

Abstract
It is unresolved whether recently transmitted human immunodeficiency viruses (HIV) have genetic features that specifically favour their transmissibility. To identify potential “transmission signatures”, we compared 20 full-length HIV-1 subtype C genomes from primary infections, with 66 sampled from ethnically and geographically matched individuals with chronic infections. Controlling for recombination and phylogenetic relatedness, we identified 39 sites at which amino acid frequency spectra differed significantly between groups. These sites were predominantly located within Env, Pol and Gag (14/39, 9/39 and 6/39 respectively) and were significantly clustered (33/39) within known immunoreactive peptides. Within 6 months of infection we detected reversion-to-consensus mutations at 14 sites and potential CTL escape mutations at seven. Here we provide evidence that frequent reversion mutations probably allows the virus to recover replicative fitness which, together with immune escape driven by the HLA alleles of the new hosts, differentiate sequences from chronic infections from those sampled shortly after transmission.
Keywords: HIV, subtype C, primary infection, immune escape, reversion
Introduction
It is well established that HIV populations experience extreme bottlenecks during sexual transmission (Derdeyn et al., 2004; Wolfs et al., 1992) with approximately 80% to 90% of infections being a consequence of a single transmitted variant (Abrahams et al., 2009; Haaland et al., 2009; Keele et al., 2008). The strongest evidence that “transmission sieves” have been a major factor in HIV evolution is that, relative to viruses sampled during chronic infections, recently transmitted HIV-1 subtype C genetic variants are in general more sensitive to neutralization and tend to have both shorter V1-V2 and V1-V4 loops and fewer glycosylation sites (Derdeyn et al., 2004; Li et al., 2006; Rong et al., 2007). Differences in sites under selection have also been identified between the envelope glycoproteins (gp41) of viruses from the primary and chronic infection phases suggesting the existence of different selective pressures during these different infection phases (Bandawe et al., 2008).
However, outside of studies on env, there is limited information on features which distinguish recently transmitted viruses from those found in chronic infections. Viruses from chronic infections have usually undergone strong cytotoxic T lymphocyte (CTL) driven selection pressures and are therefore expected to have accumulated immune escape mutations. These CTL escape mutations, while highly adaptive within the context of immune environments where hosts have the appropriate HLA-alleles (Brumme et al., 2007; Brumme, et al., 2008; Kelleher et al., 2001; Rousseau et al., 2008), can also seriously diminish viral replicative fitness (Allen et al., 2005; Brockman et al., 2007; Liu et al., 2006; Liu et al., 2007; Martinez-Picado et al., 2006; Miura et al., 2009). Although there is some evidence from mother to child transmission pair studies that fitter virus variants are selectively transmitted (Kong et al., 2008), other studies have shown that genetic variants that carry attenuating CTL escape mutations are also transmitted (Chopera et al., 2008;Goepfert et al., 2008).
Following transmission, viruses generally accumulate both immune evasion mutations and reversion mutations that recoup replicative fitness losses experienced due to deleterious escape mutations accrued in previous hosts (Brumme et al., 2008; Leslie et al., 2004; Liu et al., 2007; Matthews et al., 2008; Rousseau et al., 2008). The rate at which such reversion mutations occur is most likely dependant on the magnitude of their effects on replicative fitness (Brumme et al., 2008; Matthews et al., 2008). The clinical importance of viruses carrying attenuating CTL escape mutations is that the recipients of such viruses will, in some cases at least, have lower set-point viral loads, higher CD4 counts and, possibly, better survival prospects (Chopera et al., 2008;Goepfert et al., 2008).
A successful HIV vaccine will need to effectively combat viruses during the earliest stages of infection. Identifying the specific genetic features that might predispose particular HIV variants to being more transmissible than others, and understanding the evolutionary processes at play during the early evolution of successfully transmitted variants, are therefore both important for defining potential targets for vaccine induced immunity. As events during acute HIV-1 infections are thought to have a disproportionately large influence on both long-term disease outcomes (deWolf et al., 1997; Lavreys et al., 2006) and global HIV evolution in general (Rambaut et al., 2004), understanding the transmission bottleneck and the subsequent evolution of successfully transmitted variants are probably key to identifying and understanding the viral and host determinants of HIV pathogenesis.
To identify genetic features that are characteristic of recently transmitted viruses, we developed a phylogeny and recombination aware method to compare amino acid mutation spectra between groups of sequences. We used this approach to identify amino acid sites that differentiated between full-length HIV-1 subtype C genomes sampled during primary and chronic infections. We then examined longitudinally sampled sequences to infer the processes that might underlie the amino acid frequency differences observed in viruses from the different infection phases.
Results
Classification of infection stages
A cohort of twenty women experiencing primary HIV-1 infections was recruited as part of the CAPRISA 002 acute infection study (van Loggerenberg et al., 2008) (Table 1). These women were estimated to have been infected for a median of 39 days (range 22 to 62 days) at enrolment. Most participants had high viraemia with a median viral load of 110 900 copies per ml (range from 610 to 621 000 copies/ml; Table 1).
Table 1.
Summary of participants’ clinical markers, laboratory staging, and full-length genome template diversity.
| Participants | Sample Date (month-day-year) |
aDays post infection |
Viral load (copies/ml) |
CD4 count (cells/μl) |
bLaboratory stage |
cSequence template diversity (V1V2) |
|---|---|---|---|---|---|---|
| CAP8 | 05-17-2005 | 23 | 373000 | 360 | V | 1 |
| CAP30 | 10-27-2004 | 35 | 10200 | 989 | V | 1 |
| CAP45 | 05-11-2005 | 35 | 236000 | 974 | V | ND |
| CAP61 | 12-20-2004 | 57 | 610 | 389 | VI | 1 |
| CAP63 | 01-26-2005 | 34 | 202000 | 584 | dV | 2 |
| CAP65 | 09-06-2005 | 42 | 90800 | 243 | VI | 2 |
| CAP84 | 02-28-2005 | 22 | 9140 | 636 | V | 2 |
| CAP85 | 06-22-2005 | 23 | 621000 | 419 | V | 2 |
| CAP88 | 02-17-2005 | 36 | 29400 | 963 | VI | 1 |
| CAP174 | 10-04-2005 | 28 | 474000 | 353 | VI | 1 |
| CAP206 | 07-12-2005 | 41 | 368000 | 365 | VI | 1 |
| CAP210 | 05-25-2005 | 36 | 127000 | 461 | V | 1 |
| CAP228 | 05-18-2005 | 53 | 2360 | 851 | dVI | 1 |
| CAP229 | 07-19-2005 | 48 | 126000 | 558 | ND | 1 |
| CAP239 | 08-10-2005 | 36 | 95800 | 845 | V | 2 |
| CAP244 | 05-23-2005 | 58 | 19200 | 557 | VI | 1 |
| CAP248 | 05-24-2005 | 62 | 55000 | 420 | V | 1 |
| CAP255 | 06-21-2005 | 54 | 196000 | 693 | VI | 1 |
| CAP256 | 09-05-2005 | 42 | 56500 | 689 | VI | 1 |
| CAP257 | 09-12-2005 | 49 | 276000 | 450 | V | 2 |
Infection date was estimated as the midpoint between the last negative and first positive antibody test or as 14 days if the sample was PCR positive, antibody negative sample. ND = not done
No. of bands on heteroduplex tracking assay gel
determined on samples from a week before.
Characterization of full-length HIV-1 genomes
Full-length genomes were amplified and genetic homogeneity in V1V2 of the template, indicative of amplification from a single genome, was confirmed for 13 out of 20 amplicons. Heterogeneity was identified in each of the remaining seven samples (Table 1). Amplicons were cloned and sequenced from each of the 20 study participants. All 20 of the full length genome sequences clearly belonged to HIV-1 subtype C and none were detectably inter-subtype recombinants (supplementary figure 1).
To identify polymorphisms associated with recently transmitted viruses, we compiled from public databases a dataset of subtype C chronic sequences which were closely matched to our acute infection dataset for geographical origin, host population and mode of transmission. As we were interested in identifying genetic features that differed between viruses sampled during primary and chronic infections, it was necessary to ensure that there were no obvious sampling biases. The mean genetic distances between the env genes of viruses within each dataset was similar: 11.5% (range 8.2% - 14.8%) in the primary infection dataset compared to 10.9% (range 6% - 15.1%) in the chronic infection dataset. In addition, there was no obvious evidence of close epidemiological linkage as the sequences were generally dispersed throughout a subtype C phylogenetic tree containing viruses sampled world-wide (Supplementary S1). A comparison of the 86 sequences used in this study showed limited structure in the phylogenetic tree (Figure 1) with only seven lineages displaying bootstrap support above 75%. Of these seven lineages, six consisted of only two sequences each. Most lineages contained a mixture of acute and chronic sequences. Thus, despite a common geographic origin, there was no obvious evidence of close genetic and phylogenetic relationships within or between primary and chronic sequences.
Figure 1.

Maximum Likelihood tree of env gene sequences from primary (n = 20) and chronic (n = 66) infection HIV-1 subtype C strains. The HXB2 subtype B strain was used as root and 100 bootstrap replicates were done. Primary infection strains are indicated in blue squares and chronic strains as unlabelled tips. Subclusters indicated in red had bootrap values ≥85%. Scale bar = 0.05.
Envelope glycoprotein variable loop length and N-linked glycosylation
Previous studies have shown statistically significant differences in both the lengths of variable loops and the numbers of N-linked glycosylation (PNGs) sites found in the envelope glycoproteins of viruses sampled during primary and chronic infections (Derdeyn et al., 2004; Li et al., 2006). Consistent with these studies we found significantly fewer PNGs in the V1V2 loop regions of the viral Env sequences sampled during primary infections (p = 0.025) (Figure 2a). We did not, however, find any significant differences between the two datasets with respect to either the number of PNGs across the entire V1V4 region (median of 20 for both primary and chronic) or in the lengths of the V1V2 (median of 67 and 68 amino acids in the primary and chronic datasets, respectively) and V1V4 regions (median of 280.5 and 280 amino acids in the primary and chronic datasets respectively).
Figure 2.

(a) Number of potential N-linked glycosylation sites (PNGs) in the V1-V2 variable domains of gp120 from HIV-1 subtype C strains from primary and chronic infection. (b) Amino acid positions that displayed a significant difference between primary and chronic infection subtype C sequences are showed graphically across the HIV-1 proteome (p<0.025).
Site-specific differences in amino acid frequencies between the primary and chronic infection datasets
We used a phylogenetic approach to test for more subtle differences between the primary and chronic infection datasets. Our method accounts for detectable signals of recombination and controls for founder effects in the underlying evolution of these sequences (Bhattacharya et al., 2007; Scheffler et al., 2006). The method infers the amino acid states of ancestral viruses, and evaluates the difference in the mutational patterns between two groups of sequences, at each site along a protein sequence alignment (See Materials and Methods for details). Intra-subtype recombination breakpoints were identified in gag, pol, env and nef genes. However, no recombination breakpoints were found in vif, vpr, vpu and tat using the GARD method (Pond et al., 2006;http://www.datamonkey.org).
Amino acid frequency spectra in the primary and chronic infection datasets differed most notably at 39 amino acid sites. These 39 sites were identified using a phylogenetically corrected test with a multiple testing uncorrected one tail p-value cut-off of 0.025. We used a permutation test to investigate the impact of multiple hypothesis testing on our results. We permuted the sample labels (i.e. primary versus chronic infection) randomly 1,000 times and counted the number of sites in each permuted dataset that differed significantly (p < 0.025) between the permuted primary and chronic groups. While in the observed (unpermuted) data there were 39 sites with a p-value below 0.025, among the 1,000 permuted samples, the mean number of sites with a p-value below 0.025 was 9.3 and there were no permuted datasets with as many as 39 sites with associated p-values less than 0.025. This provided evidence that the 39 sites we identified were significantly enriched for sites displaying genuine allele frequency differences between our chronic and acute infection datasets. Specifically, we estimated that the false discovery rate amongst the 39 identified sites was approximately 24% (9.4/39 sites are false positives).
Fourteen of the 39 sites were within Env, nine in Pol, six in Gag, four in Nef, three in Vif, two in Vpr and one in Vpu (Figure 2b). We then investigated each site in detail to identify possible biological processes responsible for these differences.
Sites differentiating the primary and chronic infection datasets have higher entropy in the primary infection dataset
To better explore the nature of the changes in amino acid mutational spectra between the primary and chronic infection datasets, we examined the relative entropies of the 39 identified sites (Figure 3a). On average, the site-specific entropy was higher in the primary infection dataset (median = 0.518) than it was in the chronic infection dataset (median = 0.263, p < 0.0001, two-tailed Wilcoxon rank-sum test) (Figure 3b). Based on analysis of HIV-1 protein sequences sampled from public databases, Bansal et al. (2005) defined high entropy sites as those with an entropy score greater than 0.25 and low entropy sites as those with an entropy score less than 0.15. Whereas in the primary infection dataset all 25/39 sites had high entropy, in the chronic infection dataset only twelve had high entropy. Seventeen sites with entropies from 0.325 to 0.588 in the primary infection dataset were either fully conserved or highly conserved, in the chronic infection dataset (Figure 3c).
Figure 3.

A comparison of the 39 positions identified as significant changes (p<0.025), in amino acid spectra between sequences from primary and chronic infection showing (a) amino acid variety and relative frequency in primary and chronic infection (b) difference in median entropy (p<0.05) and (c) site-specific entropy at each position.
Sites with differential amino acid frequency spectra are significantly clustered within known CTL epitopes
It has been suggested that there is typically higher sequence entropy at amino acid positions where escape mutations occur (Liu et al., 2007) and that CTL responses during early infections mostly target peptides with high degrees of entropy (Bansal et al., 2005). To investigate whether the sites identified by our analysis were associated with CTL responses we checked the sites against the genomic positions of peptides that were immuno-reactive in Elispot assays (http://www.hiv.lanl.gov/content/immunology/hlatem/study4/index.html; Gray et al., 2009; Kiepiela et al., 2007; Matthews et al., 2008). We found that 33 of the 39 sites were located within immunoreactive peptides (Table 2). Immunoreactivity has been mapped to approximately 48% of the HIV-1 subtype C proteome. We found that the 39 sites clustered more frequently within these immunoreactive regions than is expected by chance (p= 0.006). This implied that polymorphisms at the sites differentiating the primary and chronic infection datasets are most likely associated with CTL immune pressures.
Table 2.
Amino acid positions were the frequency of gain and loss of specific amino acids at terminal branches differ significantly between HIV-1 subtype C strains from primary and chronic infection.
| Protein |
aAmino acid position (HXB2) |
P-value |
bSubtype C CTL reactive peptide sequence (site in boldface and underlined) |
bKnown HLA restriction |
|---|---|---|---|---|
| Gag p17 | 69 | 0.0037373 | EGCKQIMKQLQPALQTGT, QLQPALQTGTEELRSLY | B*0801, B*4006, A2, A*0101, B57 |
| Gag p17 | 72 | 0.0103261 | QLQPALQTGTEELRSLY | B*0801, B*4006, A2, A*0101, B57 |
| Gag p17 | 105 | 0.0131367 | EALDKIEEEQNK | A11 |
| Gag p24 | 138 | 0.0097975 | GKVSQNY/PIVQNLQGQMV | B13, A68, A*6802, A*2402 |
| Gag p24 | 228 | 0.0163842 | PVAPGQMREPRG | B35, B13 |
| Gag p2 | 371 | 0.0192564 | EAMSQANSVNIM | A2, A*0201, A2 supertype, B*4002, B*4501 |
| Pol protease | 113 | 0.0103261 | GGIGGFIKVRQYDQIL | A2, B13, Cw6 |
| Pol protease | 128 | 0.0015348 | QIPIEICGKKAIGTVLV, GKKAIGTVLVGPTPVNII | B*1503, B57, B58, B63 |
| Pol protease | 131 | 0.0103261 | GKKAIGTVLVGPTPVNII | B*1503, B57, B58. B63, A*0201 |
| Pol RT | 276 | 0.0165847 | DAYFSVPLDEGFRKYTAF | B*5702, B*5703, B35, B*3501, A11 |
| Pol RT | 447 | 0.0140015 | AKALTDIVPLTEEA | B*0702, B*1501, B*3501, B*5101, B*5301, B35, B51, B7 |
| Pol integrase | 726 | 0.0193219 | KAQEEHEKYHSNWR | B*4403 |
| Pol integrase | 756 | 0.0103261 | EIVASCDKCQLKGE | B*8101 |
| Pol integrase | 813 | 0.0103261 | PAETGQETAYYILKLAGR | A*6802, A*2601, B7, B56 |
| Pol integrase | 850 | 0.0131367 | VKAACWWAGIQQEFGIPYNPQS | A2 supertype, B*1503 |
| Vif | 46 | 0.0103261 | RHHYESRHPKVSSE | B*0702, B*4201, B7 |
| Vif | 78 | 0.0158979 | D/WHLGHGVSI/, LQTGERDWHLGHGVSIEW | B*1510, B*5703, B35 |
| Vif | 137 | 0.0103261 | HIVSPRCDYQAGHNKVGSLQYLAL | |
| Vpr | 68 | 0.0015348 | AIIRILQQL/L | A*0201, A2, A2 supertype |
| Vpr | 81 | 0.0103261 | GCQHSRIGILRQR | |
| Vpu | 33 | 0.0097975 | YIEYRKLVRQR, EYRKILRQR | A*3303 |
| Env gp120 | 106 | 0.0103261 | KNDMVDQMHEDIISLW | A*0201, B*3801, A2, |
| Env gp120 | 162 | 0.0015348 | CSFNITTELRDKKQKVYA, NCSFNISTSI | Cw8, Antibody pressure |
| Env gp120 | 171 | 0.0007625 | CSFNITTELRDKKQKVYA | |
| Env gp120 | 184 | 0.0099403 | YALFYRLDIVPLNENNSSEY | |
| Env gp120 | 340 | 0.0192564 | HCNISEAAWNKTLQQVR | A11, A*0201 |
| Env gp120 | 352 | 0.0200731 | QQVRKKLEEHFPNKTIIF | A*0201, A11 |
| Env gp120 | 476 | 0.0197145 | TFRPGGGDMRRNWRSELY, MRRNWRSELYKYKVVEI | A*2601 |
| Env gp120 | 477 | 0.0003449 | TFRPGGGDMRRNWRSELY, MRRNWRSELYKYKVVEI | A*2601 |
| Env gp120 | 485 | 0.0103261 | NWRSELYKYKVVEI | |
| Env gp41 | 535 | 0.0191913 | GSTMGAASITLTVQARQ | A2 |
| Env gp41 | 583 | 0.0015348 | GIKQLQTRVLAIERYLK, RVLAIERYLKDQQLLGIW | B*5802, B14 |
| Env gp41 | 668 | 0.0173911 | EKDLIALDKW(Q/N)NLWNWFDIT | |
| Env gp41 | 687 | 0.0165847 | WYIKIFIMIVGGLIGLR | A*2402, A2, A*0201 |
| Env gp41 | 708 | 0.0103261 | AVLSVVNRVRQGYSPLS | A*2501, A*3002, A30 |
| Nef | 5 | 0.0131367 | MGGKWSKSSIV | A2, A*2501 |
| Nef | 65 | 0.0000520 | WLRAQEEEEEVGFPVRPQV, EVGFPVRPQVPLRPMTFK | B*4501, B45, B7, A*0201, A1, B8, B35 |
| Nef | 88 | 0.0171700 | KAAFDLSFF, GAFDLSFFL | B57/ B*5801, A*0205, B60, B62, A2, Cw8, Cw*0802 |
| Nef | 169 | 0.0103261 | LLHPMSQHGMDDPER | B35 |
Sites identified with a p-value <0.025 are reported.
Reactive peptides of which some contains published CTL epitopes were obtained from the Los Alamos HIV database (http://www.hiv.lanl.gov/content/immunology/hlatem/study4/index.html, http://www.hiv.lanl.gov/content/immunology/ctl_search )
Longitudinal monitoring of evolution at amino acid sites which differed between primary and chronic phases of infection
To more directly determine the nature of discordant amino acid mutation spectra in our primary and chronic infection datasets, we obtained longitudinal samples from 18 of the 20 study participants at between three and six months after our initial samples were taken. We were specifically interested in determining whether increased entropy at the sites identified in our analysis was due to (i) viruses sampled in primary infections carrying transient immune evasion mutations that they had carried over from former hosts (reversion), (ii) viruses accumulating novel immune evasion mutations in response to changes in the immune environment following transmission (escape), or (iii) a combination of both (i) and (ii).
Here we defined probable CTL escape mutations as amino acid substitutions within epitopes restricted by patient HLA alleles (or in immediately adjacent amino acids) where changes were from amino acids found in ≥50% in the population (i.e. consensus or wild-type states) to amino acids found in <50% of the population (i.e. mutant states; Allen et al., 2005; Liu et al., 2006; Li et al., 2007). Conversely we defined probable reversion mutations as being amino acid substitutions within known CTL epitopes that were not targeted by a patients HLA alleles in which low frequency amino acids were replaced with high frequency ones such as those corresponding with the subtype consensus sequence (Allen et al., 2005; Liu et al., 2006; Li et al., 2007).
Amino acid changes were seen in 13 participants at 20 sites, of which four sties were associated with both escape and reversion (Tables 3 & 4). Evolution from low to high frequency amino acids (putative reversion) occurred at 14 sites, and evolution to low frequency amino acids (putative CTL escape) were seen at seven sites. At three sites we saw escape followed by reversion to the original wild-type amino acid within six months (transient escape) post infection. In total, eight putative escape events were identified at seven sites (Vif 78, Env 162, Env 352, and Nef 65) in six individuals with one individual (CAP256) showing escape at three sites (Gag 371, Pol 113, and Vif 78). Seven sites associated with escape evolved from high frequency (median frequency of 0.775) to low frequency (median frequency of 0.168) amino acids. At position 65 in Nef a change from the consensus E (frequency of 0.865) to a D is associated with CTL escape in HLA-B*45/ B*4501 positive individuals (Rousseau et al., 2008). However in CAP63 position 65 in Nef evolved from a low frequency amino acid (G = 0.046) to another low frequency amino acid (D = 0.07). Although it is slightly more frequent, this new amino acid polymorphism was classified as an escape mutation. It is also possible that the original G polymorphism was itself also an early escape mutation as the first sample recorded for this patient was only obtained approximately 34 days post infection. Mutation to D at this site may have simply provided more selectively beneficial escape than was provided by the intermediate G state. In three participants the Vif and Nef sites were located in peptides restricted by the host HLA (B*1503 and HLA-B*45 respectively) providing further evidence that these sites were associated with evasion of CTL responses. The one putative escape in Env 162 reverted to consensus at 29 weeks with concomitant escape at an adjacent site. This oscillation of amino acids within nine–mer CTL epitopes is commonly observed in the early stages of escape prior to the selective expansion of viruses carrying in most cases just the single highest fitness escape mutation (Borrow et al., 1997; Delport et al., 2008; Iversen et al., 2006). However as this site was located in an N-linked glycosylation motif, it is also possible that antibody pressures played some role in its selective value.
Table 3.
Putative escape mutations within CTL epitopes
| Site | PID | Weeks post infection |
Putative epitopes aligned to matching test peptides |
Amino acid frequency change | HLA restricted |
|---|---|---|---|---|---|
| Gag 371 | CAP256 | 6 13 30 |
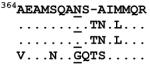 |
77.48> 8.96 N>N>G |
Affinity = 6.24 B*1503 |
| Pol 113 | CAP256 | 6 13 30 |
 |
97.68> 2.09 R>R>K |
Affinity = 457.37 B1503 |
| Pol 756 | CAP45 | 2 5 12 |
 |
98.84> 0.46 D>D>G |
No |
| Vif 78 | CAP256 | 6 13 30 |
 |
0.386 → 0.061 E→A |
B*1503 |
| Vpr 81 | CAP239 | 5 11 22 |
 |
100> 0 I>I>M |
No |
| Env 352 | CAP244 | 8 12 28 |
 |
0.727 → 0.168 E→K |
No |
| Nef 65 | CAP85 | 5 13 29 |
 |
0.865 → 0.07 E → D |
B *4501 |
| CAP63 | 5 11 29 |
 |
0.046 → 0.07 G → D |
B *4501 | |
| Transient escape at sites within 6 months post infection | |||||
|---|---|---|---|---|---|
| Vif 46 | CAP45 | 2 5 12 |
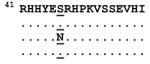 |
96.33> 0.73> 96.33 S>N>S |
Affinity = 137.72, A*2902 |
| Env 162 | CAP63 | 5 11 29 |
 |
0.979 → 0.006 → 0.979 T→S→T |
No |
| Nef 88 | CAP257 | 7 14 30 |
 |
76.28> 23.21 S>G>S |
Affinity = 183.39 B*4202 |
Table 4.
Putative reversion mutations
| Site | PID | Weeks post infection |
Putative epitopes aligned to matching test peptides |
Amino acid frequency change |
aFold frequency increase |
HLA restricted |
|---|---|---|---|---|---|---|
| Gag 69 | CAP256 | 6 13 30 |
 |
0.157 → 0.823 K → Q |
5.24 | No |
| Gag 228 |
CAP256 | 6 13 30 |
 |
0.0024 → 0.981 I → I → M |
409 | No |
| Pol 131 | CAP174 | 4 28 |
0 → 1.00 A →V |
B*5802 | ||
| Pol 447 | CAP255 | 8 13 |
 |
0.0534 → 0.944 V → I |
17.7 | No |
| Pol 850 | CAP61 | 8 11 33 |
 |
0.0139 → 0.981 V → I → I |
70.6 | No |
| CAP174 | 4 28 |
 |
0.0046 → 0.981 T → I |
213.3 | No | |
| Vif 137 | CAP256 | 6 13 30 |
 |
0.0073 → 0.993 T →T → A |
136 | A*2902 |
| Vpr 81 | CAP206 | 8 15 33 |
 |
0 → 1.00 V → I → I |
*No | |
| Env 106 | CAP239 | 5 11 22 |
 |
0.0019 → 0.932 K → K → E |
490.5 | No |
| Env 162 | CAP206 | 8 15 33 |
 |
0.004 → 0.979 A → T |
245 | No |
| Env 352 | CAP256 | 6 13 30 |
 |
0.168 → 0.727 K → E |
4.33 | No |
| Env 477 | CAP85 | 5 13 29 |
 |
0.070 → 0.927 N → D |
13.2 | No |
| CAP256 | 6 13 30 |
 |
0.070 → 0.927 N → D |
13.2 | No | |
| Env 535 | CAP256 | 6 13 30 |
 |
0.063 → 0.852 M → M → I |
13.5 | No |
| Env 668 | CAP256 | 6 13 30 |
 |
0.173 → 0.781 S → N |
4.5 | No |
| Nef 65 | CAP30 | 5 11 29 |
 |
0.070 → 0.865 D → E |
12.4 | B*4501 |
| CAP84 | 3 14 19 |
 |
0.070 → 0.865 D → E |
12.4 | No | |
Fold frequency increase: the difference in frequency of an amino acid at an alignment position compared to the frequency of another amino acid at the same alignment position
In total 17 potential reversion mutations were identified at 14 sites, within viruses sampled from nine of the study participants. Longitudinally sampled viruses from CAP256 showed putative reversion at 7/14 sites with CAP174 and CAP206 each having putative reversion mutations at two sites. Reversion mutations involved substitutions of low frequency amino acids (median population-wide frequencies = 0.058 at the site in question) with higher frequency amino acids (median population-wide frequencies = 0.930). These potential reversion mutations were distributed throughout the genome with six occurring in Env, three in Pol, two in Gag, and one each in Vif, Vpr and Nef (Table 4). There was no predicted HLA association for 14 out of the 17 reversion mutations providing further evidence that these sites were associated with reversion of CTL escape mutations that had occurred in former hosts which had different HLA alleles than the virus’ current hosts. The one exception was a probable reversion mutation located at Nef 65 within the CTL epitope restricted by one participant’s HLA-B*4501 allele. Importantly, escape mutations were also seen at adjacent positions (63, 64) within this putative CTL epitope. The potential reversion at amino acid position 162 in Env is probably associated with regain-of-function as this site is almost invariably a threonine (T) residue (HIV-1 subtype C population-wide frequency = 0.979) and resides within a potential N-linked glycosylation site motif in the V2 loop.
In summary, a total of 28 evolutionary events were observed in 13 participants at 20 sites of which three events were associated with transient escape (10.7%), eight with putative escape (28.6%) and 17 with putative reversion (60.7%). Thus the longitudinal evolutionary changes observed at these sites were mainly associated with reversion to high frequency amino acids during primary infection with a minority of changes potentially being associated with CTL escape.
Discussion
HIV transmission is associated with a severe virus population bottleneck and there is some evidence that certain genotypic and phenotypic properties of the viral envelope are selectively advantageous during transmission (Derdeyn et al., 2004; Rong et al., 2007; Wolfs et al., 1992). However, open questions remain as to whether this holds true both for genome regions other than the envelope, and for all HIV-1M subtypes. To further explore this concept, we generated full-length genomes from 20 recently HIV-1 subtype C infected individuals and compared these sequences to those sampled during chronic infections. Similar to Li et al. (2006) we also found fewer glycosylation sites but not shorter variable loop lengths within the envelopes of viruses sampled during primary infections. However, in an analysis corrected for founder effects and recombination, we found that site-specific amino acid mutational differences across the full-length proteome are almost exclusively associated with signals of virus adaptation to the new host rather than with signatures obviously associated with preferential transmission.
Our study describes full-length HIV-1 subtype C genomes sampled from individuals during the primary phase of infection. We identified thirty-nine sites within the proteomes of these viruses that differentiated them from viruses sampled during chronic infections. Through longitudinal analysis of amino acid frequency changes that occurred during the first six months of infection, together with data on host HLA alleles, we provide evidence that approximately 28.6% of site-specific differences in amino acid frequency spectra between primary and chronic infection proteomes are potential immune escape mutations. The remaining 60.7% of frequency differences between the two groups are probably due to defunct immune evasion substitutions reverting to consensus amino acid states following transmission. This data provides further understanding of processes determining the genomic and immunogenic properties of viruses during early infections which is important if we are to understand HIV pathogenesis sufficiently well to design protective vaccines against the virus.
Almost all of the sites displaying substantially different amino acid frequency spectra between viruses sampled during primary and chronic infections were located within peptides that have known immuno-reactivity. Most of these sites were in Env followed by Pol and Gag containing more sites than any of the remaining proteins.
We further investigated the nature of the immune selection operating on these sites through analysis of sequence sampled longitudinally from the study participants. Based on changes in amino acid frequencies relative to the global HIV database, we found that the amino acid frequency variations in 14/39 of the identified sites were consistent with high rates of reversion mutations being associated with either transmission or primary infection. The frequency spectra differences at 7/39 sites were consistent with early escape from CTL responses during primary infection. The timing of escape may well be crucial, as none of the individuals had IFNg responses to subtype C-based peptide pools containing the presumptive immunoreactive epitopes screened using the ELISPOT assay (Gray et al., data not shown). It is also possible, however, that these assays may have missed responses due to mismatches between the peptide sequences used and the infecting virus.
Our observation that reversion mutations are potentially more common than CTL escape mutations during the early stages of HIV infections is broadly in agreement with that of Li et al. (2007) but is at odds with those of Goonetilleke et al. (2009) and Kearney et al. (2009). What differentiates ours and the Li et al study from those of Goonetilleke et al. and Kearney et al., is that the latter studies examined sequences sampled pre-seroconversion (Fiebig I/II and III). We and Li et al. sampled sequences post-seroconversion, in Fiebig V, VI and beyond. Although CTL escape mutations were only believed to be detectable 30 or more days after peak vireamia (Borrow et al., 1997; Liu et al., 2006), Goonetilleke et al. (2009) have recently described the appearance of CTL escape mutations as early 14 days post infection. Thus we may potentially have underestimated the numbers of CTL escape mutations in viruses which were only sampled a median of 39 days post infection. This possibly indicates that despite a more rapid initial accumulation of novel CTL escape mutations during the first weeks of an infection, over the following months the rates at which successful CTL escape mutations emerge trails off to the point where their frequency is surpassed by that of reversion mutations.
Nevertheless our classification of reversion and escape mutations was supported by the fact that some of the sites predicted to be associated with escape (in Vif and Nef) were located in peptides reported to be restricted by the HLA-alleles of the relevant study participants, whereas 14/17 evolutionary events at fourteen sites we classified as being potentially associated with high frequency reversion mutations which were not restricted by the HLA-alleles of the relevant study participants. It must be pointed out, however, that certain HLA-epitope associations may have been missed as it has previously been shown that CTL responses are poorly predicted in subtype C sequences due, in part, to lack of detailed characterisation of HLA alleles in African populations (Ngandu et al., 2007). This result, based on full-length genomes, supports previous studies based on Gag, Pol and Nef (Brumme et al., 2008; Li et al., 2007) which suggest that most of the high entropy sites displaying amino acid frequency differences between viruses sampled in primary and chronic HIV infections represent defunct escape mutations accumulated in former hosts that had different HLA alleles from the virus’ current hosts.
During early infection many CTL evasion mutations accumulated within previous hosts revert to their consensus states because these “wild-type” polymorphisms provide a greater degree of replicative fitness (Brumme et al., 2008; Martinez-Picado et al., 2006). At the same time that defunct CTL evasion mutations are reverting, viruses are forced to escape the immune pressures exerted by the immune environment of their new hosts. CTL escape during early infections has been associated with oscillation of amino acids within CTL targeted epitopes prior to their convergence on more stable states (Borrow et al., 1997; Delport et al., 2008; Iversen et al., 2006). This oscillation may either be due to the negative replicative fitness effects of some CTL evasion mutations, or due to some mutations only providing partial escape from CTL responses due to, for example, their influencing epitope processing rather than recognition (Borrow et al., 1997). By the chronic phase of infections many of these changes may have reached a degree of equilibrium. It is possible, therefore, that in our study we have detected different stages of this oscillation process in the different infection stages. The increased entropy within targeted CTL epitopes in early infections may be due to amino acid switching or toggling within targeted epitopes as the viruses try to balance the survival benefits of CTL escape with the replicative fitness costs incurred by many CTL evasion mutations (Delport et al., 2008; Goonetilleke et al., 2009;Iversen et al., 2006).
We provide evidence that the innate potential of particular genetic variants to mutationally respond to the selective constraints imposed by new hosts underlie virtually all detectable differences in amino acid frequency spectra between viruses sampled during primary and chronic infections. These data provide valuable insights into unique virological and immunological events during primary infection. We provide evidence which suggests that during the early stages of HIV infections adaptation to the immune environment of new hosts is perhaps secondary to the mutational recovery of replicative fitness losses incurred during CTL escape in former hosts. Our discovery that early infections are primarily characterised by reversion mutations adds to an accumulating body of evidence suggesting the transience of many immune evasion mutations during global population-wide HIV evolution. It is becoming increasingly apparent that CTL escape mutations often have complex evolutionary costs and benefits such that many are likely to have subtle and difficult to predict influences on long-term HIV pathology, epidemiology and evolution. Given that the mutational accessibility and fitness benefits of reversion mutations that occur during early infections should strongly impact the broader effects of CTL evasion mutations, our study emphasizes the importance of studying the evolutionary changes occurring in HIV during the very earliest stages of infection. Although our data suggest that the majority of the amino acid frequency spectrum differences we have observed between viruses sampled during acute and chronic infections are rationally attributable to evolutionary processes at play post-transmission, it would nevertheless be of great interest to determine whether all such signals are generated de novo at the onset of infections. Evidence of even a small proportion of these signals having being generated prior to transmission would provide valuable support for the notion of an evolutionarily relevant “transmission sieve”.
Materials and Methods
Study subjects
Plasma samples were obtained from twenty women who had been recently infected through heterosexual contact, and had been enrolled within three months of infection from prospective cohorts of high-risk HIV negative individuals as part of the CAPRISA 002 Acute Infection study (Table 1) (van Loggerenberg et al., 2008). The time of infection was estimated as the midpoint between the last seronegative and first seropositive sample or as 14 days if diagnostic tests were antibody negative but RNA-positive. Classification of HIV-1 infection stages was carried out as in (Fiebig et al., 2003) Briefly, individuals classified with stage I HIV were HIV RNA positive but p24 antigen negative, those in stage II were HIV RNA and p24 antigen positive, those in stage III were antibody-enzyme immuno assay (EIA) positive but Western blot negative, those in stage IV were antibody-EIA positive with an indeterminate Western blot, those in stage V were Western blot positive but with no p31 band and those in stage VI were Western blot positive with a p31 band. All study participants were antiretroviral therapy naïve.
All samples were collected with informed consent and research ethics approval was obtained from the Universities of Kwa-Zulu/Natal, Witwatersrand and Cape Town (REC 025/2004).
Assembly of a chronic infection dataset
We assembled a reference HIV-1 subtype C chronic infection dataset consisting of 63 publicly available subtype C full-length sequences (Kiepiela et al., 2004); http://hiv.lanl.gov/components/sequence/HIV/search/search.html) and an additional 3 full-length sequences sampled from participants of a sex-worker cohort (Van Damme et al., 2002). Similar to the sequences from primary infection, the chronic infection sequences were obtained from heterosexually infected women with the same ethnic background (Xhosa/ Zulu) and from the same geographic location (KwaZulu-Natal, South Africa). Sequences from participants with AIDS as defined by CD4+ counts less than 200 cells per μl were excluded. In addition, sequences from participants with viral loads >200 000 copies/ml were also excluded to minimize inadvertent inclusion of primary infection sequences in the chronic infection dataset.
Whole genome amplification
Full-length genome sequences were generated from a minimum number of cDNA template molecules in order to both increase the efficiency of full-length genome amplification (Rousseau et al., 2006) and reduce the probability of in vitro recombination during PCR (Fang et al., 1998; Edmonson & Mullins, 1992). RNA was extracted from plasma obtained from peripheral blood using the QIAamp® Viral RNA mini spin kit and protocol (Qiagen, Valencia, CA, USA). Near full-length HIV-1 genomes were amplified as a single fragment using a modified limiting dilution reverse transcription mediated nested PCR approach as described previously (Rousseau et al., 2006). Amplified full-length genomes were gel purified and cloned into the XL-TOPO rapid ligation vector (Invitrogen, GmbH, Karlsruhe, Germany). Cloned genomes were sequenced in both directions using primer-walking.
Diversity following limiting dilution was assessed using a heteroduplex tracking assay (HTA). V1V2 env gene fragments were amplified from the outer PCR reactions used to generate full-length genomes and were probed with a radioactively labelled env gene (V1V2 region) probe generated from the subtype C isolate Du151 using methods described by (Kitrinos et al., 2003).
DNA sequencing
DNA sequencing reactions were performed using the ABI PRISM Dye Terminator Cycle sequencing kit V3.1 (Applied Biosystems, Foster City , CA, USA) using both the primers described by which are specifically optimised for HIV-1 subtype C sequencing, and those described by . The CAPRISA sequence assembly pipeline tool (www.tools.caprisa.org) employing the Phred, Phrap and Cross_match software packages was used to assemble full-length genome sequences. Assembled sequences and chromatograms were viewed and edited using Consed.
Phylogenetic analysis
A neighbor-joining tree was constructed in MEGA 4 (Tamura et al., 2007) for all full genome HIV-1 subtype C sequences from this study and from the HIV sequence database (total n = 421) using a maximum composite likelihood model with a gamma distribution rate (α = 2) determined using the FindModel tool which is based on MODELTEST (http://www.hiv.lanl.gov, Posada & Crandall, 1998). The primary and chronic infection full-length genome datasets were aligned using ClustalW as implemented in BioEdit with manual editing in BioEdit (Hall, 1999). Full-length genome sequences were split into individual gene fragments for gene-specific analyses. A maximum likelihood phylogenetic tree for the env gene were inferred using PHYML (Guindon & Gascuel, 2003) as implemented in RDP3.26 (Heath et al., 2006) ,using the General Time Reversible nucleotide substitution model with gamma correction for site-to-site rate variation (α = 2) selected by the FindModel tool (http://www.hiv.lanl.gov, Posada & Crandall, 1998).
Phylogeny-aware comparison of amino acid mutational spectra
As recombination can seriously confound phylogenetic analyses, we sought to account for recombination by performing separate analyses for different alignment partitions as defined by identified recombination breakpoints. Recombination breakpoints were identified in different HIV gene alignments using the RDP (Martin & Rybicki, 2000), GENECONV (Padidam et al., 1999), BOOTSCAN (Martin et al., 2005a), MAXCHI , CHIMAERA (Martin & Rybicki, 2000; Martin et al., 2005b) and SISCAN (Gibbs et al., 2000) methods implemented in RDP3. Default settings were used throughout and only potential recombination events detected by two or more of the above methods (with associate Bonferroni corrected P-values <0.05) coupled with phylogenetic evidence of recombination were considered significant. The gag, pol, env and nef genes were partitioned at breakpoint positions. Vif, vpr, vpu, and tat genes were not detectably recombinant. Recombination could also not be detected in these genes using the GARD (Genetic Algorithm for Recombination Detection) method implemented on the Datamonkey webserver (Pond et al., 2006; http://www.datamonkey.org). Overlapping reading frames, variable regions in env as well as insertions or deletions were removed before genes and partition fragments were translated to amino acids. Neighbor-joining trees for protein alignments (without bootstrapping) were inferred with MEGA 4 (Tamura et al., 2007) using the Poisson correction distance model which assumes equal substitution rates and equal amino acid frequencies. Rev was not analysed as it is completely embedded in overlapping reading frames and was therefore unsuitable for analysis.
For each alignment partition defined by identified recombination breakpoints we inferred the sequences at the ancestral nodes of the corresponding tree (Edwards & Shields, 2004;Edwards & Shields, 2005) and, for each site, designated the amino acid at the root of the tree as the ancestral amino acid for that site.. Each terminal branch with a mutation towards the ancestral amino acid was assigned a score of +1; terminal branches with a mutation from the ancestral amino acid to any other amino acid were assigned a score of −1 and terminal branches for which no amino acid replacement was inferred were assigned a score of 0. We then compared the numbers of −1, 0 and +1 scores in terminal branches leading to sequences sampled during primary infection to the corresponding numbers from chronic infection sequences using a two-tailed Wilcoxon rank-sum test with a p-value cut-off of 0.025. We then carried out a permutation test in order to investigate the impact of multiple hypothesis testing on our results. We randomly shuffled the sample labels (primary/ chronic infection) 1,000 times and for each randomization we repeated the test and evaluated the number of sites with a p-value below the significance threshold.
Site-by-site Shannon entropy estimation
The average entropy was used to estimate the variability at amino acid sites at each alignment position of the primary and chronic datasets at signature positions (Yusim et al., 2002; Korber et al., 1994;http://www.hiv.lanl.gov/tmp/ENTROPY/). HIV-1 subtype C sequences available on the HIV sequence database were used to determine the database frequency of amino acids at alignment positions for gp41 (n = 508); Gp120 (n = 531); Gag (n=413); Nef (n= 586), Rev (n = 457 and n = 562 for exons 1 and 2 respectively), Vif (n = 409), Vpr (n = 401), and Pol (n = 412) (http://hiv.lanl.gov/components/sequence/HIV/).
Variable loop length and N-linked glycosylation sites (PNGS)
The length (number of amino acids) of env variable loops and the total number of PNGS were determined with the N-Glycosite tool on the HIV sequence database (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html, (Zhang et al., 2004).
Screening for possible CTL epitopes
Motif Scan (a program that uses known HLA-1 restricted CTL epitope binding motifs to predict HLA-peptide binding sites; http://www.hiv.lanl.gov/content/immunology/motif_scan/), the CTL epitope database (http://www.hiv.lanl.gov/content/immunology/ctl_search), the Los Alamos HIV Molecular Immunology Compendium 2006/2007 and the NetMHCpan tool (http://www.cbs.dtu.dk/services/NetMHCpan/) which use HLA and peptide sequence information to predict the affinity (nM) of peptide-HLA interactions (Nielsen et al., 2007) were used to identify putative CTL epitopes predicted to be restricted by each study participant’s particular HLA alleles.
HLA-I A, B and C typing was carried out at high resolution by sequencing using the Atria AlleleSeqr (Abbott Diagnostics) and Assign-SBT 3.5 (Conexio Genomics) kits as described in Chopera et al. (2008).
We calculated the proportion of immunoreactive peptides with respect to the complete HIV-1 subtype C proteome, by mapping reported immunoreactive peptides in subtype C infections onto the viral proteins.
Statistical analyses
The non-parametric Wilcoxon rank-sum test was used to identify differences between the primary and chronic infection datasets with respect to both the numbers of N-linked glycosylation sites (PNGs) and the lengths of the variable loops in env. Differences in entropy scores between primary and chronic strains at each identified position were evaluated using the two-tailed Wilcoxon rank-sum test. These statistical tests were carried out using GraphPad Prism® 5.0 (GraphPad Software Inc., CA, USA).
The 2×2 Chi-square test (http://faculty.vassar.edu/lowry/tab2x2.html) was used to determine whether or not sites with significant allele frequency spectrum differences between viral isolates from primary and chronic infections, clustered within immunoreactive regions of the HIV-1 proteome (Fisher’s exact one-tailed T test was used to measure significance).
Nucleotide sequence accession numbers
All near full-length sequences were submitted to GenBank under accession numbers GQ999972 to GQ999991.
Supplementary Material
Acknowledgments
We would like to thank the staff and participants involved in the CAPRISA 002 Acute Infection study for willingness to participate and the provision of specimens. This work was funded by the National Institute of Allergy and infectious Disease (NIAID), National Institutes of Health (NIH) and the US Department of Health and Human Services (DHHS) (grant# AI51794) and the National Research Foundation under grant # 67385. F.K Treurnicht is a Fogarty AITRP fellow (TWO-02). D.P. Martin is supported by the Wellcome Trust. The CAPRISA sequence assembly pipeline was developed by Winston Hide, Adam Dawe, Allan Kamau, Ruby van Rooyen, Alan Powell, Anelda Boardman and Heikki Lehvaslaiho at the South African National Bioinformatics Institute, University of the Western Cape, South Africa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. Quantitating the Multiplicity of Infection with Human Immunodeficiency Virus Type 1 Subtype C Reveals a Non-Poisson Distribution of Transmitted Variants. Journal of Virology. 2009;83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O’Sullivan KM, DeSouza I, Feeney ME, Eldridge RL, Maier EL, Kaufmann DE, Lahaie MP, Reyor L, Tanzi G, Johnston MN, Brander C, Draenert R, Rockstroh JK, Jessen H, Rosenberg ES, Mallal SA, Walker BD. Selective escape from CD8(+) T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. Journal of Virology. 2005;79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandawe GP, Martin DP, Treurnicht F, Mlisana K, Karim SSA, Williamson C. Conserved positive selection signals in gp41 across multiple subtypes and difference in selection signals detectable in gp41 sequences sampled during acute and chronic HIV-1 subtype C infection. Virology Journal. 2008;5 doi: 10.1186/1743-422X-5-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Gough E, Sabbaj S, Ritter D, Yusim K, Sfakianos G, Aldrovandi G, Kaslow RA, Wilson CM, Mulligan MJ, Kilby JM, Goepfert PA. CD8 T-cell responses in early HIV-1 infection are skewed towards high entropy peptides. Aids. 2005;19:241–250. [PubMed] [Google Scholar]
- Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, Carlson J, Yusim K, McMahon B, Gaschen B, Mallal S, Mullins JI, Nickle DC, Herbeck J, Rousseau C, Learn GH, Miura T, Brander C, Walker B, Korber B. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science. 2007;315:1583–1586. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- Borrow P, Lewicki H, Wei XP, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MBA, Shaw GM. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nature Medicine. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, DeSouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. Escape and compensation from early HLA-1357-Mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. Journal of Virology. 2007;81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Carlson J, Streeck H, John M, Eichbaum Q, Block BL, Baker B, Kadie C, Markowitz M, Jessen H, Kelleher AD, Rosenberg E, Kaldor J, Yuki Y, Carrington M, Allen TM, Mallal S, Altfeld M, Heckerman D, Walker BD. Marked epitope- and allele- specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. Journal of Virology. 2008;82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, Kadie C, Bhattacharya T, Chui C, Szinger J, Mo T, Hogg RS, Montaner JSG, Frahm N, Brander C, Walker BD, Harrigan PR. Evidence of differential HLA class I-Mediated viral evolution in functional and Accessory/Regulatory genes of HIV-1. Plos Pathogens. 2007;3:913–927. doi: 10.1371/journal.ppat.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Karim SA, Gray CM, Williamson C. Transmission of HIV-1CTL escape variants provides HLA-mismatched recipients with a survival advantage. Plos Pathogens. 2008;4 doi: 10.1371/journal.ppat.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Scheffler K, Seoighe C. Frequent toggling between alternative amino acids is driven by selection in HIV-1. Plos Pathogens. 2008;4:e1000242. doi: 10.1371/journal.ppat.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- deWolf F, Spijkerman I, Schellekens PT, Langendam M, Kuiken C, Bakker M, Roos M, Coutinho R, Miedema F, Goudsmit J. AIDS prognosis based on HIV-1 RNA, CD4+ T-cell count and function: Markers with reciprocal predictive value over time after seroconversion. AIDS. 1997;11:1799–1806. doi: 10.1097/00002030-199715000-00003. [DOI] [PubMed] [Google Scholar]
- Edmonson PF, Mullins JI. Efficient amplification of HIV half-genomes from tissue DNA. Nucleic Acids Research. 1992;20:4933. doi: 10.1093/nar/20.18.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RJ, Shields DC. GASP: Gapped ancestral sequence prediction for proteins. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RJ, Shields DC. BADASP: predicting functional specificity in protein families using ancestral sequences. Bioinformatics. 2005;21:4190–4191. doi: 10.1093/bioinformatics/bti678. [DOI] [PubMed] [Google Scholar]
- Fang GW, Zhu G, Burger H, Keithly JS, Weiser B. Minimizing DNA recombination during long RT-PCR. Journal of Virological Methods. 1998;76:139–148. doi: 10.1016/s0166-0934(98)00133-5. [DOI] [PubMed] [Google Scholar]
- Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, Heldebrant C, Smith R, Conrad A, Kleinman SH, Busch MP. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. Aids. 2003;17:1871–1879. doi: 10.1097/00002030-200309050-00005. [DOI] [PubMed] [Google Scholar]
- Gibbs MJ, Armstrong JS, Gibbs AJ. Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000;16:573–582. doi: 10.1093/bioinformatics/16.7.573. [DOI] [PubMed] [Google Scholar]
- Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang JM, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJR, Hunter E. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. Journal of Experimental Medicine. 2008;205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonetilleke N, Liu MKP, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EM, Salazar MG, Weinhold KJ, Moore S, CHAVI Clinical Core B. Letvin N, Haynes BF, Cohen MS, Harber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. The first T cell response to transmitted/Founder virus contributes to the control of acute viremia in HIV-1 infection. Journal of Experimental Medicine. 2009 doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond KSL, Posada D, Gravenor MB, Woelk CH, Frost SD. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006;22(24):3096–8. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- Gray CM, Mlotshwa M, Riou C, Mathebula T, Rosa DD, Mashishi T, Seoighe C, Ngandu N, van Loggerenberg F, Morris L, Mlisana K, Williamson C, Karim SA. Human Immunodeficiency Virus-Specific Gamma Interferon Enzyme-Linked Immunospot Assay Responses Targeting Specific Regions of the Proteome during Primary Subtype C Infection Are Poor Predictors of the Course of Viremia and Set Point. Journal of Virology. 2009;83:470–478. doi: 10.1128/JVI.01678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haaland RE, Hawkins PA, Salazar-Gonzalez J, Johnson A, Tichacek A, Karita E, Manigart O, Mulenga J, Keele BF, Shaw GM, Hahn BH, Allen SA, Derdeyn CA, Hunter E. Inflammatory Genital Infections Mitigate a Severe Genetic Bottleneck in Heterosexual Transmission of Subtype A and C HIV-1. Plos Pathogens. 2009;5 doi: 10.1371/journal.ppat.1000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–98. [Google Scholar]
- Heath L, van der Walt E, Varsani A, Martin DP. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. Journal of Virology. 2006;80:11827–11832. doi: 10.1128/JVI.01100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen AKN, Stewart-Jones G, Learn GH, Christie N, Sylvester-Hviid C, Armitage AE, Kaul R, Beattie T, Lee JK, Li YP, Chotiyarnwong P, Dong T, Xu XN, Luscher MA, MacDonald K, Ullum H, Klarlund-Pedersen B, Skinhoj P, Fugger L, Buus S, Mullins JI, Jones EY, van der Merwe PA, McMichael AJ. Conflicting selective forces affect T cell receptor contacts in an immunodominant human immunodeficiency virus epitope. Nature Immunology. 2006;7:179–189. doi: 10.1038/ni1298. [DOI] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun CX, Grayson T, Wang SY, Li H, Wei XP, Jiang CL, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. Identification and characterisation of transmitted and early founder virus envelopes in primary HIV-1 infection. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. Journal of Experimental Medicine. 2001;193:375–385. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, Zimbwa P, Moore S, Allen T, Brander C, Addo MM, Altfeld M, James I, Mallal S, Bunce M, Barber LD, Szinger J, Day C, Klenerman P, Mullins J, Korber B, Coovadia HM, Walker BD, Goulder PJR. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–774. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. CD8(+) T-cell responses to different HIV proteins have discordant associations with viral load. Nature Medicine. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- Kitrinos KM, Hoffman NG, Nelson JAE, Swanstrom R. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. Journal of Virology. 2003;77:6811–6822. doi: 10.1128/JVI.77.12.6811-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong XH, West JT, Zhang H, Shea DM, M’soka TJ, Wood C. The Human Immunodeficiency Virus Type 1 Envelope Confers Higher Rates of Replicative Fitness to Perinatally Transmitted Viruses than to Nontransmitted Viruses. Journal of Virology. 2008;82:11609–11618. doi: 10.1128/JVI.00952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber BT, Kuntsman K, Patterson B, Furtado M, McEvilly M, Levy R, Wolinsky S. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain derived sequences. Journal of Virology. 1994;68:7467–81. doi: 10.1128/jvi.68.11.7467-7481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavreys L, Baeten JM, Chohan V, McClelland RS, Hassan WM, Richardson BA, Mandaliya K, Ndinya-Achola JO, Overbaugh J. Higher set point plasma viral load and more-severe acute HIV type 1 (HIV-1) illness predict mortality among high-risk HIV-1-infected African women. Clinical Infectious Diseases. 2006;42:1333–1339. doi: 10.1086/503258. [DOI] [PubMed] [Google Scholar]
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJR. HIV evolution: CTL escape mutation and reversion after transmission. Nature Medicine. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- Li B, Decker JM, Johnson RW, Bibollet-Ruche F, Wei XP, Mulenga J, Allen S, Hunter E, Hahn BH, Shaw GM, Blackwell JL, Derdeyn CA. Evidence for potent autologous neutralizing antibody titers and compact envelopes in early infection with subtype C human immunodeficiency virus type 1. Journal of Virology. 2006;80:5211–5218. doi: 10.1128/JVI.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gladden AD, Altfeld M, Kaldor JM, Cooper DA, Kelleher AD, Allen TM. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. Journal of Virology. 2007;81:193–201. doi: 10.1128/JVI.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Cao JH, Zhao H, Genowati I, Wong K, McLaughlin S, McSweyn MD, Diem K, Stevens CE, Maenza J, He HX, Nickle DC, Shriner D, Holte SE, Collier AC, Corey L, McElrath MJ, Mullins JI. Selection on the human immunodeficiency virus type 1 proteome following primary infection. Journal of Virology. 2006;80:9519–9529. doi: 10.1128/JVI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Zhao H, Tebit DM, Troyer RM, McSweyn M, Ghosh AK, Shriner D, Arts EJ, McElrath MJ, Mullins JI. Evolution of human immunodeficiency virus type I cytotoxic T-lymphocyte epitopes: Fitness-balanced escape. Journal of Virology. 2007;81:12179–12188. doi: 10.1128/JVI.01277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- Martin DP, Posada D, Crandall KA, Williamson C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. Aids Research and Human Retroviruses. 2005;21:98–102. doi: 10.1089/aid.2005.21.98. [DOI] [PubMed] [Google Scholar]
- Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. Journal of Virology. 2006;80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews PC, Prendergast A, Leslie A, Crawford H, Payne R, Rousseau C, Rolland M, Honeyborne I, Carlson J, Kadie C, Brander C, Bishop K, Mlotshwa N, Mullins JI, Coovadia H, Ndung’u T, Walker BD, Heckerman D, Goulder PJR. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. Journal of Virology. 2008;82:8548–8559. doi: 10.1128/JVI.00580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, Rothchild AC, Li B, Trocha A, Cutrell E, Frahm N, Brander C, Toth I, Arts EJ, Allen TM, Walker BD. HLA-B57/B*5801 Human Immunodeficiency Virus Type 1 Elite Controllers Select for Rare Gag Variants Associated with Reduced Viral Replication Capacity and Strong Cytotoxic T-Lymphotye Recognition. Journal of Virology. 2009;83:2743–2755. doi: 10.1128/JVI.02265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngandu NG, Bredell H, Gray CM, Williamson C, Seoighe C. CTL response to HIV type 1 subtype C is poorly predicted by known epitope motifs. Aids Research and Human Retroviruses. 2007;23:1033–1041. doi: 10.1089/aid.2007.0024. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. Plos One. 2007;8:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padidam M, Beachy RN, Fauquet CM. A phage single-stranded DNA (ssDNA) binding protein complements ssDNA accumulation of a geminivirus and interferes with viral movement. Journal of Virology. 1999;73:1609–1616. doi: 10.1128/jvi.73.2.1609-1616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Posada D, Crandall KA, Holmes EC. The causes and consequences of HIV evolution. Nature Reviews Genetics. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- Rong R, Gnanakaran S, Decker JM, Bibollet-Ruche F, Taylor J, Sfakianos JN, Mokili JL, Muldoon M, Mulenga J, Allen S, Hahn BH, Shaw GM, Blackwell JL, Korber BT, Hunter E, Derdeyn CA. Unique mutational patterns in the envelope alpha 2 amphipathic helix and acquisition of length in gp120 hypervariable domains are associated with resistance to autologous neutralization of subtype C human immunodeficiency virus type 1. Journal of Virology. 2007;81:5658–5668. doi: 10.1128/JVI.00257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau CM, Birditt BA, Mckay AR, Stoddard JN, Lee TC, McLaughlin S, Moore SW, Shindo N, Learn GH, Korber BT, Brander C, Goulder PJR, Kiepiela P, Walker BD, Mullins JI. Large-scale amplification, cloning and sequencing of near full-length HIV-1 subtype C genomes. Journal of Virological Methods. 2006;136:118–125. doi: 10.1016/j.jviromet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN, Maust BS, Learn GH, Nickle DC, Coovadia H, Ndung’u T, Frahm N, Brander C, Walker BD, Goulder PJR, Bhattacharya T, Heckerman DE, Korber BT, Mullins JI. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype C proteome: Immune escape and viral load. Journal of Virology. 2008;82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Martin DP, Seoighe C. Robust inference of positive selection from recombining coding sequences. Bioinformatics. 2006;22:2493–2499. doi: 10.1093/bioinformatics/btl427. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Van Damme L, Ramjee G, Alary M, Vuylsteke B, Chandeying V, Rees H, Sirivongrangson P, Mukenge-Tshibaka L, Ettiegne-Traore V, Uaheowitchai C, Karim SSA, Masse B, Perriens J, Laga M. Effectiveness of COL-1492, a nonoxynol-9 vaginal gel, on HIV-1 transmission in female sex workers: a randomised controlled trial. Lancet. 2002;360:971–977. doi: 10.1016/s0140-6736(02)11079-8. [DOI] [PubMed] [Google Scholar]
- van Loggerenberg F, Mlisana K, Williamson C, Auld SC, Morris L, Gray CM, Karim QA, Grobler A, Barnabas N, Iriogbe I, Karim SA, for the CAPRISA 002 Acute Infection Study Team Establishing a cohort at high risk of HIV infection in South Africa: Challenges and experiences of the CAPRISA 002 acute infection study. Plos One. 2008;3:e1954. doi: 10.1371/journal.pone.0001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfs TFW, Zwart G, Bakker M, Goudsmit J. Hiv-1 Genomic RNA Diversification Following Sexual and Parenteral Virus Transmission. Virology. 1992;189:103–110. doi: 10.1016/0042-6822(92)90685-i. [DOI] [PubMed] [Google Scholar]
- Zhang M, Gaschen B, Blay W, Foley B, Haigwood N, Kuiken C, Korber B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology. 2004;14:1229–1246. doi: 10.1093/glycob/cwh106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.