

Abstract
Microbial cellulose degradation is a central part of the global carbon cycle and has great potential for the development of inexpensive, carbon-neutral biofuels from non-food crops. Clostridium phytofermentans has a repertoire of 108 putative glycoside hydrolases to break down cellulose and hemicellulose into sugars, which this organism then ferments primarily to ethanol. An understanding of cellulose degradation at the molecular level requires learning the different roles of these hydrolases. In this study, we show that interspecific conjugation with Escherichia coli can be used to transfer a plasmid into C. phytofermentans that has a resistance marker, an origin of replication that can be selectively lost, and a designed group II intron for efficient, targeted chromosomal insertions without selection. We applied these methods to disrupt the cphy3367 gene, which encodes the sole family 9 glycoside hydrolase (GH9) in the C. phytofermentans genome. The GH9-deficient strain grew normally on some carbon sources such as glucose, but had lost the ability to degrade cellulose. Although C. phytofermentans upregulates the expression of numerous enzymes to break down cellulose, this process thus relies upon a single, key hydrolase, Cphy3367.
Introduction
Cellulose is the most abundant renewable, biological energy source on earth (Leschine, 1995), and its degradation is a key part of the global carbon cycle. Cellulose has tremendous potential as a biofuel feedstock; more than a billion tons of lignocellulosic plant biomass could be used each year for liquid biofuels in North America (Perlack et al., 2005). The major challenge of producing biofuel from cellulose is the recalcitrance of cellulosic fibres to break down into sugars. The current cost of converting cellulosic biomass to sugar doubles the carbohydrate purchase cost, nullifying the advantage of biomass relative to corn (Lynd et al., 2008). Microbes both that secrete enzymes to break down cellulosic biomass and that ferment the resulting saccharides can make cellulosic biofuels more efficient by obviating feedstock pre-treatment and raising conversion efficiencies. However, the development of robust microbial strains for conversion of biomass to fuels has been hindered by our inability to genetically manipulate these organisms.
Clostridium phytofermentans, a mesophilic anaerobe isolated from forest soil (Warnick et al., 2002), is an ideal microbe to study the direct conversion of cellulosic biomass to ethanol. It grows on both of the two main components of plant biomass, cellulose and hemicellulose, by secreting enzymes to cleave these polysaccharides and then fermenting the resulting hexose (glucose, galactose, mannose) and pentose (xylose, arabinose) sugars to ethanol. Among clostridial genomes sequenced to date, C. phytofermentans has the highest number of genes encoding enzymes for the modification and breakdown of complex carbohydrates. It contains genes for 161 carbohydrate-active enzymes (CAZy), which include 108 glycoside hydrolases spread across 39 families (Cantarel et al., 2009). This abundance of hydrolases highlights how cellulosic biomass is a complex substrate whose degradation likely requires the concerted action of many enzymes. Cellulolytic clostridia usually have numerous genes encoding family 9 glycoside hydrolases (GH9): the Clostridium thermocellum ATCC 27405 genome has 16 GH9 genes, Clostridium cellulolyticum H10 has 13 GH9 genes, and Clostridium cellulovorans has five GH9 genes. In contrast, C. phytofermentans has only a single GH9-encoding gene, cphy3367. GH9 proteins are all β-1,4-glucanases, but members of the GH9 family may have different substrate specificities and end-products (Arai et al., 2006). There are subfamilies of GH9 proteins with different molecular architectures (Fig. 1A). Cphy3367 belongs to the subfamily, called theme B (Gilad et al., 2003), in which the hydrolytic module is fused to a family 3 carbohydrate-binding module (CBM) (Fig. 1B).
Fig. 1.
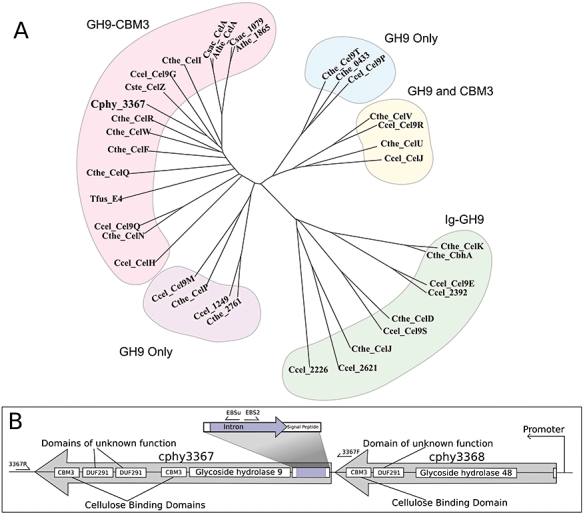
A. Unrooted dendrogram of family 9 glycoside hydrolase amino acid sequences shows that Cphy3367 belongs to the subfamily in which a CBM potentiates the adjacent hydrolytic domain. Abbreviations: Cphy, Clostridium phytofermentans; Cthe, Clostridium thermocellum; Cste, Clostridium stercorarium; Ccel, Clostridium cellulolyticum; Tfus, Thermomonospora fusca; Csac, Caldicellulosiruptor saccharolyticus; Athe, Anaerocellum thermophilum. B. Diagram of the cphy3367–cphy3368 putative operon showing the protein domains, the site of intron insertion in cphy3367 and the location of primers used to map the intron insertion by PCR.
Cellulolytic bacteria secrete a battery of glycoside hydrolases to depolymerize cellulosic biomass. The different roles of these hydrolases could be uncovered by targeted gene inactivation. Although cellulolytic clostridia have been studied for decades, targeted mutagenesis in these organisms has remained challenging, likely due to highly active DNases and inefficient homologous recombination. Previously, cellulolysis-deficient strains of C. cellulolyticum were isolated by spontaneous mutation of the scaffoldin gene, which anchors the cellulolytic enzymes to the cell surface (Maamar et al., 2003; 2004), and by reduction of CelF48 expression with antisense-RNA (Perret et al., 2004). In this study, we developed a general system for targeted, chromosomal gene inactivation in C. phytofermentans. Efficient transfer of DNA into C. phytofermentans is achieved by conjugation with Escherichia coli expressing the broad-host-range RP4 conjugal apparatus. Conjugation has been used transfer plasmids to several mesophilic clostridia including Clostridium acetobutylicum (Williams et al., 1990), C. perfringens (Lyras and Rood, 1998), C. cellulolyticum (Jennert et al., 2000) and C. difficile (Purdy et al., 2002). To make targeted chromosomal insertions in C. phytofermentans, we modified a Gram-positive conjugal shuttle vector (Trieu-Cuot et al., 1991) to contain a strong C. phytofermentans promoter driving expression of a group II intron that was designed to inactivate cphy3367.
Group II introns are catalytic RNAs that insert into dsDNA in a site-specific manner called retrohoming (reviewed by Lambowitz and Zimmerly, 2004). The intron used in this study is derived from the Lactococcus lactis Ll.LtrB group II intron (Karberg et al., 2001). It consists of a 0.9 kb Ll.LtrB-deltaORF intron flanked by short exon sequences and a downstream ltrA gene that encodes a protein with endonuclease and reverse transcriptase activity (Guo et al., 2000). The intron inserts into the genome by splicing into a 13–16 bp DNA recognition sequence. The LtrA protein then cleaves the opposite DNA strand and uses the 3′ DNA as a primer to reverse transcribe the inserted intron RNA. Because the DNA binding specificity of the group II intron is conferred by short sequences, the intron can be easily modified to integrate into a desired DNA target site. Group II introns can be made to insert into virtually any DNA sequence with frequencies in E. coli of 0.1–22% (Karberg et al., 2001). In theory, group II introns can function in any bacterial taxa into which plasmid DNA can be delivered because intron insertion does not require host-supplied factors. Group II introns have been used to make mutations in both Gram-negative bacteria (Yao and Lambowitz, 2007) and Gram-positive bacteria, including clostridia (Heap et al., 2007). Previous experiments in the Gram-positive Staphylococcus aureus found that this mobile group II intron made targeted gene disruptions in 37–100% of colonies (Yao et al., 2006).
Broadly, our goal is to dissect the set of cellulolytic enzymes in C. phytofermentans in order to identify key genes for the degradation of cellulosic biomass. The C. phytofermentans genome differs from other cellulolytic clostridia in encoding only a single GH9, Cphy3367. In this study, we disrupted the cphy3367 gene with a targeted group II intron, which was delivered into C. phytofermentans by conjugation with E. coli. Inactivation of cphy3367 resulted in a strain that grew normally on glucose, cellobiose and hemicellulose, but had lost the ability to degrade crystalline cellulose. mRNA expression supports that cphy3367 is among the most highly upregulated CAZy genes during growth on cellulose. These findings reveal a central role played by Cphy3367 in cellulose degradation. Generally, these results show that targeted gene inactivation can be used to identify key enzymes for cellulose degradation in C. phytofermentans.
Results
A group II intron plasmid for use with C. phytofermentans
The plasmid we developed for gene inactivation in C. phytofermentans is derived from pAT19 (Trieu-Cuot et al., 1991). This plasmid has an RP4 conjugal origin of transfer, origins of replication both for E. coli (pUC) and for Gram-positive bacteria (Enterococcus pAMβ1) and an erythromycin resistance gene from Streptococcus pneumoniae Tn1545 that functions in both Gram-negative and Gram-positive bacteria. This plasmid was modified to disrupt cphy3367 with a group II intron in three steps: addition of a strong C. phytofermentans promoter to pAT19, insertion of the intron (Ll.LtrB-deltaORF and ltrA gene) downstream of the C. phytofermentans promoter, and targeting the intron to cphy3367. In the first step, the promoter from C. phytofermentans pyruvate ferredoxin oxidoreductase (cphy3558) was cloned into pAT19 to yield pQexp (Fig. 2A). Pyruvate ferredoxin oxidoreductase functions in central carbon metabolism to couple the reduction of ferredoxin with the oxidative decarboxylation of pyruvate to acetyl-CoA and CO2. Genome-wide studies of C. phytofermentans mRNA (J. Blanchard, pers. comm.) support that cphy3558 is among the most highly expressed genes in the genome on all tested carbon sources. The gene upstream of cphy3558 in the genome is transcribed in the opposite direction, showing that cphy3558 is transcribed from its own promoter. The high, constitutive expression of cphy3558 supports that this promoter can be used in pQexp for strong expression of the group II intron, or other genes, in C. phytofermentans under diverse physiological conditions.
Fig. 2.
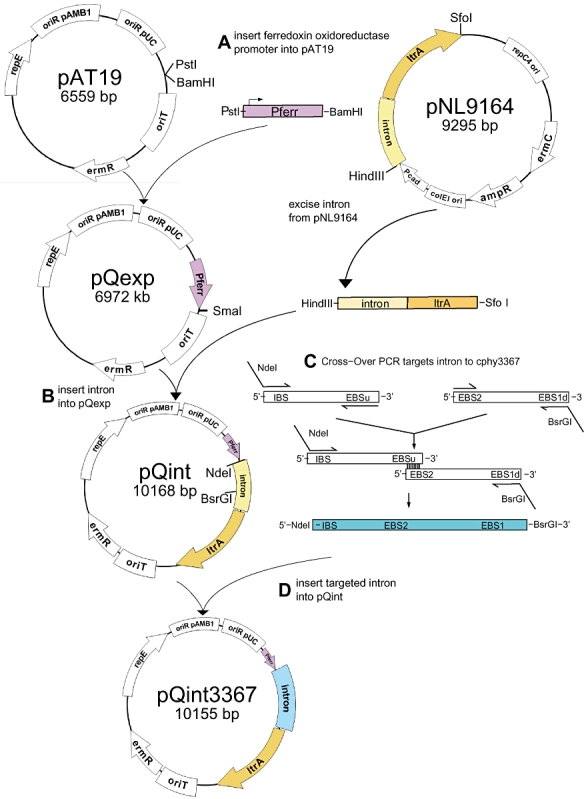
Construction of a plasmid for inactivation of cphy3367 with a group II intron. A. The C. phytofermentans pyruvate ferrodoxin oxidoreductase promoter (Pferr) was inserted into pAT19 to make pQexp. B. The intron cassette from pNL9164 was inserted downstream of Pferr in pQexp to make pQint. C. The intron cassette was retargeted to cphy3367 by two-step, cross-over PCR. D. The retargeted intron was inserted into pQint to make pQint3367. Plasmid diagrams show restriction sites used in cloning.
The group II intron and ltrA gene from pNL9164 (Yao et al., 2006) were inserted into pQexp downstream of the cphy3558 promoter to produce pQint (Fig. 2B). The DNA binding region of the intron in pQint was retargeted to cphy3367 by a two-step, cross-over PCR (Fig. 2C). An intron insertion site in cphy3367 was selected that was both in the antisense orientation and near the start codon (Fig. 1B). An antisense site was used because introns integrated in the sense orientation can splice out from the RNA transcript in vivo with the help of LtrA (Yao et al., 2006). An antisense insertion in cphy3367 would yield an unconditional disruption, which was predicted to grow normally on media containing a simple carbon source such as glucose that does not require cleavage by a β-1,4-glucanase. The antisense insertion site nearest to the start codon was used in order to have the highest probability of disrupting cphy3367 expression. This site is 37 bp downstream of the predicted start codon and resides in the putative secretion signal sequence (other potential sites in Supporting information S1). As cphy3367 is the downstream member of a putative two gene operon (Fig. 1B), disruption of cphy3367 should not result in polar transcriptional effects. The retargeted intron fragment was inserted into pQint to make pQint3367 (Fig. 2D). The complete sequences of pQexp, pQint and pQint3367 are in Supporting information S2.
DNA is efficiently transferred to C. phytofermentans by conjugation with E. coli
pQint3367 was introduced into C. phytofermentans ISDg (ATCC 700394) by conjugal transfer from E. coli. The conjugal donor was E. coli strain 1100-2 (Bandrin et al., 1983) containing pRK24 (Meyer et al., 1977), the plasmid encoding the RP4 conjugal apparatus, and pQint3367. Cells were mated on solid G2 medium overnight. Following mating, C. phytofermentans transconjugants were isolated by selecting against both E. coli and C. phytofermentans cells that did not receive pQint3367. As C. phytofermentans, but not E. coli 1100-2, is naturally resistant to nalidixic acid and trimethoprim, these antibiotics were used to prevent growth of E. coli 1100-2 after mating. If other E. coli strains that are less sensitive to these antibiotics are used for matings, additional steps may be needed to isolate pure C. phytofermentans colonies. For example, if plates are supplemented with X-gal and IPTG, lacZ+E. coli strains will form blue colonies whereas C. phytofermentans colonies will be white. Alternatively, liquid cultures can be inoculated with phage T7 before plating to selectively remove E. coli (Tolonen et al., 2006). Relative to C. phytofermentans colony counts on plates lacking erythromycin, colony counts on plates containing erythromycin showed that the efficiency of plasmid transfer to C. phytofermentans was ∼1 transconjugant per 106 recipient cells. This transfer rate is typically efficient enough to isolate 10–20 C. phytofermentans transconjugants per plate. In this study, 10 independent transconjugants were isolated, hereafter called AT02-1 to AT02-10. When erythromycin-resistant C. phytofermentans colonies were picked, the absence of residual E. coli cells was confirmed by plating an aliquot on solid LB medium and incubating aerobically at 37°C overnight.
Genomic intron insertions in C. phytofermentans are accurate and specific
The presence of pQint3367 was examined in C. phytofermentans transconjugants by PCR amplification of the ermR gene. The ermR PCR product was absent in wild-type C. phytofermentans cultures, but was present in AT02-1 to AT02-10. The expected intron insertion, 37 bp downstream of the start codon cphy3367 in the antisense direction (Fig. 1B), was confirmed by PCR mapping of the cphy3367 locus in the AT02 isolates. PCR of the cphy3367 coding region yielded a 3.1 kb product in wild type and a 4 kb product in AT02-1, supporting the expected 0.9 kb intron insertion in cphy3367 (Fig. 3A, lanes 1 and 2). PCR of the 5′ and the 3′ intron–genome junction regions using one primer in the genome and the other in the intron (3367_F/Ebs2_3367 for the 5′ junction and 3367_R/Ebs_univ for the 3′ junction) resulted in products in AT02-1 (Fig. 3A, lanes 4 and 6), but not wild type (Fig. 3A, lanes 3 and 5). These PCR products were sequenced to confirm that AT02-1 bears the expected intron insertion in cphy3367 (Fig. S1). Similarly, PCR screening of the other nine AT02 isolates confirmed the correct insertion in cphy3367, supporting that the Ll.LtrB group II intron is efficient and accurate in C. phytofermentans.
Fig. 3.
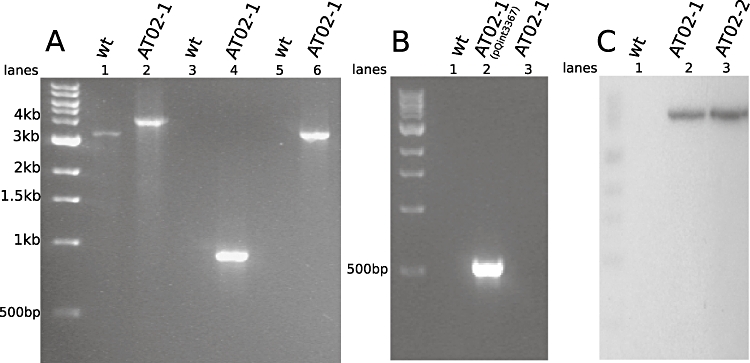
The intron insertion in cphy3367 of AT02 is both accurate and specific. A. PCR of the cphy3367 locus in wild-type and AT02-1 strains. The cphy3367 gene in AT02-1 contains a 900 bp insertion relative to wild type (lanes 1 and 2). Primers to amplify the genome–intron junctions yield PCR products in AT02-1, but not wild type, for both the 5′ junction (lanes 3 and 4) and the 3′ junction (lanes 5 and 6). B. PCR of the ermR gene from pQint3367 shows acquisition of the plasmid in AT02-1 following conjugation (lanes 1 and 2) and its loss by plasmid curing (lane 3). C. Southern blot probed with a 32P-labelled intron probe reveals no band in wild type (lane 1) and a single intron insertion in two independent transconjugants, AT02-1 and AT02-2 (lanes 2 and 3). The single band shown in the AT02 lanes was the only one visible on the blot (100 bp to 10 kb).
Once the correct intron insertion in cphy3367 had been confirmed, the loss of pQint3367 was induced in AT02-1 and AT02-2 by diluting cultures 1:100 in GS2 (−erm) medium and growing to late log phase (OD600 1.0) through five serial transfers. AT02-1 was then plated on solid GS2 (−erm) medium and 10 colonies were tested by PCR, both for the presence of the ermR gene in pQint3367 and for the retention of an intron insertion in cphy3367 with primers 3367_F and Ebs2_3367. Transfer in medium lacking erythromycin resulted in rapid loss of pQint3367 (Fig. 3B). In total, 8 of 10 colonies had lost pQint3367, while all 10 had retained the intron insertion in cphy3367 (Fig. S2A). Further, plating on GS2 medium containing erythromycin showed that strains that had lost pQint3367 were erythromycin-sensitive (Fig. S2B). After an additional five transfers in the absence of erythromycin selection, it was again confirmed that all 10 AT02-1 cultures still retained the genomic intron insertion in cphy3367. The pQint plasmid can thus be used to make genomic insertions in C. phytofermentans that are stable in the absence of selection. Because the pQint plasmid can be efficiently lost in the absence of selection and the intron does not contain an antibiotic resistance gene, multiple genes in the same C. phytofermentans strain can be inactivated by sequentially introducing pQint plasmids targeted to different genes.
Southern blotting showed that AT02-1 and AT02-2 do not harbour any additional intron insertions elsewhere in the genome (Fig. 3C). The Southern blot showed no bands in wild type (Fig. 3C, lane 1) and a single band in both transconjugants, AT02-1 and AT02-2, at the expected size of 5855 bp (Fig. 3C, lanes 2 and 3). A Southern blot of AT02-1 prior to loss of pQint3367 showed a second band of 7799 bp due to the plasmid-born intron. The Ll.LtrB group II intron can thus be used for targeted gene disruptions in C. phytofermentans without making additional, unintended genomic insertions.
AT02-1 grows normally on glucose, cellobiose and hemicellulose, but is unable to degrade filter paper cellulose
Growth rates (OD600) of AT02-1 and wild-type strains were compared in GS2 media containing either glucose, cellobiose or hemicellulose xylan as the sole carbon source. As expected, wild type and AT02-1 showed similar growth rates on glucose (Fig. 4A). The comparable growth rates of wild type and AT02-1 on cellobiose (Fig. 4B) supports that the glucanase activity of the Cphy3367 protein is not required to break the 1,4-β bond between glucose subunits of cellobiose. Further, wild type and AT02-1 grew at similar rates on hemicellulose (Fig. 4C). The hemicellulose used in this study was birchwood xylan, which is a polysaccharide of 89.3% xylose, 1% arabinose, 1.4% glucose and 8.3% anhydrouronic acid (Kormelink et al., 1993). Although this assay does not establish whether Cphy3367 is active on β-1,4-d-xylose bonds, the robust growth of AT02-1 on hemicellulose shows that Cphy3367 is not required for C. phytofermentans to metabolize hemicellulose.
Fig. 4.
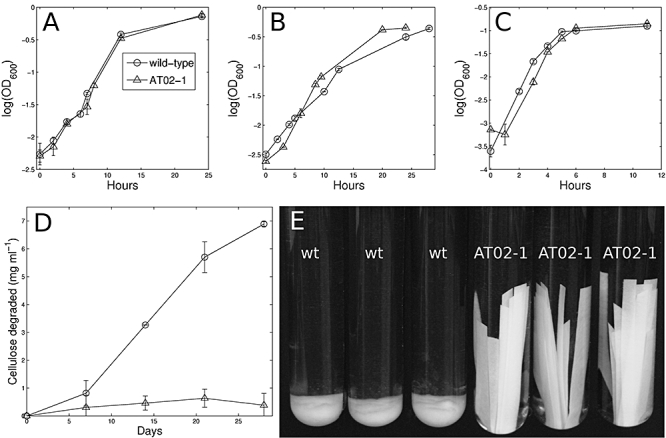
A–C. Strain with disruption of cphy3367 (AT02-1) had similar growth rates as wild type on glucose (A), cellobiose (B) and hemicellulose xylan (C). Growth curves are means of triplicate cultures. Error bars show one standard deviation and are smaller than the symbols where not apparent. D. Disruption of cphy3367 results in inability to degrade filter paper cellulose. Cellulose degradation was measured as dry mass of cellulose remaining in culture. E. After 4 weeks, cellulose strips in wild-type (wt) tubes had broken down while strips in AT02-1 cultures appeared unchanged.
The cellulose degradation rates of AT02-1 and wild type were measured in cultures of GS2 medium with filter paper cellulose as the sole carbon source. We found that wild type consumed cellulose at a mean rate of 0.24 mg ml−1 day−1, while cellulose consumption by AT02-1 was negligible (Fig. 4D). The composition of the cellulose in wild-type and AT02-1 cultures was visibly different after a few days. Following the 4-week experiment, the remaining cellulose in wild-type cultures was a viscous pulp, while the cellulose strips in AT02-1 cultures remained unchanged (Fig. 4E). Disruption of cphy3367 thus renders C. phytofermentans unable to degrade filter paper cellulose.
Wild-type and AT02-1 expression of cphy3367 and other genes on cellulosic carbon sources
The inability of AT02-1 to degrade cellulose prompted us to examine the expression patterns of cphy3367 and related genes. Initially, we compared the expression of cphy3367 with those of surrounding genes in the genome to see which genes were coexpressed with cphy3367. The cphy3367 gene is 76 bp downstream of cphy3368, a family 48 glycoside hydrolase, and is 341 bp upstream of cphy3366, a MoxR-like ATPase (Fig. 5A). Sequence analysis suggests that cphy3366 is transcribed from its own promoter; disruption of cphy3367 would thus not result in polar transcriptional effects. A putative promoter upstream of cphy3368 led us to hypothesize that cphy3367 is the second gene in a putative two gene operon (Fig. 1B). In support of this hypothesis, we found that in wild-type cells cphy3367 and cphy3368 mRNA transcripts are upregulated on hemicellulose (Fig. 5B) and cellulose (Fig. 5C) relative to expression on glucose, while the surrounding genes did not increase in expression. Similar expression changes were observed for the Cphy3366–Cphy3369 proteins on hemicellulose (Fig. 5D) and cellulose (Fig. 5E). The cphy3367–cphy3368 putative operon is thus upregulated independent of the surrounding genes, which do not appear to be involved in acclimation to growth on cellulosic biomass. Further, the much higher upregulation of cphy3367 on cellulose than on hemicellulose supports that this gene is more directly involved in the degradation of cellulose.
Fig. 5.
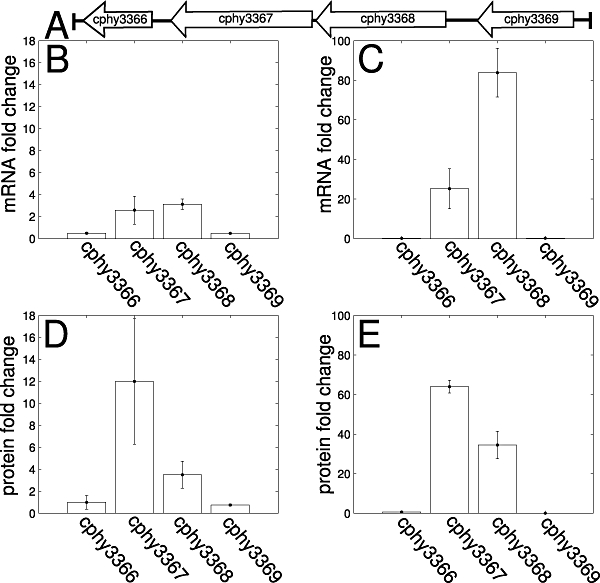
Expression changes of cphy3367 and surrounding genes (A) in the genome on different carbon sources. Wild-type mRNA (B and C) and protein (D and E) expression of cphy3367 and cphy3368 increased in hemicellulose (B and D) and cellulose (C and E) cultures compared with glucose while expression of the surrounding genes did not increase. The mRNA expression was measured by qRT-PCR. Bars show mean fold change calculated as 2−ΔΔCt. Protein expression was quantified by mass spectrometry. Bars show mean number of peptides detected from each protein in hemicellulose or cellulose cultures divided by the number of peptides detected in glucose cultures.
The mRNA expression of cphy3367 on cellulosic carbon sources was also compared with the expression of 39 other CAZy genes by qRT-PCR. The set of CAZy genes we examined included 33 glycoside hydrolases spread across 20 families, two carbohydrate esterases, three glycosyl transferases and two polysaccharide lyases (see Supporting information S3 for a complete list of genes). Among these 40 CAZy genes, 14 genes were greater than twofold upregulated on hemicellulose (Fig. 6A) and 13 genes were upregulated on cellulose (Fig. 6B) relative to their expression during growth on glucose. Moreover, there were widespread differences in the mRNA expression of CAZy genes between hemicellulose and cellulose (shaded bars in Fig. 6A and B). C. phytofermentans can thus differentiate between the components of cellulosic biomass and respond by transcriptionally altering the set of CAZy genes that are expressed. Only three other CAZy enzymes (cphy1799, cphy1800, cphy3368) were more highly upregulated than cphy3367 on cellulose (Fig. 6B), while a dozen CAZy genes were more highly upregulated than cphy3367 on hemicellulose (Fig. 6A), further supporting that Cphy3367 is more directly involved in the breakdown of cellulose than hemicellulose. As the acclimation to growth on cellulosic carbon sources in C. phytofermentans involves the increased expression of numerous CAZy genes, the breakdown of cellulosic substrates likely requires the concerted action of additional enzymes to Cphy3367.
Fig. 6.
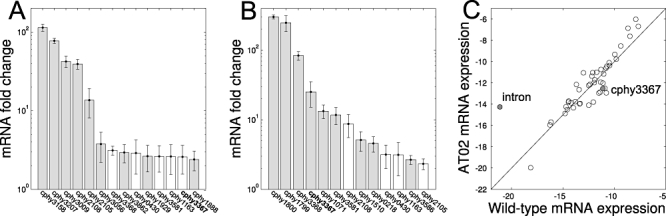
mRNA expression changes of genes encoding carbohydrate-active enzymes. A and B. Genes that were greater than twofold upregulated in wild-type cells grown on hemicellulose (A) and cellulose (B) relative to expression on glucose are shown. Genes that were also greater than twofold upregulated on hemicellulose relative to cellulose in (A) or vice versa in (B) are shaded. C. The mRNA expression of genes for carbohydrate-active enzymes was similar in wild-type and AT02-1 strains on hemicellulose, supporting that disruption of cphy3367 does not result in widespread changes in the expression of other enzymes. The mRNA expression was measured by qRT-PCR. Bars in (A) and (B) show mean fold change calculated as 2−ΔΔCt; circles in (C) show −ΔCt. Errors show one standard deviation. Expression of cphy3367 and the intron integrated in cphy3367 in AT02-1 are shaded in (C).
The mRNA expression of CAZy genes in wild-type and AT02-1 cells growing on hemicellulose was examined to determine if disruption of cphy3367 affected the expression of additional genes. While cphy3367 expression was elevated on hemicellulose (Fig. 5B), the mRNA expression of CAZy genes in AT02-1 and wild-type cells was highly similar (Fig. 6C). Proper expression of cphy3367 is thus not required for the induction of other CAZy genes, supporting that the inability of strain AT02-1 to degrade cellulose is specifically due to loss of the Cphy3367 protein. The expression of cphy3366, the gene downstream of cphy3367 in the genome, was moderately lower in AT02-1 than in wild type when growing on hemicellulose, but these changes were less than twofold. Further, we found that mRNA expression of the intron was similar to cphy3367 in AT02-1, but was not detected in wild-type cells (Fig. 6C).
Discussion
Although the C. phytofermentans genome encodes over a hundred glycoside hydrolases and many of these genes are upregulated on cellulosic carbon sources, this study shows that the inactivation of a single hydrolase, Cphy3367, results in an inability to degrade cellulose. To enable the genetic analysis of cellulose degradation by C. phytofermentans, the initial contribution of this study was to develop a system to make targeted insertions in the C. phytofermentans chromosome. We showed that interspecific conjugation with E. coli can be used to reliably transfer foreign DNA into C. phytofermentans. The plasmid we transferred to C. phytofermentans established that the ermR gene from S. pneumoniae Tn1545 gives high erythromycin resistance in this organism. Further, the Enterococcus pAMβ1 origin of replication replicates stably in C. phytofermentans under antibiotic selection, but can be easily cured from strains in the absence of selection. Finally, the L. lactis Ll.LtrB group II intron makes accurate (Fig. 3A) and specific (Fig. 3C) chromosomal insertions in C. phytofermentans. We found that when the strong C. phytofermentans cphy3558 promoter is used to drive expression of the intron, it inserts into the genome with such a high efficiency that mutants can be easily isolated without selecting for integration. As the intron insertions are stable in the absence of selection, the intron need not contain a resistance gene. Once the plasmid is cured from C. phytofermentans, no antibiotic resistance genes remain. Multiple intron insertions can thus be made in the same strain without needing independent resistance markers. This is particularly helpful because so few resistance markers are known to function in clostridia. It is, however, possible that intron insertions in certain regions of the genome might occur with lower efficiency such that a direct selection for integration would be advantageous. Group II introns have been modified to contain Retrotransposition-Activated Markers (RAM) to positively select for integration (Zhong et al., 2003). RAM consist of an antibiotic resistance gene that is interrupted by a group I intron. When the group II intron integrates, the group I intron is excised from the resistance gene. RAM have been used to positively select for intron insertions in other clostridia (Heap et al., 2007) and would likely be useful in C. phytofermentans to obtain strains with intron insertions in low-efficiency positions in the genome.
We applied these methods to study the genetic basis of cellulose degradation by disrupting cphy3367, encoding the only GH9 in C. phytofermentans. The GH9-deficient strain, AT02-1, grows normally on glucose, cellobiose and hemicellulose (Fig. 4A–C) but has lost the ability to degrade crystalline cellulose (Fig. 4D and E). Several lines of evidence support that this phenotype resulted specifically from the inactivation of Cphy3367. AT02-1 did not have any additional intron insertions (Fig. 3C). The gene expression patterns of cphy3367 and surrounding genes (Fig. 5) support that cphy3367 is the downstream member of a two-gene operon such that inactivation of cphy3367 would not result in polar transcriptional effects. Finally, the expression of CAZy genes in wild type and AT02-1 appears similar, supporting that disruption of cphy3367 does not result in altered expression of other genes for the breakdown of cellulose.
In contrast to other clostridia such as C. thermocellum, C. cellulolyticum and C. cellulovorans, the glycoside hydrolases in C. phytofermentans do not appear to multimerize into an extracellular cellulosome. The C. phytofermentans genome does not appear to encode a cellulosomal scaffoldin to anchor the cellulases to the cell surface and the hydrolases do not have dockerin domains to attach to a scaffoldin. The inability of AT02-1 to degrade cellulose is unlikely to result from the disruption of an extracellular, cellulosome-like complex. Although C. phytofermentans is different from other cellulolytic clostridia in having only one family 9 hydrolase, numerous other C. phytofermentans hydrolases are upregulated on hemicellulose and cellulose (Fig. 6), suggesting that the breakdown of cellulose requires the co-ordinated action of multiple enzymes.
How do we explain the critical dependence of C. phytofermentans upon Cphy3367 for cellulose degradation? There are well-studied members of the same GH9 subfamily as Cphy3367 (Fig. 1A). The protein with the highest sequence similarity to Cphy3367 in the NCBI database is Clostridium stercorarium CelZ, with which Cphy3367 shares 65% amino acid identity. Cphy3367 and CelZ have the same domain organization and the CelZ catalytic residues D84 and E447 (Riedel and Bronnenmeier, 1999) are conserved in Cphy3367 as D83 and E446. Degradation of crystalline cellulose is typically achieved by a synergy between endocellulases to liberate cellulose chains from the cellulose surface and exocellulases to cleave these chains into oligosaccharides. However, GH9 proteins in the subfamily with Cphy3367, such as C. stercorarium CelZ, are unusual in that they act both as endoglucanases and as exoglucanases (Jauris et al., 1990). Purified C. thermocellum CelI is sufficient to solubilize filter paper in vitro (Gilad et al., 2003). Our results build upon this finding by showing that Cphy3367 is required by C. phytofermentans to solubilize filter paper in vivo. Cphy3367 may have the central role in cellulose degradation, with other cellulases having accessory functions. Alternatively, some or all of the C. phytofermentans cellulases may have required non-redundant functions to break down cellulose such that inactivation of these genes would have similar phenotypes to AT02 (Fig. 7A). Future gene inactivation studies will reveal the relative contributions of other C. phytofermentans hydrolases to cellulose degradation.
Fig. 7.

Non-exclusive models for the roles of Cphy3367 in cellulose degradation. A. The set of C. phytofermentans cellulolytic enzymes has members, such as Cphy3367, with key hydrolytic functions for which there is no redundancy. B. The CBM on Cphy3367 acts as a wedge that is required to free cellulose chains such that they can be cleaved by Cphy3367 and other cellulases. C. Attachment of the cell to the cellulosic substrate by Cphy3367 is required for efficient cellulose degradation. The Cphy3367 DUF291 domain binds to the cell and the CBM binds to cellulose.
Besides the GH9 hydrolytic module, Cphy3367 has additional domains that play important roles in cellulose degradation. Cphy3367 has two family 3 CBMs (CBM3): one adjacent to the hydrolytic domain and one at the C-terminus (Fig. 1B). CBMs promote polysaccharide degradation by bringing the enzyme into prolonged, close association with the surface of the carbohydrate. Although CBMs are generally non-catalytic, some CBMs have been shown to actively disrupt the polysaccharide structure (Din et al., 1994). In the GH9 subfamily with Cphy3367, the CBM adjacent to the GH9 domain has been shown to potentiate the catalytic activity while the C-terminal CBM anchors the enzyme to the insoluble, cellulosic substrate (Sakon et al., 1997). Both CBMs are required for the enzyme to have full activity on crystalline cellulose (Gilad et al., 2003). It has been proposed that the hydrolase domain and adjacent CBM of CelZ structurally modify the substrate by freeing cellulose chains to make them accessible to other cellulases (Riedel and Bronnenmeier, 1999). In the Hypocrea jecorina Cel7A hydrolase, the CBM and the catalytic domain are proposed to function as a molecular machine (Mulakala and Reilly, 2005) where the CBM is a wedge to pry cellulose chains from the crystalline cellulose surface. The CBM then functions as a Brownian ratchet to pass the freed chain to the GH domain for cleavage. Similarly, C. phytofermentans may require Cphy3367 to free cellulose chains from the cellulose fibril for attack by other cellulases (Fig. 7B).
Cphy3367 also has two, tandem C-terminal domains of unknown function (Fig. 2B) that are alternatively called DUF291, hydrophilic domains (Pages et al., 1999), or X2 modules (Mosbah et al., 2000). In addition to being found in Cphy3367, DUF291 domains are found in Cphy2128 (GH26), Cphy3202 (GH5) and Cphy3368 (GH48). Homologous domains are found in the scaffoldin proteins of C. cellulolyticum (CipC), C. cellulovorans (CbpA) and C. josui (CipA) as well as the hydrolases CelY and CelZ from C. stercorarium. The CbpA DUF291 domains bind both cellulose and the cell wall (Kosugi et al., 2004), and have thus been proposed both to promote cellulose degradation and to anchor the cellulosome to the cell surface. Similarly, the DUF291 domains of Cphy3367 may be critical to degrade cellulose or to attach Cphy3367 to the cell surface while the CBM domains bind Cphy3367 to the cellulosic substrate (Fig. 7C). The inability of AT02 to degrade cellulose could be due, at least in part, to an inability to retain the cell in close enough proximity to the cellulose substrate so that hydrolases can function efficiently.
Microbial cellulose degradation is of global ecological importance to recycle photosynthetically fixed carbon. Further, a mechanistic understanding of how microbes break down cellulose will facilitate the development of cellulosic biofuels as renewable, carbon-neutral alternatives to fossil fuels. This study demonstrates that we can now study the genetic basis of cellulose degradation using targeted chromosomal insertions in C. phytofermentans. If the intron is modified to contain a gene of interest, genomic insertion of the intron would allow stable expression of this gene on the chromosome. As these introns do not rely upon host-encoded factors for insertion, they may be generally applicable to the study of cellulolytic clostridia. In this study, we applied these introns for targeted disruption of a glycoside hydrolase gene, cphy3367. Among the many C. phytofermentans glycoside hydrolase genes that are upregulated during growth on cellulosic substrates, it is encouraging that the inactivation of a single gene had a strong effect upon cellulose degradation because it suggests that this process can be engineered using reverse genetics focusing on individual genes. Future genetic studies of C. phytofermentans will untangle the roles of additional hydrolases to provide a mechanistic understanding of cellulose degradation.
Experimental procedures
Culture conditions
Escherichia coli was cultured in LB medium supplemented with 50 μg ml−1 carbenicillin or 200 μg ml−1 erythromycin, as appropriate. C. phytofermentans was cultured anaerobically in GS2 (Johnson et al., 1981) with either 3 g l−1 glucose, 3 g l−1 cellobiose, 3 g l−1 hemicellulose xylan or 12 g l−1 cellulose. The hemicellulose used in this study was birchwood hemicellulose xylan (Sigma X0502), which consists primarily of poly β-1,4-d-xylopyranose. Cellulose consisted of 0.5 × 5 cm strips of Whatman #1 filter paper (Cat 1001-090). Degradation of cellulose was quantified as the dry mass of cellulose remaining in culture. Cellulose was collected on filters by vacuum filtration, dried overnight at 65°C and weighed.
Plasmid construction
The cphy3558 promoter was inserted into pAT19 (Trieu-Cuot et al., 1991) by PCR of the 400 bp region upstream of the cphy3558 start codon using primers Pferr_F and Pferr_R and cloning this fragment between the PstI and BamHI sites in pAT19 such that the promoter was directed towards the BamHI site. In addition to containing a 5′ BamHI site, the primer used to amplify the downstream portion of the cphy3558 promoter (Pferr_R) also contains an NdeI site just 3′ of the BamHI site. The NdeI site was used in a later step to target the intron to cphy3367 and can be used to clone other genes for expression from the cphy3558 promoter. The sequence of the cphy3558 promoter in pQexp was verified using primers PferrSeq_F and PferrSeq_R. To insert the intron into pQexp, a fragment containing the intron and ltrA gene was excised from pNL9164 (Yao et al., 2006) with HindIII and SfoI. The HindIII overhang was blunted using Klenow and the intron was cloned into pQexp at the SmaI site, which was directly downstream of the BamHI site used to clone the cphy3558 promoter. The intron borders in pQint were verified by sequencing. The intron integration site was chosen by calculating all potential sites for intron insertion into the 5′ 750 bp of cphy3367 using a probabilistic model (Perutka et al., 2004) that is implemented on the Targetron website (http://www.sigma-aldrich.com). An antisense integration site 37 bp downstream from the start codon was chosen. PCR primers were designed to promote base pairing between cphy3367 and the EBS1, EBS2, and gamma sequences of the intron RNA. The IBS1 and IBS2 sites in the 5′ exon of the intron were made complementary to the EBS1 and EBS2 sites for efficient RNA splicing. To retarget the intron to cphy3367, a 350 bp NdeI–BsrGI intron fragment containing the EBS, gamma and IBS sites was made by two-step cross-over PCR (Fig. 2C) using external primers Ibs_3367 and Ebs1D_3367 and internal primers Ebs2_3367 and Ebs_univ. The intron targeted to cphy3367 was inserted between the NdeI and BsrGI of pQint to form pQint3367. The intron sequence in pQint3367 was sequence verified using primers 3367intron_F and 3367intron_R. The sequences of primers used in this study are shown in Table 1.
Table 1.
PCR primers, plasmids and strains used in this study.
| Primer/plasmid/strain | Primer sequence/plasmid features/strain genotype | Function/source |
|---|---|---|
| Primer | ||
| Pferr_F | TGCATGCTGCAGTTTGGTCAACATTTAACCTC | Forward primer to clone cphy3558 promoter |
| Pferr_R | ACGTACGGATCCCATATGGTTTGAATATCCTCCTT | Reverse primer to clone cphy3558 promoter |
| PferrSeq_F | ATTAATGCAGCTGGCACGAC | Forward primer to sequence cphy3558 promoter in pAT19 |
| PferrSeq_R | CTGCAAGGCGATTAAGTTGG | Reverse primer to sequence cphy3558 promoter in pAT19 |
| Ebs_univ | CGAAATTAGAAACTTGCGTTCAGTAAAC | EBS universal primer to target pQint to cphy3367 |
| Ibs_3367 | AAAACATATGATAATTATCCTTAATGGACATCAGAGTGCGCCCAGATAGGGTG | IBS primer to target pQint to cphy3367 |
| Ebs2_3367 | TGAACGCAAGTTTCTAATTTCGGTTTCCATCCGATAGAGGAAAGTGTCT | EBS2 primer to target pQint to cphy3367 |
| Ebs1d_3367 | CAGATTGTACAAATGTGGTGATAACAGATAAGTCATCAGAAGTAACTTACCTTTCTTTGT | EBS1d primer to target pQint to cphy3367 |
| 3367intron_F | TTCGCCAGAAAACAAAAGAAA | Forward primer to sequence targeted intron in pQint3367 |
| 3367intron_R | ACTGTACCCCTTTGCCATGT | Reverse primer to sequence targteted intron in pQint3367 |
| 3367_F | ATTGGAACAAGGCAACTGCT | Forward primer upstream of cphy3367 |
| 3367_R | TAGCACTATTCGCGGACGAT | Reverse primer downstream of cphy3367 |
| erm_F | TGGAACAGGTAAAGGGCATT | Forward primer for internal ermR fragment from pAT19 |
| erm_R | GCGTGTTTCATTGCTTGATG | Reverse primer for internal ermR fragment from pAT19 |
| Plasmid | ||
| pAT19 | ermR, pAMbeta1 origin, pUC origin, RP4 oriT | Trieu-Cuot et al. (1991) |
| pNL9164 | Ll.LtrB-deltaORF intron, ltrA | Yao et al. (2006) |
| pRK24 | RP4 conjugal genes, tetR, ampR | Meyer et al. (1977) |
| pQexp | pAT19 with cphy3558 promoter | This study |
| pQint | pQexp with Ll.LtrB-deltaORF intron, ltrA | This study |
| pQint3367 | pQint with intron targeted to cphy3367 | This study |
| Strain | ||
| E. coli 1100-2 | mcrA0, endA1, mcrB9999 | Bandrin et al. (1983) |
| C. phytofermentans ISDg | Wild type | ATCC 700394 |
| C. phytofermentans AT02 | cphy3367::intron | This study |
Restriction enzyme sites in primers are underlined.
Conjugation
The conjugal donor used in matings was E. coli strain 1100-2 (Bandrin et al., 1983) transformed with the RP4 conjugal plasmid pRK24 (Meyer et al., 1977) and pQint3367. To mate E. coli and C. phytofermentans, a 10 ml culture of the E. coli donor was grown overnight in LB medium containing 50 μg ml−1 carbenicillin to select for pRK24 and 200 μg ml−1 erythromycin to select for pQint3367. The culture was washed twice with LB medium to remove residual antibiotics and re-suspended in 100 μl of LB medium. C. phytofermentans was cultured anaerobically in GS2 glucose medium overnight to late log phase (0.9 OD600). Approximately 0.5 ml of the C. phytofermentans culture was then centrifuged and re-suspended in 100 μl of GS2 medium. The E. coli and C. phytofermentans were mixed, aliquoted on solid GS2 medium as a series of 10 μl spots, and allowed to mate overnight at 32°C in anaerobic jars. Parallel, mock matings of C. phytofermentans and E. coli conjugal donors lacking pQint3367 were conducted to ensure that erythromycin resistance in C. phytofermentans required transfer of pQint3367. After mating, the cells were re-suspended from the plates into 5 ml of liquid GS2 medium containing 40 μg ml−1 nalidixic acid and 10 μg ml−1 trimethoprim and were cultured anaerobically for 3 h. These cultures did not contain erythromycin to permit the C. phytofermentans to recover from the mating. The cultures were centrifuged, re-suspended in 50 μl of GS2, and plated on GS2 solid medium containing 40 μg ml−1 nalidixic acid, 10 μg ml−1 trimethoprim and 40 μg ml−1 erythromycin. The plates were incubated anaerobically for 6 days at 32°C. Erythromycin-resistant C. phytofermentans colonies were picked, transferred to liquid GS2 medium containing 200 μg ml−1 erythromycin and cultured anaerobically. Once cultures had grown to OD600 > 0.5, the absence of residual E. coli cells was confirmed by plating a 50 μl aliquot on solid LB medium and incubating aerobically at 37°C overnight.
Southern blotting
To prepare a Southern blot, C. phytofermentans genomic DNA was isolated from 3 ml of cultures using a Qiagen Genomic Tip 20/G (Cat 10223). One microgram of genomic DNA was digested with HindIII, which does not cut in the intron insertion, and resolved by electrophoresis. The DNA was transferred to a nylon membrane (Perkin Elmer Genescreen, Cat NEF972001PK) and cross-linked using a Stratagene UV Stratalinker 2400. The blot was probed with a 32P-labelled intron PCR product that was amplified from pQint3367 with primers Ibs_3367 and Ebs1d_3367.
qRT-PCR
Cultures of wild-type and AT02 C. phytofermentans strains were grown in GS2 medium containing either hemicellulose xylan, glucose or cellulose as a sole carbon source. Cells were collected by centrifugation in mid-log phase for glucose and hemicellulose cultures; samples were taken from cellulose cultures after 2 weeks of growth. RNA was extracted using Ribopure Bacteria Kit (Ambion AM1925) and residual DNA was removed using DNase I (Ambion AM2222). Total RNA yields were approximately 5 μg of RNA per ml of culture. One microgram RNA was reverse transcribed to single-stranded DNA using the Superscript First Strand cDNA Synthesis Kit (Invitrogen 11904018). Real-time PCR amplification was conducted using a MJ Research DNA Engine Opticon II machine by monitoring incorporation of SYBR green I (Invitrogen S7563). PCR primers were designed to amplify 100 bp products from each gene (see Supporting information S3 for primer sequences). Relative gene expression was quantified using the comparative CT method (Schmittgen and Livak, 2008) with the 16S ribosomal sequence serving as the internal control gene. Expression levels represent the means of duplicate measurements taken from duplicate cultures (see Supporting information S3 for qRT-PCR data files).
Mass spectrometry
Protein lysates were prepared by French press from cultures of wild-type C. phytofermentans grown in GS2 medium containing either glucose, hemicellulose xylan or filter paper cellulose as the sole carbon source. Proteins were precipitated by adding one-fourth volume of 100% (w/w) trichloroacetic acid in water, re-suspended in 1% SDS, 0.2 M NaOH and 10 mM DTT and incubated for 60 min at 37°C. Cysteine residues were then derivatized with iodoacetic acid (30 mM) at room temperature in the dark for 60 min. Five hundred micrograms of protein was resolved on 4–12% Bis-Tris SDS-PAGE gels (Invitrogen NP0335BOX). Gels were sliced into eight sections and proteins were in-gel digested as described in Shevchenko et al. (1996). The resulting peptides were subjected to C18 solid-phase extraction using Waters SepPak cartridges (Waters WAT054960). Purified peptides were analysed by microcapillary liquid chromatography tandem mass spectrometry using an in-house prepared 125-μm-ID C18 reversed-phase column and a hybrid linear quadrupole/FT-ICR mass spectrometer (LTQ FT, Thermo Scientific, Bremen, Germany) as described in Haas et al. (2006).
Acquired MS/MS spectra were searched against a concatentated target/decoy protein sequence database consisting of the predicted proteins from C. phytofermentans (NCBI NC_010001.faa), common contaminants such as trypsin and human keratins, and the reversed sequences of these proteins used as decoy component (Elias and Gygi, 2007). Database searches were performed using the SEQUEST algorithm (Eng et al., 1994). Peptide assignments for each treatment were filtered to a protein false discovery rate of smaller than 1%. The number of peptides that could be uniquely mapped to cphy3366–9 were defined based upon peptide sequence, charge state and the gel section from which they were identified. Peptide counts per protein represent the mean of duplicate cultures for each treatment.
Acknowledgments
We thank A.L. Sonenshein, W. Haas, F. Winston and D. Spatt for technical expertise, S. Leschine, T. Warnick and J. Blanchard for their insights and for sharing strain ISDg, and J. Aach, K. Lemon and M. Umbarger for helpful discussions. This work was funded by a DOE GtL grant to G.M.C. and an SRA with Qteros, Inc.
References
- Arai T, Kosugi A, Chan H, Koukiekolo R, Yukawa H, Inui M, Doi RH. Properties of cellulosomal family 9 cellulases from Clostridium cellulovorans. Appl Microbiol Biotechnol. 2006;71:654–660. doi: 10.1007/s00253-005-0249-6. [DOI] [PubMed] [Google Scholar]
- Bandrin SV, Rabinovich PM, Stepanov AI. Three linkage groups of genes involved in riboflavin biosynthesis in Escherichia coli. Genetika (Sov Genet) 1983;19:1419–1425. [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Din N, Damude HG, Gilkes NR, Miller RCJ, Warren RA, Kilburn DG. C1-Cx revisited: intramolecular synergism in a cellulase. Proc Natl Acad Sci USA. 1994;91:11383–11387. doi: 10.1073/pnas.91.24.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Gilad R, Rabinovich L, Yaron S, Bayer EA, Lamed R, Gilbert HJ, Shoham Y. Cell, a noncellulosomal family 9 enzyme from Clostridium thermocellum, is a processive endoglucanase that degrades crystalline cellulose. J Bacteriol. 2003;185:391–398. doi: 10.1128/JB.185.2.391-398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Karberg M, Long M, Jones JP, Sullenger B, Lambowitz AM. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science. 2000;289:452–457. doi: 10.1126/science.289.5478.452. [DOI] [PubMed] [Google Scholar]
- Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, et al. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods. 2007;70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Jauris S, Rücknagel KP, Schwarz WH, Kratzsch P, Bronnenmeier K, Staudenbauer WL. Sequence analysis of the Clostridium stercorarium celZ gene encoding a thermoactive cellulase (Avicelase I): identification of catalytic and cellulose-binding domains. Mol Gen Genet. 1990;223:258–267. doi: 10.1007/BF00265062. [DOI] [PubMed] [Google Scholar]
- Jennert KC, Tardif C, Young DI, Young M. Gene transfer to Clostridium cellulolyticum ATCC 35319. Microbiology. 2000;146(Part 12):3071–3080. doi: 10.1099/00221287-146-12-3071. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Madia A, Demain AL. Chemically defined minimal medium for growth of the anaerobic cellulolytic thermophile Clostridium thermocellum. Appl Environ Microbiol. 1981;41:1060–1062. doi: 10.1128/aem.41.4.1060-1062.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karberg M, Guo H, Zhong J, Coon R, Perutka J, Lambowitz AM. Group II introns as controllable gene targeting vectors for genetic manipulation of bacteria. Nat Biotechnol. 2001;19:1162–1167. doi: 10.1038/nbt1201-1162. [DOI] [PubMed] [Google Scholar]
- Kormelink FJ, Gruppen H, Viëtor RJ, Voragen AG. Mode of action of the xylan-degrading enzymes from Aspergillus awamori on alkali-extractable cereal arabinoxylans. Carbohydr Res. 1993;249:355–367. doi: 10.1016/0008-6215(93)84100-k. [DOI] [PubMed] [Google Scholar]
- Kosugi A, Amano Y, Murashima K, Doi RH. Hydrophilic domains of scaffolding protein CbpA promote glycosyl hydrolase activity and localization of cellulosomes to the cell surface of Clostridium cellulovorans. J Bacteriol. 2004;186:6351–6359. doi: 10.1128/JB.186.19.6351-6359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S. Mobile group II introns. Annu Rev Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- Leschine SB. Cellulose degradation in anaerobic environments. Annu Rev Microbiol. 1995;49:399–426. doi: 10.1146/annurev.mi.49.100195.002151. [DOI] [PubMed] [Google Scholar]
- Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, et al. How biotech can transform biofuels. Nat Biotechnol. 2008;26:169–172. doi: 10.1038/nbt0208-169. [DOI] [PubMed] [Google Scholar]
- Lyras D, Rood JI. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid. 1998;39:160–164. doi: 10.1006/plas.1997.1325. [DOI] [PubMed] [Google Scholar]
- Maamar H, de Philip P, Bélaich J, Tardif C. ISCce1 and ISCce2, two novel insertion sequences in Clostridium cellulolyticum. J Bacteriol. 2003;185:714–725. doi: 10.1128/JB.185.3.714-725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamar H, Valette O, Fierobe H, Bélaich A, Bélaich J, Tardif C. Cellulolysis is severely affected in Clostridium cellulolyticum strain cipCMut1. Mol Microbiol. 2004;51:589–598. doi: 10.1046/j.1365-2958.2003.03859.x. [DOI] [PubMed] [Google Scholar]
- Meyer R, Figurski D, Helinski DR. Physical and genetic studies with restriction endonucleases on the broad host-range plasmid RK2. Mol Gen Genet. 1977;152:129–135. doi: 10.1007/BF00268809. [DOI] [PubMed] [Google Scholar]
- Mosbah A, Belaïch A, Bornet O, Belaïch JP, Henrissat B, Darbon H. Solution structure of the module X2 1 of unknown function of the cellulosomal scaffolding protein CipC of Clostridium cellulolyticum. J Mol Biol. 2000;304:201–217. doi: 10.1006/jmbi.2000.4192. [DOI] [PubMed] [Google Scholar]
- Mulakala C, Reilly PJ. Hypocrea jecorina (Trichoderma reesei) Cel7A as a molecular machine: a docking study. Proteins. 2005;60:598–605. doi: 10.1002/prot.20547. [DOI] [PubMed] [Google Scholar]
- Pages S, Bélaïch A, Fierobe HP, Tardif C, Gaudin C, Bélaïch JP. Sequence analysis of scaffolding protein CipC and ORFXp, a new cohesin-containing protein in Clostridium cellulolyticum: comparison of various cohesin domains and subcellular localization of ORFXp. J Bacteriol. 1999;181:1801–1810. doi: 10.1128/jb.181.6.1801-1810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlack RD, Wright LL, Graham RL, Stokes BJ, Erbach DC. ) Biomass as feedstock for a bioenergy and bioproducts industr: the technical feasibility of a billion-ton annual supply. USDA/DOE joint report, 1–78.
- Perret S, Maamar H, Bélaich J, Tardif C. Use of antisense RNA to modify the composition of cellulosomes produced by Clostridium cellulolyticum. Mol Microbiol. 2004;51:599–607. doi: 10.1046/j.1365-2958.2003.03860.x. [DOI] [PubMed] [Google Scholar]
- Perutka J, Wang W, Goerlitz D, Lambowitz AM. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J Mol Biol. 2004;336:421–439. doi: 10.1016/j.jmb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Purdy D, O'Keeffe TAT, Elmore M, Herbert M, McLeod A, Bokori-Brown M, et al. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol. 2002;46:439–452. doi: 10.1046/j.1365-2958.2002.03134.x. [DOI] [PubMed] [Google Scholar]
- Riedel K, Bronnenmeier K. Active-site mutations which change the substrate specificity of the Clostridium stercorarium cellulase CelZ implications for synergism. Eur J Biochem. 1999;262:218–223. doi: 10.1046/j.1432-1327.1999.00374.x. [DOI] [PubMed] [Google Scholar]
- Sakon J, Irwin D, Wilson DB, Karplus PA. Structure and mechanism of endo/exocellulase E4 from Thermomonospora fusca. Nat Struct Biol. 1997;4:810–818. doi: 10.1038/nsb1097-810. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Tolonen AC, Liszt GB, Hess WR. Genetic manipulation of Prochlorococcus strain MIT9313: green fluorescent protein expression from an RSF1010 plasmid and Tn5 transposition. Appl Environ Microbiol. 2006;72:7607–7613. doi: 10.1128/AEM.02034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu-Cuot P, Carlier C, Poyart-Salmeron C, Courvalin P. Shuttle vectors containing a multiple cloning site and a lacZ alpha gene for conjugal transfer of DNA from Escherichia coli to gram-positive bacteria. Gene. 1991;102:99–104. doi: 10.1016/0378-1119(91)90546-n. [DOI] [PubMed] [Google Scholar]
- Warnick TA, Methé BA, Leschine SB. Clostridium phytofermentans sp. nov., a cellulolytic mesophile from forest soil. Int J Syst Evol Microbiol. 2002;52:1155–1160. doi: 10.1099/00207713-52-4-1155. [DOI] [PubMed] [Google Scholar]
- Williams DR, Young DI, Young M. Conjugative plasmid transfer from Escherichia coli to Clostridium acetobutylicum. J Gen Microbiol. 1990;136:819–826. doi: 10.1099/00221287-136-5-819. [DOI] [PubMed] [Google Scholar]
- Yao J, Lambowitz AM. Gene targeting in gram-negative bacteria by use of a mobile group II intron (‘Targetron’) expressed from a broad-host-range vector. Appl Environ Microbiol. 2007;73:2735–2743. doi: 10.1128/AEM.02829-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhong J, Fang Y, Geisinger E, Novick RP, Lambowitz AM. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA. 2006;12:1271–1281. doi: 10.1261/rna.68706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Karberg M, Lambowitz AM. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 2003;31:1656–1664. doi: 10.1093/nar/gkg248. [DOI] [PMC free article] [PubMed] [Google Scholar]