer and ovarian cancer in combination with conventional cytotoxic drugs including paclitaxel, carboplatin and the antiangiogenic agent brevacizumab.10 Mechanistically, agent 1 rapidly alters HUVEC morphology causing cell contraction, membrane blebbing and disruption of endothelial monolayer permeability.11 These effects are potentially mediated by a Rho/Rho kinase stimulated increase in actin stress fibers. Prodrug 2 has also been shown to disrupt VE-cadherin signaling in HUVECs.12 Most recently, it was shown to rapidly upregulate connective tissue growth factor (CTGF) in microvascular endothelial cells in a cell density dependent manner.13 Upregulation of CTGF, a pleotropic endothelial cell growth factor, was observed only in subconfluent endothelial cell cultures or in endothelial cells growing as capillary-like tubules. Prodrug 2-mediated activation of the CTGF might explain why thin, unstabilized tumor blood vessels are so susceptible to 1 while other mature vessels are unaffected. It is not known which of these compound 1-mediated signaling events are responsible for the specific antivascular effects leading to rapid catastrophic tumor vascular shutdown.
Figure 1.

Structures of combretastatin-A4 (CA-4, 1), its water-soluble derivate (CA-4P, 2), its amino analogs (3 and 4), combretastatin-A1 (CA-1, 5), combretastatin-A1 diphosphate (CA-1P, 6), along with the pyrazole (7) and pyrazoline (8) analogs. Two general types of oxadiazoline analogs were generated, including the Type I compounds 9a–l and the Type II compounds 10a–l.
Significant efforts have been undertaken to develop analogs of compound 1 that retain the biological actions of the parent molecule but provide improved pharmacokinetic properties and better water solubility.14 In addition to prodrug 2, notable analogs undergoing clinical evaluations include AVE 8062 (3), the amino acid derivative of AC-7739 (4) and the CA-1 (5) derivative CA-1P (6) (OXI-4503).10, 15 Our work in this area has yielded several new classes of analogs including pyrazoles16 (7) and pyrazolines (8, Figure 1) of 1.17 These derivatives demonstrate a wide range of potencies. A significant drop in activity was observed in the pyrazole derivatives as compared to 1. X-ray crystallography studies of a pyrazole analog of compound 7 showed that it adopted a planar conformation, as opposed to the non-planar geometry of compound 1.16 The structure of 1 contains two substituted benzene rings, referred to as the A and B rings, which fit into the A and B pockets within the colchicines binding site on tubulin. Modeling studies of 1 within the colchicines binding site of tubulin suggest that a geometrically planar analog of 1 would not allow optimal interactions with this binding site.18, 19 It is reasonable to assume that the pyrazole’s loss of potency can be attributed to its incompatible conformation within the colchicines binding site of tubulin. Consistent with this relationship, pyrazolines have a less planar geometry than the pyrazoles and are more potent inhibitors of cell proliferation and anti-tubulin effects.
A new series of 2,5-diaryl-1,3,4-oxadiazoline analogs were designed to retain the geometric features of 1. Compared to the pyrazolines, (8), the oxadiazoline moiety should provide an optimal conformational geometry for interaction with the colchicine binding site while increasing the number of heteroatoms in the core structure. The net effect is an increase in the polarity of the molecule, which should enhance water solubility. Accordingly, twenty-four specific oxadiazoline analogs were synthesized and they are divided into two types (Figure 1). Type I compounds (oxadiazoline 9a–l) contain a 3,4,5-trimethoxyphenyl group on the A ring and Type II compounds (oxadiazoline 10a–l) contain a 4-methoxyphenyl moiety on the A ring. These analogs were synthesized and evaluated for biological activities. The most potent compounds were evaluated in mechanistic studies and attempts have been made to understand the structure activity relationships (SAR) among these compounds.
Chemistry
Oxadiazoline analogs were synthesized by adapting a procedure reported by Ali, Amer, and Abdel-Rahman.20 As an example, the reaction of a hydrazide (11) with a benzaldehyde (12) in reflux with water and glacial acetic acid in ethanol for 5 h produced the hydrazone intermediate 13 (Scheme 1). The reaction mixture was poured into ice-water and the precipitate was collected and dried. When needed, the hydrazones were purified by recrystallization in aqueous methanol. The yields of the hydrazones ranged between 60–98%. The chemical structures of the hydrazones were confirmed using 400 MHz 1H-NMR and IR. The oxadiazoline products were synthesized by refluxing the appropriate hydrazone intermediate in acetic anhydride for 1 h under dry conditions (Scheme 1). The reaction mixture was poured into ice-water and the solid product was collected and washed with water. Most products were found to be homogeneous by TLC and 400 MHz 1H-NMR analyses, but when needed, heterogeneous products were readily purified by silica gel column chromatography using a methanol/chloroform eluent. Oxadiazoline product yields ranged from 25%–70%. The structures of compounds 9a–l and 10a–l were confirmed using NMR, IR, mass spectrometry, and elemental analysis. The compounds that showed a small amount (<5 %) of rotational isomers (about the amide bond) were analyzed by reversed phase HPLC (Nova Pak C18 3.6 × 150 mm column using methanol as the eluent). Each of the compounds showed a single peak with a retention time of 2–3 minutes, confirming the purity of the compounds.
Scheme 1.

Representation of hydrazone intermediate and oxadiazoline synthesis.
To gain insight into the conformation of the oxadiazoline analogs, molecular modeling studies on compound 10i were conducted using MacSpartan, version ’04. The structure and conformation of 10i was first optimized using molecular mechanics (MMFF) and molecular dynamics (equilibrium conformer search option and molecular mechanics). The optimal structure was further optimized using Hartree-Fock (equilibrium geometry optimization using the 3–21 G basis set) followed by density function theory (equilibrium geometry optimization using the B3LYP and 6–31 G* basis set) calculations. The resulting conformation of analog 10i is shown in Figure 2A. It is evident that the oxadiazoline-containing compound adopts a non-planar geometry similar to the parent compound 1, unlike the previously reported pyrazole molecules. This result is consistent with earlier results from single x-ray diffraction studies indicating that the oxadiazoline structure adopts a puckered conformation.21 In addition, the conformation of cis-4,3’,4’,5’-tetramethoxystilbene, a dehydroxy analog of 1, was also ascertained using the aforementioned modeling method, and a model is given in Figure 2B. It is worthy to note that both 10i and cis-4,3’,4’,5’-tetramethoxystilbene possess a non-planar or “twisted” geometry similar to the pyrazolines (8),17 and they show resemblance to the conformation of 1 itself.16
Figure 2.

Molecular ball and stick model of oxadiazoline 10i (A) and cis-4,3’,4’,5’-tetramethoxystilbene (B). Black represents carbon, blue nitrogen, gray hydrogen, and red oxygen.
Biological Results and Discussion
Antiproliferative effects of oxadiazoline compounds on murine cancer cells
The antiproliferative effects of the 24 compounds were evaluated in murine L1210 leukemia cells and the IC50 values determined (Table 1). Additionally, the compounds were evaluated in murine B16 melanoma cells (data not shown) with general concordance with L1210 data. The results reveal several interesting points including the broad range of potencies in this series. The novel oxadiazoline compounds are substantially less potent than compound 1 itself, which has reported IC50 values ranging from 0.003–0.007 µM 3–5 for L1210 and B16 cells. A correlation between the potencies of the Type I and Type II compounds could be expected, but this pattern was not observed. Not only is there no trend in L1210 potency with A ring alterations but a single adjustment to the B ring rarely affects a Type I compound in the same way that it affects a Type II compound. For example, a 3’-acetyl, 4’-methoxy B ring substitution in 9e results in a compound with an IC50 of 3.9 µM while this same substitution in the Type II compound (10e) results in a totally inactive compound (IC50 > 100 µM). To explain these inconsistencies, one possibility is to consider that compound 1 can bind in two orientations within the colchicine binding site of tubulin. For this reason the SAR of analogs of 1 gains complexity, as the B ring could orient within the standard A ring pocket of the binding site, if it results in a more energetically favorable conformation. Consequently, the effect of a single ring manipulation was less predictable and the methodical alterations of the A and B ring substituents were considered.
Table 1.
Sensitivity of L1210 cells to oxadiazoline analogs.
| Type I | IC50 (µM) | Type II | IC50 (µM) | ||
|---|---|---|---|---|---|
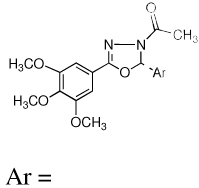 Ar = |
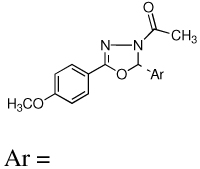 Ar = |
||||
| 9a | 4.2 ± 0.5 | 10a | 68.0 ± 4.9 | ||
| 9b | 39.0 ± 2.0 | 10b | 47.0 ± 2.9 | ||
| 9c | 58.0 ± 0.7 | 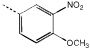 |
10c | >100 | |
| 9d | 57.0 ± 5.1 | 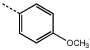 |
10d | >100 | |
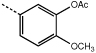 |
9e | 3.9 ± 0.4 | 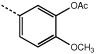 |
10e | >100 |
| 9f | 45.0 ± 8.6 | 10f | 41.0 ± 6.4 | ||
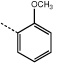 |
9g | 48.0 ± 1.0 | 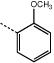 |
10g | 5.8 ± 0.6 |
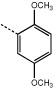 |
9h | 55.0 ± 4.5 | 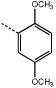 |
10h | 0.5 ± 0.05 |
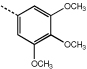 |
9i | >100 | 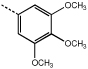 |
10i | 0.5 ± 0.06 |
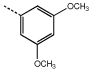 |
9j | 5.9 ± 1.6 | 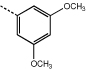 |
10j | 0.5 ± 0.1 |
| 9k | 6.4 ± 1.2 | 10k | >100 | ||
| 9l | 0.6 ± 0.7 | 10l | 37.0 ± 6.0 | ||
The standard deviation for the IC50 values is within 5–20% of the reported values.
Compound 9i, with 3”,4”,5”-trimethoxy A ring and 3’,4’,5’-trimethoxy B ring substitutions was found to be entirely inactive (IC50 >100 µM), suggesting that this symmetric compound with two trimethoxy phenyl groups does not interact well within the colchicine binding site. Compound 1,with the 3”,4”,5”-trimethoxy A ring is very potent,4 implying the unfavorable nature of the 3’,4’,5’-trimethoxy B ring. At the other extreme, 10d, a Type I compound with only a 4” methoxy on the A ring and a 4’-methoxy substitution on the B ring is also inactive (IC50 >100 µM).Interestingly, shifting the 4’-methoxy to the 3’ position (10f) on the Type II compound improves potency (IC50 41.0 µM). The methoxy group in the 2’ position gives the highest potency of these three positions (10g, IC50 5.8 µM). Together, these data suggest that a 4-methoxy substituted ring interacts poorly with one pocket of the binding site. Because of the potency of 10i (IC50 0.5 µM), which contains a trisubstituted methoxy ring that is predicted to only favorably interact with the A site, the single methoxy-substituted ring must favorably interact with the B site. This indicates that Type II compounds might position the 4”-methoxy A ring in the B ring pocket of the colchicine binding site on tubulin.
Compounds 10h and 10j have a 2’, 5’-dimethoxy and a 3’,5’-dimethoxy substituted B ring, respectively. These compounds are potent, with IC50 values of 500 nM. Compounds 9h and 9j have very different potencies (9h, IC50 55.0 µM; 9j, IC50 5.9 µM). The 2’,5’-dimethoxy B ring on 9h would be expected to interact poorly with the B ring pocket whereas the 3’,5’-dimethoxy B ring on 9j favorably interacts with the binding site.
The pattern observed with 10d, 10f, and 10g is not observed in the Type I compounds; 9d, 9f, and 9g and all have low potencies with IC50s of 57 µM, 45 µM and 48 µM, respectively. The 4’-methoxy B ring substitution is the least potent in both Type I and Type II compounds. In an attempt to improve 9d and 10d, the 4’-methoxy substitution was replaced with a 4’-nitro group (9a, 10a). This resulted in a modest improvement in potency of the Type II compound (10a, IC50 68.0 µM). For the Type I compound this change resulted in a dramatic, 13-fold increase in potency (9a IC50 4.2 µM). The Type I compound (9a) demonstrates that the 4’-nitro group substitution is substantially more favorable than the 4’-methoxy in the B ring pocket. Shifting the nitro group to the 3’ position, 10a to 10b, results in a slight increase in potency (10b, IC50 47.0 µM). In the Type I compound, however, this change in the position of the nitro group decreased potency 9-fold (9b, IC50 39.0 µM) suggesting that in the B ring pocket a p-nitro substitution is more favorable than an m-nitro substitution. The 3’-nitro substitution on compound 10b may orient into the A ring-pocket, improving interaction and biological potency. This is consistent with the pattern observed with 10d, 10f, and 10g, which suggests a preferable A pocket interaction with an ortho or meta substitution as compared to the para substitution.
Compounds 9c and 10c, with a 3’-nitro, 4’-methoxy B ring, are less potent than a single 3’-nitro substituted B ring. Replacing the 3’-nitro with a 3’-bromo group produces a more potent Type I compound (9b, IC50 39.0 µM; 9l, IC50 0.6 µM). In the Type II compounds, the 3’-nitro a 3’-bromo group (10b, IC50 47.0 µM; 10l, IC50 37 µM) have similar activities. Compounds 9k and 10k have no substitution on the B ring and they have dramatically different potencies (9k, IC50 6.4 µM; 10k, IC50 >100 µM). Compound 9k could favorably position the non-substituted ring in the B ring pocket, however the low potency of 10k (> 100 µM) is consistent with the notion that the Type I and Type II compounds might orient differently within the colchicine binding site.
Antiproliferative effects against MDA-MB-435
In the development of chemical agents with potential application in human anticancer therapeutics, it was important to determine the activity of the most potent oxadiazolines in human cancer cells. The five most potent inhibitors of L1210 cell growth were evaluated in our reference human cell line, MDA-MB-435, a cell line originally designated as originating from breast but which has been shown definitively to be derived from the M14 melanoma cell line.22 The results, presented in Table 2 show that there is excellent correlation between the L1210 and MDA-MB-435 potencies, with 4 of the 5 compounds having sub-micromolar potency against human cancer cells. These compounds were evaluated further in mechanistic assays to determine if they retain the microtubule disrupting and antimitotic effects of the combretastatins.
Table 2.
Antiproliferative and microtubule depolymerizing effects of oxadiazoline analogs
| Compound | IC50 (µM) | EC50 (µM) | Ratio |
|---|---|---|---|
| ± SD | Microtubule | EC50/IC50 | |
| MDA-MB-435 | Depolymerization | ||
| 9e | 3.3 ± 0.2 | 10.2 | 3.1 |
| 9l | 0.15 ± 0.01 | 1.5 | 10.0 |
| 10h | 0.20 ± 0.02 | 1.3 | 6.5 |
| 10i | 0.14 ± 0.01 | 0.5 | 3.6 |
| 10j | 0.11 ± 0.01 | 1.0 | 9.1 |
| 1 | 0.003 ± .0002 | 0.007 | 2.3 |
The antiproliferative actions (IC50) of the oxadiazoline analogs and compound 1 were measured using the SRB assay (n=3–5) and EC50 values using indirect immunofluorescence techniques in at least 3 independent experiments.
Microtubule depolymerizing activity
With the goal of ascertaining the mechanism of action of the oxadiazoline compounds, the effects of 9e and 9l, 10h, 10i, 10j on interphase cellular microtubules were evaluated in A-10 smooth muscle cells. This cell line is valuable because the cells are relatively large and flat and they arrest in interphase in response to tubulin binding antimitotics, consistent with the responses of other smooth muscle cells to these drugs.23 All of the compounds caused a dose dependent loss of cellular microtubules, similar to the effects of 1. Representative normal interphase microtubules and the microtubule depolymerizing effects of compound 10i are shown in Figure 3. A range of concentrations was evaluated for each compound and the EC50, the concentration required to cause 50% loss of cellular microtubules, as visualized by microscopy, was calculated (Table 2). The ratio of EC50 for microtubule depolymerization and the IC50 for inhibition of proliferation provides an indication of tight linkage of antiproliferative effects and the compound’s tubulin-dependent mechanisms of action.24, 25 Compound 10i had a ratio of 3.5, 9e a ratio of 3.1, 10h a ratio of 6.5 and 1 a ratio of 2.3 (Table 2). The ratios are within the ranges we have seen with 2-methoxyestradiol analogs24 and a large subset of polysubstituted pyrrole compounds.25 Interestingly, the other potent antiproliferative compounds, and 10j and 9l did not demonstrate correspondingly potent microtubule depolymerization effects, and their ratios of EC50/IC50 ratios were 8.8 and 10 respectively (Table 2). This suggests that these compounds might exert cytotoxicity through mechanisms in addition to tubulin inhibition as has been shown with similar molecules that can bind directly to DNA.26
Figure 3.

Effects of 10i on cellular microtubules. A-10 cells were treated for 18 h with vehicle (A) or 1 µM 10i (B) and cellular microtubule structures visualized with indirect immunofluorescence techniques. Normal interphase microtubules are visible in panel A. The 10i-induced loss of interphase microtubules is shown in panel B.
Interaction of compound 10i with purified bovine brain tubulin
The potent antiproliferative and microtubule depolymerizing effects prompted further mechanistic studies on 10i. The effects of 10i on purified tubulin polymerization were evaluated in a biochemical assay. In this assay, 10i caused a concentration-dependent inhibition of tubulin polymerization (Figure 4). The concentration required to cause 50% inhibition of tubulin polymerization was 9.6 µM. In similar assays 2–3 µM of 1 is needed to cause this effect.4 The tubulin data are consistent with the lower potency of 10i for cellular microtubule and antiproliferative effects and strongly implicate a direct interaction of 10i with tubulin.
Figure 4.

In vitro tubulin polymerization assays. The polymerization of bovine brain tubulin was monitored turbidimetrically by absorbance at 340 nm at 37°C in the presence of a range of 10i concentrations.
Antimitotic effects of compound 10i
In addition to causing the loss of cellular microtubules, colchicine, compound 1 and their analogs initiate the formation of highly abnormal mitotic spindles.27–29 The effects of 10i on mitotic spindles were evaluated in HeLa cells. HeLa cells, unlike the smooth muscle cells, arrest in the G2/M phase of the cell cycle in response to tubulin binding drugs.28, 29 Microtubules were visualized 18 h after treatment with vehicle or 0.5 µM 10i. Normal bipolar mitotic spindles emanating from γ-tubulin-containing centrosomes were observed and they aligned the chromosomes in the metaphase plate (Figure 5A). In contrast, compound 10i initiated the formation of highly aberrant, multipolar mitotic spindles (Figure 5B). At least 7 mitotic spindles, 3 of which contained γ-tubulin, were visible in a single representative focal plane (Figure 5B). The DNA was not well organized into a metaphase plate, suggesting these cells did not have the potential to successfully divide into two equivalent daughter cells with normal complements of DNA.
Figure 5.

Effects of 10i on mitotic spindles and cell cycle distribution. HeLa cells were treated with vehicle (A) or 0.50 µM 10i (B) for 18 h, fixed and cellular structures visualized by indirect immunofluorescence. Cellular microtubules were visualized with a β-tubulin antibody (green), centrosomes using a γ-tubulin antibody (red), and DNA using DAPI (blue). For the cell cycle distribution studies, MDA-MB-435 cells were treated for 18 h with vehicle (A) or 0.50 µM 10i then stained with Kirshan’s reagent and evaluation by flow cytometry.
The cell cycle distribution of cells treated with 10i was further evaluated by flow cytometry to determine whether the formation of abnormal mitotic spindles caused cells to accumulate in the G2/M phase of the cell cycle. MDA-MB-435 cells were treated for 18 h with vehicle or 0.5 µM 10i. A normal cell cycle distribution for MDA-MB-435 cells was seen in the vehicle treated cells (Figure 5C). In contrast, in the 10i-treated cells complete G2/M accumulation occurred, indicative of mitotic blockade. Paclitaxel caused an identical effect (data not shown). These data, combined with the cellular microtubule depolymerizing effects and the ability of 10i to directly inhibit purified tubulin assembly, provide strong evidence that this oxadiazoline retains the cellular anti-tubulin mechanisms of action of 1.
Compound 10i can circumvent clinically relevant mechanisms of drug resistance
One major limitation of anticancer drugs is intrinsic or acquired multidrug resistance. Tumor expression of the ATP-dependent drug efflux pump, P-glycoprotein (Pgp) is associated with anticancer treatment failure30. A second clinically relevant mechanism of drug resistance to tubulin-targeted agents, including the taxanes and vinorelbine, is the expression of the βIII isotype of tubulin.31 Drugs that can overcome these mechanisms of resistance might have advantages in the treatment of drug resistant tumors. The ability of the oxadiazoline 10i to overcome these mechanisms of resistance was tested in isogenic cell line pairs. The sensitivity of the Pgp expressing SK-OV-3-MDR-6-1-6/6 cell line was compared to the sensitivity of the parental cell line, SK-OV-3.32 IC50s were measured for each cell line and relative resistance (Rr) values were calculated by dividing the IC50 of the genetically engineered cell line by the IC50 of the parental cell line. In this cell line pair, the Pgp substrate paclitaxel has a Rr of 860 and the non-Pgp substrate 2-methoxyestradiol had an Rr value of 2.6.32 As shown in Table 3, Compound 1 has a Rr of 1.8 and compound 10i a Rr of 1.3, suggesting that 10i, like 1, is a poor substrate for transport by Pgp. The effect of βIII tubulin expression was evaluated using the WTβIII and HeLa cell line pair.32 In this cell line pair paclitaxel, which is susceptible to βIII-mediated resistance, has a Rr of 4.7 while 2-methoxyestradiol, which is unaffected by βIII expression, has a Rr of 1.0.32 The Rr value for compound 10i was 1.2 and a Rr of 1.1 was measured for compound 1 in these cell lines (Table 3). These data suggest that the oxadiazoline 10i retains the ability of 1 to overcome drug resistance mediated by the expression of Pgp and the βIII isotype of tubulin. Therefore, these drugs would be expected to have advantages in the treatment of multidrug resistant tumors.
Table 3.
Sensitivity of cell lines expressing Pgp and βIII tubulin
| Cell line | IC50 (µM) ± SD | IC50 (µM) ± SD |
|---|---|---|
| 1 | 10i | |
| SK-OV-3 | 0.0033 ± 0.0002 | 0.34 ± 0.01 |
| SK-OV-3-MDR 1–6/6a | 0.0035 ± 0.0005 | 0.45 ± 0.01 |
| Rrc | 1.8 | 1.3 |
| HeLa | 0.0029 ± 0.0003 | 0.23 ± 0.03 |
| WTβIIIb | 0.0033 ± 0.0008 | 0.27 ± 0.02 |
| Rrc | 1.1 | 1.2 |
SK-OV-3-MDR 1–6/6 cells were generated from SK-OV-3 by adenoviral expression of MDR-1.
HeLa cells were transfected with the βIII tubulin isotype to generate the WTβIII cells.
The Rr values were calculated by dividing the IC50 of the genetically engineered line by the IC50 of the parental cell line.
Analytical measurement to estimate aqueous solubility
One major limitation of the combretastatins compounds 1 and 6 (CA-1) is low aqueous solubility; this led to the synthesis and use of the more water soluble prodrugs 2 and 6. Excellent water solubility would provide a significant advantage for new analogs of 1. There was a good expectation that the increased number of heteroatoms in the core structure of the oxadiazoline analogs would facilitate aqueous solubility. An analytical assay derived from the shake-flask solubility method was used to evaluate the relative aqueous solubility of the most potent oxadiazoline analogs. This assay is rapid and highly reproducible, requires only small quantities of material and provides good, but not perfect, correlation with the shake-flask solubility method. The results, (Table 4) show that there was a wide range of aqueous solubilities in this subset of compounds. Compounds 10h and 10i have aqueous solubilities in the same range as compound 1, while compounds 9e, 9l and 10j have lower aqueous solubility.
Table 4.
Relative aqueous solubility of oxadiazoline analogs.
| Compound | Aqueous Solubility (µM) |
|---|---|
| 9e | 112 |
| 9l | 66 |
| 10h | 259 |
| 10i | 257 |
| 10j | 94 |
| 1 | 350 |
The relative aqueous solubilities were measured using a protocol based on Millipore’s MultiScreen aqueous solubility filter plate assay. Maximal solubility in this assay is 500 µM.
In summary, the synthesis of novel 2,5-diaryl-1,3,4-oxadiazolines as anti-tubulin, antimitotic cytotoxins is described. Compound 10i exhibits promising antiproliferative activities and a mechanism of action identical to prodrug 2 in vitro. The promising activities of this compound will be investigated further with in-vivo murine antitumor experiments, which will be reported in due course.
Experimental Section
Solvents and organic reagents were purchased from Aldrich or Fisher, and were used without further purification. Melting points (mp) were performed using a Mel-temp instrument and the results were uncorrected. Infrared (IR) spectra were recorded with a Midac M1700 FT-IR instrument as films on KBr discs unless stated otherwise. Proton NMR spectra were recorded on a Varian INOVA 400 MHz Fourier transform spectrometer using an internal deuterium lock. Chemical shifts are quoted in parts per million downfield from tetramethylsilane. High-resolution mass spectra (HRMS) and low-resolution mass spectra (LRMS) were provided by the Mass Spectrometry Laboratory, University of South Carolina, Columbia, SC. Elemental analyses were performed by Midwest Microlab, LLC, Indianapolis, IN. Reactions were monitored using thin-layer chromatography (TLC) using commercially available pre-coated plates (Merck Kieselgel 60 F254 silica). Visualization was achieved with UV light at 254 nm or I2 vapor staining. In addition to NMR and elemental analysis, HPLC analysis was used to determine the purity (>95%) of the compounds. Compounds were dissolved in methanol (1.5 mL). A reversed phase Nova Pak C18 3.9 × 150 mm column attached to a Perkin Elmer Series 200 pump coupled to a Hitachi uv-vis detector was used. Each sample was injected at a volume of 5 µL, the eluent was methanol and the flow rate was 0.5 mL/min.
Representative experimental procedure for the synthesis of oxadiazoline 9d via hydrazone intermediate 13
To a stirred solution of hydrazide 11 (226 mg, 1 mmol) and p-anisaldehyde (0.12 mL, 1 mmol) in ethanol (25 mL), water (5 mL) was added followed by drop-wise addition of glacial acetic acid (0.2 ml). The resulting mixture was refluxed for 5 hours, after which the solution was poured into ice water. The mixture was stirred until a precipitate formed which was collected using suction filtration and dried followed by recrystallization in aqueous methanol gave hydrazone 13 as a pale yellow solid (0.19 g, 54%); RF [methanol:chloroform (2:98)] 0.47; m.p. 74–76°C; δH (400 MHz, CDCl3) 8.66 (1 H, s br), 8.26 (1 H, s), 7.53 (2 H, d, J = 8.4), 7.24 (1 H, s), 7.06 (1 H, s), 6.94 (2 H, d, J = 8.4), 3.93 (9 H, s) and 3.86 (3 H, s); υmax (CH2Cl2)/cm−1 3484, 1645, 1501, 1332, 1126 and 735; LRMS (EI+) m/z (rel intensity) 344 (20%, M+), 211 (35%) and 195 (100%); HRMS Found 344.1373 (M+, C18H20N2O5+ requires M+ 344.1372). 2-(4’-Methoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline 9d was synthesized by addition of acetic acid (2 mL) to hydrazone 13 (100 mg, 0.29 mmol), the resulting solution was heated to reflux for 1 hour. The reaction mixture was poured into ice water and the resulting solid product filtered and washed with copious amounts of water followed by drying to give 9d as a white solid (74 mg, 66%); RF [methanol:chloroform (2:98)] 0.31; m.p.117–119 °C; δH (400 MHz, CDCl3) 7.41 (2 H, dt, J = 8.8, 3.2), 7.11 (2 H, s), 7.02 (1 H, s), 6.92 (2 H, dt, J = 8.8, 3.2), 3.91 (9 H, s), 3.81 (3 H, s) and 2.35 (3 H, s); υmax (KBr disc)/cm−1 2939, 1667, 1583, 1462, 1416, 1233, 1127, 1001, 843 and 694; LRMS (EI+) m/z (rel intensity) 386 (30%, M+) and 195 (100%); HRMS 386.1484 (M+, C20H22N2O5+ requires 386.1478).
2-(4’-Nitrophenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9a)
Pale yellow solid (47 mg, 73%); RF [methanol:chloroform (2:98)] 0.27; m.p. 141–142 °C; δH (400 MHz, CDCl3) 8.28 (2 H, d, J = 8.8), 7.69 (2 H, d, J = 8.8), 7.14 (1 H, s), 7.11 (2 H, s), 3.95 (3 H, s), 3.94 (6 H, s) and 2.38 (3 H, s); υmax (KBr disc)/cm−1 2940, 2847, 1666, 1584, 1525, 1505, 1416, 1175, 1233, 1129 1020, 910 and 731; LRMS (EI+) m/z (rel intensity) 401 (25%, M+), 195 (100%).
2-(3’-Nitrophenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9b)
White powder (73 mg, 65%); RF [methanol:chloroform (2:98)] 0.44; m.p. 64–66 °C; δH (400 MHz, CDCl3) 8.35 (1 H, s), 8.28 (1 H, d, J = 8.4), 7.87 (1 H, d, J = 8.4), 7.65 (1 H, t, J = 8.4), 7.14 (1 H, s), 7.12 (2 H, s), 3.92 (6 H, s), 3.91 (3 H, s) and 2.39 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1662, 1584, 1536, 1504, 1462, 1416, 1351, 1130, 1001, 733 and 633; LRMS (EI+) m/z 401 (25%, M+), 359 (20%) and 195 (100%).
2-(3’-Nitro-4-methoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9c)
White solid (85 mg, 75%); RF [methanol:chloroform (2:98)] 0.35; m.p. 176–178 °C; δH (400 MHz, CDCl3) 7.95 (1 H, d, J = 2.4), 7.71 (1 H, dd, J = 8.4, 2.4), 7.14 (1 H, d, J = 8.4), 7.10 (2 H, s), 7.04 (1 H, s), 3.98 (3 H, s), 3.92 (6 H, s), 3.91 (3 H, s) and 2.37 (3 H, s); υmax (KBr disc)/cm−1 2917, 2846, 1666, 1622, 1578, 1540, 1500, 1468, 1420, 1359, 1250, 1233, 1135, 1001 and 733; LRMS (ES+) m/z 441 (25%, M+), 389 (15%) and 195 (100%).
2-(3’-Acetoxy-4’-methoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9e)
White powder (73 mg, 65%); RF [methanol:chloroform (2:98)] 0.35; m.p. 117–119 °C; δH (400 MHz, CDCl3) 7.38 (1 H, d, J = 8.4), 7.14 (1 H, s), 7.11 (2 H, s), 7.01 (1 H, s), 6.98 (1 H, d, J = 8.4), 3.92 (6 H, s), 3.91 (3 H, s), 3.83 (3 H, s), 2.35 (3 H, s) and 2.30 (3 H, s); υmax (KBr disc)/cm−1 3100, 2940, 2841, 1769, 1666, 1583, 1513, 1463, 1416; 1271, 1200, 1126, 1002 and 733; LRMS (ES+) m/z (rel intensity) 444 (30%, M+), 402 (15%) and 195 (100%). Anal. (C22H24N2O8.H2O) C. H.
2-(3’-Methoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9f)
Yellow solid (16 mg, 14%); RF [methanol:chloroform (2:98)] 0.45; m.p. 135–137 °C; δH (400 MHz, CDCl3) 7.36 (1 H, s), 7.33 (1 H, t, J = 8.0), 7.11 (2 H, s), 7.09 (1 H, d, J = 8.0), 7.03 (1 H, d, J = 8.0), 6.94 (1 H, d, J = 8.0), 3.91 (6 H, s), 3.90 (3 H, s), 3.82 (3 H, s) and 2.39 (3 H, s); υmax (KBr disc)/cm−1 2917, 2848, 1662, 1613, 1583, 1515, 1463, 1416, 1130, 1033, 732 and 696; LRMS (EI+) m/z (rel intensity) 386 (25%, M+), 344 (15%) and 195 (100%).
2-(2’-Methoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9g)
Yellow powder (63 mg, 56%); RF [methanol:chloroform (2:98)] 0.35; m.p 151–153 °C; δH (400 MHz, CDCl3) 7.38 (1 H, t, J = 8.0), 7. 37 (1 H, s), 7.32 (1 H, d, 8.0), 7.10 (2 H, s), 6.96 (1 H, d, J = 8.0), 6.98 (1 H, t, J = 8.8), 3.91 (6 H, s), 3.90 (3 H, s), 3.88 (3 H, s) and 2.38 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1666, 1583, 1463, 1416, 1248, 1129, 911, 732 and 668; LRMS (EI+) m/z (rel intensity) 386 (M+, 35%) and 195 (100%).
2-(2’,5’-Dimethoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9h)
White powder (40 mg, 36%); RF [methanol:chloroform (2:98)] 0.37; m.p 187–189 °C; δH (400 MHz, CDCl3) 7.33 (1 H, s), 7.09 (2 H, s), 6.90 (2 H, s), 6.87 (1 H, s), 3.94 (3 H, s), 3.91 (6 H, s), 3.89 (3 H, s), 3.75 (3 H, s) and 2.38 (3 H, s); υmax (KBr disc)/cm−1 2917, 2848, 1665, 1613, 1583, 1515, 1462, 1416, 1130, 1033 and 732; LRMS (EI+) m/z (rel intensity) 416 (M+, 20%) and 195 (100%). Anal. (C21H24N2O7) C. H.
2-(3’,4’,5’-trimethoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9i)
White solid (79 mg, 68%); RF [methanol:chloroform (2:98)] 0.25; m.p. 162–163 °C; δH (400 MHz, CDCl3) 7.12 (2 H, s), 6.87 (1 H, s), 6.70 (2 H, s), 3.93 (3 H, s), 3.91 (6 H, s), 3.87 (6 H, s), 3.85 (3 H, s) and 2.39 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1666, 1582, 1503, 1331, 1234, 1127, 911 and 730; LRMS EI m/z (rel intensity) 466 (M+, 25%) and 195 (100%).
2-(3’,5’-dimethoxyphenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9j)
White powder (22 mg, 20%); RF [methanol:chloroform (2:98)] 0.46; m.p. 80–82 °C; δH (400 MHz, CDCl3) 7.11 (2 H, s), 6.99 (1 H, s), 6.64 (2 H, d, J = 2.0), 6.48 (1 H, t, J 2.0), 3.93 (6 H, s), 3.91 (3 H, s), 3.79 (6 H, s) and 2.37 (3 H, s,, Ac); υmax (KBr disc)/cm−1 2917, 2848, 2359, 2342, 1662, 1583, 1416, 1250, 1158,1173, 909 and 732; LRMS (ES+) m/z (rel intensity) 416 (M+, 29%) and 195 (100%).
2-Phenyl-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9k)
White solid (77 mg, 77%); RF [methanol:chloroform (2:98)] 0.75; m.p. 175–177 °C; δH (400 MHz, CDCl3) 7.49 (2 H, dd, J = 7.6, 2.4), 7.42 (3 H, m), 7.13 (2 H, s), 7.07 (1 H, s), 3.94 (6 H, s), 3.91 (3 H, s) and 2.37 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1667, 1582, 1462, 1416, 1250, 1127 and 762; LRMS (EI+) m/z 356 (rel intensity) (M+, 35%), 314 (15%) and 195 (100%).
2-(3’-Bromophenyl)-5-(3”,4”,5”-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9l)
Off-white solid (60 mg, 54%); RF [methanol:chloroform (2:98)] 0.77; m.p. 134–136 °C; δH (400 MHz, CDCl3) 7.61 (1 H, s), 7.54 (1 H, d, J = 8.0), 7.44 (1 H, d, J = 8.0), 7.29 (1 H, t, J 8.0), 7.11 (2 H, s), 7.01 (1 H, s), 3.92 (6 H, s), 3.91 (3 H, s) and 2.38 (3 H, s); υmax (KBr disc)/cm−1 2916, 2850, 1585, 1557, 1504, 1416, 1331, 1233, 1128 and 733; LRMS (EI+) m/z 434 ( M+, 17%), 392 (10%) and 195 (100%); HRMS 434.0470 (M+, C19H19BrN2O5+ requires 434.0478). Anal. (C19H19Br N2O5) C. H.
2-(4’-Nitrophenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10a)
Yellow powder (50 mg, 44%); RF [methanol:chloroform (2:98)] 0.69; m.p. 99–101 °C; δH (400 MHz, CDCl3) 8.26 (2 H, d, J = 8.9), 7.84 (2 H, d, J = 8.9), 7.69 (2 H, d, J = 8.8), 7.13 (1 H, s), 6.97 (2 H, d, J = 8.8), 3.89 (3 H, s) and 2.36 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1666, 1608. 1520, 1536, 1414, 1367, 1257, 1174, 1066 and 1027; LRMS (EI+) m/z 341 (M+, 12%), 299 (10%) and 135 (100%).
2-(3’-Nitrophenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10b)
Yellow solid (82 mg, 73%); RF [methanol:chloroform (2:98)] 0.51; m.p. 144–146 °C; δH (400 MHz, CDCl3) 8.34 (1 H, s), 8.26 (1 H, d, J = 8.0), 7.87 (1 H, d, J = 8.0), 7.85 (2 H, d, J = 8.8), 7.61 (1 H, t, J = 8.0), 7.13 (1 H, s), 6.97 (2 H, d, J = 8.8), 3.88 (3 H, s, OCH3; ArB) and 2.37 (3 H, s, CH3; Ac); υmax (KBr disc)/cm−1 2916, 2848, 2360, 1665, 1620, 1585, 1540, 1420, 1367, 1275, 1250, 1240, 1130, 1011 and 735; LRMS m/z 341 (M+, 12%), 299 (10%) and 135 (100%).
2-(3’-Nitro-4’-methoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10c)
White powder (27 mg, 24%); RF [methanol:chloroform (2:98)] 0.47; m.p. 146–148 °C; δH (400 MHz, CDCl3) 7.96 (1 H, s), 7.83 (2 H, d, J = 8.8), 7.72 (1 H, d, J = 8.6), 7.13 (1 H, d, J = 8.6), 6.98 (1 H, s), 6.97 (2 H, d, J = 8.8), 3.98 (3 H, s), 3.88 (3 H, s) and 2.36 (3 H, s, CH3); υmax (KBr disc)/cm−1 2920, 2850, 1667, 1622, 1583, 1536, 1462, 1416, 1279, 1249, 1230, 1087, 1011 and 735; LRMS (ES+) m/z (rel intensity) 371 (M+, 12%), 329 (8%) and 135 (100).
2-(4’-Methoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10d)
Yellow oil (108 mg, 95%); RF [methanol:chloroform (2:98)] 0.37; δH (400 MHz, CDCl3) 7.83 (2 H, d, J = 8.8), 7.41 (2 H, d, J = 8.8), 7.02 (1 H, s), 6.95 (2 H, d, J = 8.8), 6.91 (2 H, d, J = 8.8), 3.89 (3 H, s), 3.80 (3 H, s) and 2.34 (3 H, s); υmax (KBr disc)/cm−1 2919, 2849, 1630, 1461, 1255, 908 and 733; LRMS (EI+) m/z (rel intensity) 326 (M+, 18%), 282 (13%) and 135 (100%).
12-(3’-Acetoxy-4’-methoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10e)
Colorless oil (68 mg, 59%); RF [methanol:chloroform (2:98)] 0.40; δH (400 MHz, CDCl3) 7.82 (2 H, d, J = 8.9), 7.38 (1 H, d, J = 8.4), 7.16 (1 H; s), 7.02 (1 H, s), 6.96 (2 H, d, J = 8.9), 6.86 (1 H, d, J = 8.4), 3.87 (3 H, s), 3.83 (3 H, s), 2.33 (3 H) and 2.29 (3 H, s); υmax (KBr disc)/cm−1 3250, 2942, 2840, 1770, 1670, 1580, 1513, 1463, 1420, 1271, 1200, 1130, 1001 and 733; LRMS (EI+) m/z (rel intensity) 384 (M+, 15%), 342 (15%), 151 (10%) and 135 (100%). Anal. (C20H20N2O6.2H2O) C. H.
2-(3’-Methoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10f)
Colorless oil (70 mg, 61%); RF [methanol:chloroform (2:98)] 0.44; δH (400 MHz, CDCl3) 7.84 (2 H, d, J = 8.8), 7.32 (1 H, t, J = 8.0), 7.07 (1 H; d, J = 8.0), 7.02 (1 H, s), 7.01 (1 H, s), 6.94 (2 H, d, J = 8.8) 6.92 (1 H, d, J = 8.0), 3.86 (3 H, s, OCH3; ArA/B), 3.83 (3 H, s, OCH3; ArA/B) and 2.33 (3 H, s, CH3; Ac); υmax (KBr disc)/cm−1 2916, 2848, 1666, 1463, 1416, 1248, 1129, 1027, 911 and 735; LRMS (EI+) m/z (rel intensity) 326 (M+, 20%) and 135 (100%).
2-(2’-Methoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10g)
White solid (290 mg, 93%); RF [methanol:chloroform (2:98)] 0.49; m.p. 187–189 °C; 7.80 (2 H, d, J = 8.8), 7.37–7.28 (2 H, m), 6.97 (1 H, s), 6.96–6.89 (2 H, m), 6.91 (2 H, d, J = 8.8), 3.86 (3 H, s), 3.84 (3 H, s) and 2.37 (3 H, s); υmax (KBr disc)/cm−1 2916, 2848, 1666, 1583, 1463, 1416, 1249, 1129, 910, 732 and 669; LRMS (EI+) m/z (rel intensity) 326 (M+, 20%) and 135 (100%).
2-(2’,5’-Dimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10h)
Colorless oil (65 mg, 57%); RF [methanol:chloroform (2:98)] 0.56; δH (400 MHz, CDCl3) 7.80 (2 H, d, J = 8.8), 7.31 (1 H, s), 6.92 (2 H, d, J = 8.8), 6.88 (2 H, s), 6.85 (1 H, s), 3.86 (3 H, s), 3.83 (3 H, s), 3.73 (3 H, s) and 2.33 (3 H, s); υmax (KBr disc)/cm−1 2920, 2850, 1660, 1614, 1580, 1515, 1461, 1250, 1235, 1173, 1129, 909 and 732; LRMS (EI+) m/z (rel intensity) 356 (M+, 20%) and 135 (100%); HRMS 356.1365 (M+, C19H20N2O5+ requires 356.1372). Anal. (C19H20N2O5) C. H.
2-(3’,4’,5’-Trimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10i)
White powder (60 mg, 54%) RF [methanol:chloroform (2:98)] 0.31; m.p. 148–150 °C; δH (400 MHz, CDCl3) 7.84 (2 H, d, J = 8.8), 6.97 (1 H; s), 6.95 (1 H; s), 6.92 (2 H, d, J = 8.8), 6.71 (1 H, s), 3.91 (3 H, s), 3.89 (3 H, s), 3.87 (3 H, s), 3.84 (3 H, s) and 2.38 (3 H, s); υmax (KBr disc)/cm−1 2918, 2849, 1610, 1503, 1463, 1327, 1257, 1174, 1128, 908 and 733; LRMS (EI+) m/z (rel intensity) 386 (M+, 25%) and 135 (100%); HRMS 386.1484 (M+, C20H22N2O6+ requires 386.1478). Anal. (C20H22N2O6) C. H.
2-(3’,5’-Dimethoxyphenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10j)
White solid (76 mg, 73%); RF [methanol:chloroform (2:98)] 0.48; m.p. 98–100 °C; δH (400 MHz, CDCl3) 7.82 (2 H, d, J = 8.8), 6.98 (1 H, s), 6.94 (2 H, d, J = 8.8), 6.63 (2 H, d, J = 2.4), 6.47 (1 H, t, J = 2.4, CH), 3.86 (3 H, s), 3.78 (6 H, s) and 2.36 (3 H, s); υmax (KBr disc)/cm−1 2917, 2849, 1731, 1609, 1456, 1257, 1158, 908 and 732; LRMS (EI+) m/z (rel intensity) 356 (30%, M+), 314 (6%), 179 (8%) and 135 (100%); HRMS 356.1376 (M+, C19H20N2O5+ requires 356.1372).
2-Phenyl-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10k)
White powder (51 mg, 44%); RF [methanol:chloroform (2:98)] 0.65; m.p. 149–151 °C; δH (400 MHz, CDCl3) 7.85 (2 H, d, J = 8.8), 7.48 (2 H, dd, J = 4.8, 8.0), 7.41-7.28 (3 H, m), 7.07 (1 H, s), 6.95 (2 H, d, J = 8.8), 3.87 (3 H, s) and 2.36 (3 H, s); υmax (KBr disc)/cm−1 2917, 2849, 1630, 1606, 1510, 1461, 1312, 1257, 1174, 908 and 733; LRMS (EI+) (rel intensity) m/z 296 ( M+, 10%), 254 (8%), 151 (18%) and 135 (100%). Anal. (C17H16N2O3) C. H.
2-(3’-Bromophenyl)-5-(3”-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10l)
White powder (56 mg, 63%); RF [methanol:chloroform (2:98)] 0.39; m.p. 115–117 °C; δH (400 MHz, CDCl3) 7.84 (2 H, d, J = 8.8), 7.61 (1 H, s), 7.53 (1 H, d, J = 8.0), 7.45 (1 H, d, J = 8.0), 7.28 (1 H, t, J = 8.0), 7.01 (1 H, s), 6.96 (2 H, d, J = 8.8), 3.88 (3 H, s) and 2.36 (3 H, s); υmax (KBr disc)/cm−1 2917, 2849, 1644, 1557, 1509, 1462, 1425, 1364, 1316, 1256, 1174, 908 and 733; LRMS (EI+) m/z (rel intensity) 296 ( M+, 10%), 254 (8%), 151 (18%) and 135 (100%).
Inhibition of L1210 and B16 cell growth
Murine leukemia L1210 cells were obtained from American Type Tissue Culture Collection (ATCC). The murine melanoma B16-F0 cell line was obtained from the Cancer Center of the Greenville Hospital System. The cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM, Atlanta Biochemicals) supplemented with 10% fetal bovine serum, HEPES buffer (2 mM, Mediatech Cellgro), L-Glutamine (2 mM, Mediatech Cellgro), and penicillin/streptomycin (50,000 units penicillin, 50,000 mg streptomycin, Atlanta Biologicals). Cells were maintained at 37°C in a 5% humidified CO2 atmosphere. Cultured cells (8,000 cells/well) were plated into 96 well flat-bottom tissue culture plates. The drug solutions were diluted in DMEM and added to each well (5 µL/well). Quadruplicate wells were prepared for each drug concentration. The plates were incubated for 72 h at 37°C in a 5% CO2 atmosphere. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) was dissolved in PBS (5 mg/ml). After the indicated cell incubation period, 10 µL stock MTT solution was added to each well, and the plates were further incubated for 4 h at 37 °C in a 5% CO2 atmosphere. After this final incubation, 100 µL acidic isopropanol solution (16 µL 12.1 N HCl in 5 mL isopropanol) was added to each well. The contents of the wells were mixed and the plates developed at room temperature for 15 min. The plates were then read on a Dynatech Plate Reader, utilizing Dynex Revelation 3.2 software, at a test wavelenth of 570 nm and a reference wavelength of 630 nm. Dose response curves were generated and the IC50 values were extrapolated from curves.
Sulforhodamine B (SRB) Assay
The antiproliferative effects of the oxadiazoline compounds against human cancer cells were evaluated using the SRB assay33 as previously described.34 The sources of the MDA-MB-435, SK-OV-3, SK-OV-3 MDR-1–6/6, HeLa, and WTβIII cell lines and the procedures used to generate the drug resistant clones have been described.32 Cells were incubated with compounds for 48 h. Each IC50 value represents the mean of 3–5 independent experiments, each performed in triplicate.
Indirect immunofluorescence and calculation of EC50 values
Cellular structures were visualized in A-10 smooth muscle cells using indirect immunofluorescence techniques as previously described.34 Cells were treated for 18–24 h with vehicle, compound 1 or a wide range of concentrations of the oxadiazoline analogs. Microtubules were visualized with a β-tubulin antibody, centrosomes with a γ-tubulin antibody and DNA with DAPI. Images were acquired with a digital camera and compiled and colorized using NIS Elements AR 3.0 software. The EC50 values for cellular microtubule depolymerization were determined in A-10 cells by evaluating the microtubule depolymerizing effects of a range of concentrations of each compound by indirect immunofluorescence. The percent microtubule loss for each concentration was estimated visually in at least 3 independent experiments. A dose-response plot was generated from the averages of the experiments and the EC50 values were calculated by linear regression.
In vitro tubulin polymerization
The kinetics of microtubule assembly in vitro were measured using the tubulin polymerization assay kit (Cytoskeleton Inc.) according to manufacturer’s directions. Briefly, 300 µg of purified bovine brain tubulin was added to tubulin assembly buffer (80 mM Na-Pipes, pH 6.9, 1 mM EGTA, 1 mM MgCl2, 1 mM GTP and 5% glycerol) and the indicated compound in a final volume of 100 µl. Microtubule polymerization was followed by absorbance at 340 nm at 37°C in a Spectromax Plus 96 well plate spectrophotometer (Molecular Devices).
Cell cycle analysis
MDA-MB-435 cells were incubated for 24 h with 0.05 µM 10i, vehicle or 0.0125 µM paclitaxel as a positive control. The cells were harvested, stained with Krishan’s reagent and analyzed using a FacsCalibur flow cytometer. Data were plotted as propidium iodide intensity versus the number of events.
Aqueous solubility assay
Our aqueous solubility assay is a slightly modified version of the MultiScreen Solubility Filter Plate protocol developed by Millipore®. The assay is based on the shake-flask solubility method that utilizes UV-Vis spectroscopy for quantification. Maximum aqueous solubility for this assay is 500 µM. The assay is conducted at room temperature at pH 7.4. Tamoxifen, ketoconazole and furosemide span the entire range of aqueous solubilities in this assay (30–500 µM) and were used as reference standards. Standard curves, covering the range of 3.13–500 µM, were generated for each compound using a 4:1 mixture of aqueous buffer (pH 7.4) to acetonitrile. Each compound was dissolved in DMSO and the final DMSO concentration was 2.5% (v/v). Each sample was analyzed by UV-Vis spectroscopy at 10 nm increments, from 260 -800 nm to determine the optimal wavelength and standard curves constructed. To measure the aqueous solubility of the reference standards and the oxadiazoline compounds, a 500 µM solution of each compound was prepared in aqueous buffer, mixed for 6 h, and briefly centrifuged to pellet any undissolved compound. A (4:1) mixture of supernatant to acetonitrile was added to a 96 well acrylic plate and analyzed by UV-Vis spectroscopy. The drug concentration in the supernatant was determined from the standard curve.
Supplementary Material
Acknowledgements
The authors thank Conjura Pharmaceuticals, LLC for support of this project (ML) and the Cancer Therapy and Research Center of the UTHSCSA (NCI P30 CA054174) (SLM).
Abbreviations
- CA-4
combretastatin A-4
- CA-1
combretastatin A-1
- HUVEC
human umbilical vein endothelial cells
- CTGF
connective tissue growth factor
- SAR
structure activity relationships
- IC50
concentration required to cause 50% inhibition of proliferation
- EC50,
concentration required to cause 50% loss of cellular microtubules
- Pgp
P-glycoprotein
- SRB
sulforhodamine B
Footnotes
Supporting Information Available. 400 MHz 1H-NMR spectra of compounds the final compounds 9a–l and 10a–l and elemental analysis data for 9e, 9h, 9l, 10e, 10h, and 10i are provided. HPLC tracings of compounds 9b, 9c, 9f, 10c, 10d, and 10f are also included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pettit GR, Singh SB, Niven ML, Hamel E, Schmidt JM. Isolation, structure, and synthesis of combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly derived from Combretum caffrum. J. Nat. Prod. 1987;50:119–131. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- 2.Pettit GR, Cragg GM, Singh SB. Antineoplastic agents, 122. Constituents of Combretum caffrum. J. Nat. Prod. 1987;50:386–391. doi: 10.1021/np50051a008. [DOI] [PubMed] [Google Scholar]
- 3.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 4.Lin CM, Singh SB, Chu PS, Dempcy RO, Schmidt JM, Pettit GR, Hamel E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure-activity study. Mol. Pharmacol. 1988;34:200–208. [PubMed] [Google Scholar]
- 5.Dorr RT, Dvorakova K, Snead K, Alberts DS, Salmon SE, Pettit GR. Antitumor activity of combretastatin-A4 phosphate, a natural product tubulin inhibitor. Invest. New Drugs. 1996;14:131–137. doi: 10.1007/BF00210783. [DOI] [PubMed] [Google Scholar]
- 6.Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997;57:1829–1834. [PubMed] [Google Scholar]
- 7.Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, Dennis MF, Chaplin DJ. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Res. 1999;59:1626–1634. [PubMed] [Google Scholar]
- 8.Galbraith SM, Maxwell RJ, Lodge MA, Tozer GM, Wilson J, Taylor NJ, Stirling JJ, Sena L, Padhani AR, Rustin GJ. Combretastatin A4 phosphate has tumor antivascular activity in rat and man as demonstrated by dynamic magnetic resonance imaging. J. Clin. Oncol. 2003;21:2831–2842. doi: 10.1200/JCO.2003.05.187. [DOI] [PubMed] [Google Scholar]
- 9.Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, Giantonio B, Zimmer R, Petros WP, Stratford M, Chaplin D, Young SL, Schnall M, O'Dwyer PJ. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J. Clin. Oncol. 2003;21:4428–4438. doi: 10.1200/JCO.2003.12.986. [DOI] [PubMed] [Google Scholar]
- 10.Chaplin DJ, Horsman MR, Siemann DW. Current development status of small-molecule vascular disrupting agents. Curr. Opin. Investig. Drugs. 2006;7:522–528. [PubMed] [Google Scholar]
- 11.Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99:2060–2069. doi: 10.1182/blood.v99.6.2060. [DOI] [PubMed] [Google Scholar]
- 12.Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, Lam G, Bompais-Vincent H, Zhu Z, Hicklin DJ, Bohlen P, Chaplin DJ, May C, Rafii S. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J. Clin. Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samarin J, Rehm M, Krueger B, Waschke J, Goppelt-Struebe M. Up-regulation of connective tissue growth factor in endothelial cells by the microtubule-destabilizing agent combretastatin A-4. Mol. Cancer Res. 2009;7:180–188. doi: 10.1158/1541-7786.MCR-08-0292. [DOI] [PubMed] [Google Scholar]
- 14.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 15.Hinnen P, Eskens FA. Vascular disrupting agents in clinical development. Br. J. Cancer. 2007;96:1159–1165. doi: 10.1038/sj.bjc.6603694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeBlanc R, Dickson J, Brown T, Stewart M, Pati HN, VanDerveer D, Arman H, Harris J, Pennington W, Holt HL, Jr, Lee M. Synthesis and cytotoxicity of epoxide and pyrazole analogs of the combretastatins. Bioorg. Med. Chem. 2005;13:6025–6034. doi: 10.1016/j.bmc.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 17.Johnson M, Younglove B, Lee L, Leblanc R, Holt H, Jr, Hills P, Mackay H, Brown T, Mooberry SL, Lee M. Design, synthesis, and biological testing of pyrazoline derivatives of combretastatin-A4. Bioorg. Med. Chem. Lett. 2007;17:5897–5901. doi: 10.1016/j.bmcl.2007.07.105. [DOI] [PubMed] [Google Scholar]
- 18.Kong Y, Grembecka J, Edler MC, Hamel E, Mooberry SL, Sabat M, Rieger J, Brown ML. Structure-based discovery of a boronic acid bioisostere of combretastatin A-4. Chem. Biol. 2005;12:1007–1014. doi: 10.1016/j.chembiol.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Ducki S, Forrest R, Hadfield JA, Kendall A, Lawrence NJ, McGown AT, Rennison D. Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioor.g Med. Chem. Let.t. 1998;8:1051–1056. doi: 10.1016/s0960-894x(98)00162-0. [DOI] [PubMed] [Google Scholar]
- 20.Ali OM, Amer HH, Abdel-Rahman AA-H. Synthesis and antiviral evaluation of sugar uracil-1-ylmethylhydrazones and their oxadiazoline derivatives. Synthesis. 2007;18:2823–2828. [Google Scholar]
- 21.(a) Song Q-B, Zhang J, Tiekink ERT. 2-[3-Acetyl-5-(3,6-dichloropyridin-2-yl)-2,3-dihydro-1,3,4-oxadiazol-2-yl]phenyl acetate. Acta Cryst. 2006;E62:o4115–o4117. [Google Scholar]; (b) Qin Q, Xu LJ, Pan LF, Chen SQ, Song QB. 2-[3-Acetyl-5-(2-chloro-3-pyridyl)-2-methyl-2,3-dihydro-1,3,4-oxadiazol-2-yl]-4-fluorophenyl acetate. Acta Cryst. 2009;E65:o1181. doi: 10.1107/S1600536809015323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 23.Blagosklonny MV, Darzynkiewicz Z, Halicka HD, Pozarowski P, Demidenko ZN, Barry JJ, Kamath KR, Herrmann RA. Paclitaxel induces primary and postmitotic G1 arrest in human arterial smooth muscle cells. Cell Cycle. 2004;3:1050–1056. [PubMed] [Google Scholar]
- 24.Rao PN, Cessac JW, Tinley TL, Mooberry SL. Synthesis and antimitotic activity of novel 2-methoxyestradiol analogs. Steroids. 2002;67:1079–1089. doi: 10.1016/s0039-128x(02)00066-1. [DOI] [PubMed] [Google Scholar]
- 25.Tripathi A, Fornabaio M, Kellogg GE, Gupton JT, Gewirtz DA, Yeudall WA, Vega NE, Mooberry SL. Docking and hydropathic scoring of polysubstituted pyrrole compounds with antitubulin activity. Bioorg. Med. Chem. 2008;16:2235–2242. doi: 10.1016/j.bmc.2007.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupton JT, Burham BS, Krumpe K, Du K, Sikorski JA, Warren AE, Barnes CR, Hall IH. Synthesis and cytotoxicity of 2,4-disubstituted and 2,3,4-trisubstituted brominated pyrroles in murine and human cultured tumor cells. Arch. Pharm. (Weinheim) 2000;333:3–9. doi: 10.1002/(sici)1521-4184(200001)333:1<3::aid-ardp3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Tinley TL, Leal RM, Randall-Hlubek DA, Cessac JW, Wilkens LR, Rao PN, Mooberry SL. Novel 2-methoxyestradiol analogues with antitumor activity. Cancer Res. 2003;63:1538–1549. [PubMed] [Google Scholar]
- 28.Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, Hempel J, Gupton JT, Brown ML. Identification and characterization of a new tubulin-binding tetrasubstituted brominated pyrrole. Mol. Pharmacol. 2007;72:132–140. doi: 10.1124/mol.107.034876. [DOI] [PubMed] [Google Scholar]
- 29.Weiderhold KN, Randall-Hlubek DA, Polin LA, Hamel E, Mooberry SL. CB694, a novel antimitotic with antitumor activities. Int. J. Cancer. 2006;118:1032–1040. doi: 10.1002/ijc.21424. [DOI] [PubMed] [Google Scholar]
- 30.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 31.Seve P, Dumontet C. Is class III beta-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol. 2008;9:168–175. doi: 10.1016/S1470-2045(08)70029-9. [DOI] [PubMed] [Google Scholar]
- 32.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008;68:8881–8888. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 34.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Taccalonolides E and A: Plant-derived steroids with microtubule-stabilizing activity. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.