

Abstract
We used recombinant techniques to create a two-chain form (residues 1–345 and residues 346–758) of the vitamin K-dependent γ-glutamyl carboxylase, a glycoprotein located in the endoplasmic reticulum containing five transmembrane domains. The two-chain carboxylase had carboxylase and epoxidase activities similar to those of one-chain carboxylase. In addition, it had normal affinity for the propeptide of factor IX. We employed this molecule to investigate formation of the one disulfide bond in carboxylase, the transmembrane structure of carboxylase, and the potential interactions among the carboxylase’s transmembrane domains. Our results indicate that the two peptides of the two-chain carboxylase are joined by a disulfide bond. Proline 378 is important for the structure necessary for disulfide formation. Results with the P378L carboxylase indicate that noncovalent bonds maintain the two-chain structure even when the disulfide bond is disrupted. As we had previously proposed, the fifth transmembrane domain of carboxylase is the last and only transmembrane domain in the C-terminal peptide of the two-chain carboxylase. We show that the noncovalent association between the two chains of carboxylase involves an interaction between the fifth transmembrane domain and the second transmembrane domain. Results of a homology model of transmembrane domains 2 and 5 suggest that not only do these two domains associate but that transmembrane domain 2 may interact with another transmembrane domain. This latter interaction may be mediated at least in part by a motif of glycine residues in the second transmembrane domain.
The vitamin K-dependent γ-glutamyl carboxylase is a 758-residue integral membrane glycoprotein of the endoplasmic reticulum (ER).1 It catalyzes the posttranslational modification of specific glutamic acid residues of vitamin K-dependent proteins to γ-carboxyglutamic acid residues. This posttranslational modification is critical for the biological functions of the vitamin K-dependent proteins involved in blood coagulation, bone metabolism, signal transduction, and cell proliferation (1–3). Concomitant with carboxylation vitamin K hydroquinone is oxidized to vitamin K epoxide (epoxidation). The epoxide must be converted back to the hydroquinone by the enzyme vitamin K epoxide reductase, thus completing the vitamin K cycle (3).
A number of residues in the carboxylase have been implicated in function. The propeptide of vitamin K-dependent proteins appears to be the primary substrate recognition sequence for carboxylase (4). Tight binding at the site supports a processive mechanism in which multiple carboxylations occur during a single binding event (5). On the basis of affinity peptide–substrate labeling, Yamada et al. reported that the propeptide binds to the carboxylase between residues 50 and 225 (6). Propeptide cross-linking experiments from our laboratory suggested that the propeptide binding region of carboxylase is between residues 438 and 507 (7). Further studies employing site-directed mutagenesis showed that at least part of the propeptide binding region includes residues between 495 and 513 of the carboxylase (8).
Kuliopulos et al. suggested that the glutamate residue binding site is located within the first 218 amino acid residues (9). Mutucumarana et al. identified several residues in the region of a naturally occurring carboxylase mutation (L394R) that also appear to be involved in glutamate substrate binding (10). Recently, Rishavy et al. presented evidence that lysine, specifically K218, may be the essential base that deprotonates vitamin K hydroquinone to initiate the carboxylation reaction (11).
At present, the only structural model for the carboxylase is the membrane topology (12). According to this model the carboxylase spans the membrane five times with its amino terminus located in the cytoplasm and the large hydrophilic carboxyl terminus in the ER lumen. Approximately 72% and 14% of carboxylase’s sequence resides in the ER lumen and cytoplasm, respectively. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry results showed that there is a disulfide bond between cysteine residues 99 and 450 which is important for carboxylase folding and maturation (13). However, there is some disagreement on whether this disulfide bond exists in active carboxylase (14).
Recently, Schmidt-Krey et al. produced planar-tubular two-dimensional crystals of carboxylase (15). Projection map calculations reveal that the carboxylase exists as a monomer, which agrees with sedimentation equilibrium data (16). However, the present crystal data do not yet provide information about TMD packing or other details of the three-dimensional structure of the carboxylase (15).
Given the limited amount of structural information about the carboxylase, it is difficult to incorporate the information we have about residues important for function into a coherent model of how this multisubstrate enzyme simultaneously catalyzes the carboxylation and epoxidation reactions. When limited structural information about a protein is available, functional splitting and reassembly of multispanning membrane proteins can be employed to study their structure and function (17–22). It is a useful approach to study the function of protein domains, the role of individual transmembrane helices, and the helical packing within the membrane. Transmembrane helix interactions are important for the formation and maintenance of three-dimensional structure in multispanning membrane proteins. Recently, several motifs have been identified as involved in the helix–helix associations within membranes (23–25). Changing essential residues in these motifs significantly affects the helix–helix interactions.
In order to clarify issues surrounding the disulfide bond formation and transmembrane domain interactions and their role in structure formation, we have split human carboxylase in the last cytoplasmic loop between TMD4 and TMD5, and coexpressed these two peptides in insect cells. Our results show that these two peptides assemble as a fully active two-chain carboxylase molecule joined by a disulfide bond. Results from mutant two-chain carboxylases indicate that TMD5 is the last and only TMD in the C-terminal peptide. In addition, it appears that TMD2 and TMD5 and at least one other domain associate via amino acid motifs expected to support TMD interactions. Our results suggest that this two-chain carboxylase can serve as a useful model for carboxylase structure–function studies.
MATERIALS AND METHODS
Materials
All chemicals were of reagent grade. NEM, phenylmethanesulfonyl fluoride, CHAPS, and anti-FLAG M2 monoclonal antibody were obtained from Sigma (St. Louis, MO). Pentapeptide FLEEL and protease inhibitor H-D-Phe-Pro-Arg chloromethyl ketone were from Bachem (King of Prussia, PA). Aprotinin and pepstatin A were purchased from Roche Molecular Biochemicals (Indianapolis, IN). 1,2-Dioleoyl-sn-glycero-3-phosphocholine was from Avanti Polar Lipids (Alabaster, AL). Vitamin K1(20) (10 mg/mL) was from Abbott Laboratories (Chicago, IL). Vitamin K1(25) was from GLsynthesis Inc. (Worcester, MA). NaH14CO3 (specific activity, 54 mCi/mmol) was from ICN Pharmaceuticals, Inc. (Costa Mesa, CA). The peptide proFIX and the fluorescein-labeled consensus propeptide were chemically synthesized and purified by Chiron Mimotopes (Clayton, Victoria, Australia). Restriction enzymes and PNGase F were from New England Biolabs (Beverly, MA). The Bac-to-Bac baculovirus expression system, Bis-Tris NuPAGE gels, protein standards, and oligonucleotides were from Invitrogen Life Technologies (Carlsbad, CA). Pfu Turbo DNA polymerase was from Stratagene (La Jolla, CA). Immobilon-P polyvinylidene difluoride membrane was from Millipore Co. (Bedford, MA). ECL Western blotting detection reagents were from Amersham Biosciences (Piscataway, NJ). Anti-HPC4 monoclonal antibody and anti-HPC4 antibody-coupled Sepharose resin were from Dr. Charles T. Esmon (Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK).
Construction of the Two-Chain Carboxylase and Site-Directed Mutagenesis
Wild-type human carboxylase cDNA with a FLAG epitope (DYKDDDDK) at the amino terminus and a HPC4 epitope (EDQVDPRLIDGK) at the carboxyl terminus (26) was engineered into the pFastBac1 baculovirus expression vector with BamHI and EcoRI sites. The resulting plasmid was used as template for PCR amplifications. One-chain carboxylase is split at amino acid residue 345 to form the two-chain carboxylase. According to the “positive inside rule” (27, 28), more positively charged residues in the cytoplasmic loop of the C-terminal peptide will favor its correct membrane orientation. Therefore, instead of using the native tryptic cleavage site (residue 349), we moved the split site four residues toward the N-terminus. The N-terminal peptide (residues 1–345) was amplified by PCR using a sense oligonucleotide comprising a BamHI site, a start codon, and part of the FLAG tag-coding sequence as the 5′ primer and an oligonucleotide containing part of the antisense coding sequence of carboxylase ending at residues 345, a stop codon, and an EcoRI site as the 3′ primer.
The C-terminal peptide (residues 346–758) was amplified by PCR using a sense oligonucleotide comprising a BamHI site, a start codon, and part of the carboxylase coding sequence starting from residue 346 as the 5′ primer and an oligonucleotide containing part of the antisense sequence of HPC4 coding sequence, a stop codon, and an EcoRI site as the 3′ primer. For individual expression of the two peptides, the PCR-amplified coding sequence of the N-terminal peptide of carboxylase with a FLAG tag at its amino terminus or the coding sequence for the C-terminal peptide of carboxylase with a HPC4 tag at its carboxyl terminus was cloned into the pFastBac1 vector by BamHI/EcoRI in the multiple cloning site downstream of the polyhedrin promoter.
For coexpression of both peptides in insect cells, the C-terminal peptide of carboxylase was amplified by PCR using a 5′ primer flanked with a NcoI site and 3′ primer flanked with a KpnI site. The PCR product was first cloned into the coexpression vector pFastBacDual by NcoI/KpnI sites in one of the multiple cloning sites downstream of the p10 promoter. The above PCR-amplified fragment encoding the N-terminal peptide was then cloned into the pFast-BacDual vector containing the C-terminal peptide coding sequence by BamHI/EcoRI sites in another set of multiple cloning sites downstream of the polyhedrin promoter.
Site-directed mutagenesis of residues in the carboxylase for investigating TMD interactions or disulfide formation was performed by overlap PCR. The nucleotide sequences of all the constructs were verified by the DNA sequencing facility at the University of North Carolina at Chapel Hill.
Expression of Wild-Type and Mutant Carboxylases in Insect Cells
Baculovirus expression vectors that contain the one-chain carboxylase, two-chain carboxylase, or their mutants were transformed into Escherichia coli strain DH10Bac, the recombinant bacmids were screened by blue/white selection, and the positive bacmids were extracted according to the manufacturer’s instructions (Invitrogen). Recombinant baculovirus was obtained by Cellfectin-induced infection of Sf9 cells and screened by carboxylase activity assay or Western blot analysis of the cell lysate. Expression of carboxylase was performed by infection of 2 × 106 Sf9 cells/mL with the recombinant virus at a multiplicity of infection of 1. Cells were collected 48 h after infection by centrifugation for microsome preparation or stored at −80 °C until use.
Purification of Recombinant Carboxylases Using the C-Terminal Peptide Antibody Resin
Isolation of microsomes from insect cells was performed as described (29) with minor modifications. Cells harvested from 1 L of a culture were washed twice with 100 mL of ice-cold buffer A [25 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 15% glycerol]. The washed cell pellet was resuspended in 120 mL of ice-cold buffer A containing a protease inhibitor mixture (0.5 μg/mL leupeptin, 1 μg/mL pepstatin, 2 μg/mL aprotinin, and 0.1 mg/mL phenylmethanesulfonyl fluoride). Cells were broken on ice by sonication (80 pulses, 1.5 s each, 18.5 s interval between each pulse, using a Heat Systems XL2020 sonicator at a power output of 6). The homogenate was centrifuged at 4300gav at 4 °C for 15 min. The supernatant was recovered and centrifuged at 150000gav for 45 min at 4 °C. In order to avoid random disulfide bond formation during the purification and sonication, in samples for gel analyses we included 20 mM NEM. In these experiments we used 25 mM MOPS (pH 6.8) in buffer A instead of 25 mM Tris-HCl (pH 7.4), since NEM reacts more specifically with cysteines at the lower pH. Excess unreacted NEM was removed by washing the microsome pellet twice with 60 mL of ice-cold buffer A. Solubilization of the microsomal pellet and the subsequent purification of carboxylase using the C-terminal peptide antibody affinity resin were performed as described (26).
Carboxylation and Epoxidation Activity Assays
Carboxylation activity was determined by the incorporation of 14CO2 into the pentapeptide substrate FLEEL in the presence of propeptide as described (26). The concentration of active carboxylase was determined from the fraction of protein binding to the fluorescein-labeled consensus propeptide by fluorescence anisotropy as described previously (16).
Vitamin K epoxidation activity of carboxylase was determined by quantitation of the vitamin K epoxide formed during carboxylation of FLEEL as described (30).
SDS–PAGE and Western Blot Analyses
SDS–PAGE analysis was performed by the NuPAGE system (Invitrogen) under reduced or nonreduced conditions. Samples of cell lysate, microsomes, or purified enzyme were incubated with 1× SDS sample buffer [10% glycerol, 100 mM Tris-HCl (pH 8.5), 2% SDS, 1 mM EDTA, 0.1% bromophenol blue] in the presence or absence of 50 mM DTT at room temperature for 10 min unless otherwise stated. Samples were subjected to 10% NuPAGE for electrophoresis using 1× MES (50 mM MES–Tris-HCl, pH 7.2, 0.1% SDS, 1 mM EDTA) or 1× MOPS (50 mM MOPS–Tris-HCl, pH 7.7, 0.1% SDS, 1 mM EDTA) running buffer. After electrophoresis, the protein bands were visualized by silver staining or transferred to a polyvinylidene difluoride membrane for Western blot analysis. The protein bands on the membrane were probed with mouse C-terminal peptide antibody or the N-terminal peptide antibody, M2, followed by HRP-conjugated secondary antibody. Protein bands were detected using the ECL Western blotting detection reagents.
Homology Modeling of TMD2 and TMD5 Interactions
We constructed a model of the interactions between the transmembrane domains 2 and 5 based on the X-ray crystal structure (PDB entry 1JGJ) of the membrane-embedded protein, sensory rhodopsin II (SRII). SRII contains seven transmembrane helices. We used the helical segment, residues 1–20 (H1) of SRII (MVGLTTLFWLGAIGMLVGTL), to model TMD2 (MYLVYTIMFLGALGMMLGLC). TMD2 has about 75% homology (40% identity) to H1. The two helices neighboring and interacting with H1 in the SRII crystal structure, and, thus, candidates for modeling TMD5, are the C-terminal helix 7 (H7) and helix 2 (H2). H7 is antiparallel to our proposed TMD5 orientation; therefore, we used H2 to represent TMD5, even though it has somewhat lower sequence identity (only 25%) with TMD5 than does helix 7. In the X-ray crystal, the three C-terminal residues of H2 are in a turn, but in our model construction, we made them part of the helix. Appropriate amino acid substitutions were made to convert the SRII sequences to those corresponding to TMD2 and TMD5 using the software package Sybyl7.3 (Tripos, Inc.). The initial model is energy minimized using the sander module of AMBER 9.0 (31). The interaction potential was taken from the standard AMBER force field (parm99.dat) (32) with the modifications to the backbone dihedral angles introduced by Hornak et al. (33). The energy minimization was carried out in a dielectric medium with the dielectric constant 4.0.
RESULTS AND DISCUSSION
Our initial goal in this study was to evaluate a two-chain carboxylase as a tool for studying structure–function relationships in the enzyme and, in particular, to investigate the role of certain residues in formation of the single disulfide bond in the carboxylase. Wu et al. showed that, under controlled conditions, trypsin cleaved bovine liver carboxylase at residues 349 and 351, resulting in a two-chain molecule (7). The two chains are apparently joined by at least one disulfide bond. This molecule had activity similar to that of the one-chain carboxylase. Results from this study suggested to us that the two-chain carboxylase might be a good model for studying disulfide formation and other structural features of the carboxylase.
Functional Assembly of the Two-Chain Carboxylase Expressed in Insect Cells
We expressed the N-terminal carboxylase peptide (residues 1–345), the C-terminal peptide (345–758), and both of the two peptides together in insect cells. Western blot analyses of cell lysates confirmed the expression of both peptides (data not shown), but neither of the two peptides expressed alone had detectable carboxylase activity. On the other hand, when compared to the one-chain carboxylase, the purified two-chain carboxylase had 92% and 142%, respectively, of the carboxylase and epoxidase activity. The concentrations of proFIX required for half-maximal stimulation of FLEEL carboxylation by the one-chain and two-chain carboxylases were 39.1 and 75.7 nM, respectively. This result indicates that the two-chain carboxylase and the one-chain carboxylase have similar affinity for the vitamin K-dependent substrate propeptide. Taken together, these results indicate that the two-chain carboxylase is functionally assembled in vivo.
The Two-Chain Carboxylase Is Joined by a Disulfide Bond
Previously, we presented evidence that there is a disulfide bond in carboxylase between cysteine residues 99 and 450 (7, 13). However, there is some disagreement about whether the disulfide exists (34).
Our earliest work on disulfides was with purified bovine carboxylase. When the purified enzyme is cleaved with trypsin into two chains, it has full activity and migrates on SDS gel analyses as a single band until the protein is reduced; then it migrates as two bands. This result first suggested to us the existence of a disulfide bond. In our later work using recombinant proteins, we confirmed that a disulfide bond exists in the one-chain carboxylase. In that study using mass spectrometric analyses, we identified the residues involved in the disulfide as C99 and C450 (13). Also in this study we changed the only two cysteines, other than C450 in the carboxy-terminal peptide, to alanine (C598/700A) and then trypsin-cleaved this carboxylase (13). This cleaved mutant was fully active and joined by a disulfide bond, based on gel analyses. This result confirms that C450 participates in the disulfide bond (13).
In the present study, to investigate the possibility that formation of the disulfide is an artifact of our purification methods, we used our recombinant two-chain carboxylase. As mentioned above, this molecule is fully active (carboxylase and epoxidase) when purified and binds factor IXs propeptide with affinity similar to that of the wild-type carboxylase. To analyze for disulfide formation, we purified the enzyme in the presence of NEM to minimize the chances of spurious disulfide bond formation. Since the two-chain carboxylase migrates as one band unless it is treated with DTT (Figure 1, lanes 2, 3, 6, and 7), the two peptides must be joined by at least one disulfide bond.
Figure 1.

SDS gel electrophoretic analyses of purified recombinant human one-chain and two-chain carboxylases. Affinity-purified one-chain carboxylase (lanes 1 and 5), two-chain carboxylase (lanes 2 and 6), and NEM-labeled two-chain carboxylase (lanes 3 and 7) were fractionated by SDS–PAGE under nonreduced (NR) and reduced (R) conditions and visualized by silver stain. The N-terminal and C-terminal peptides are indicated by arrows.
We also analyzed this two-chain carboxylase in microsomes, to obviate any effect purification might have on disulfide formation. In this study we again included NEM in the microsome preparation to prevent disulfide formation. Western blot analyses (Figure 4B,C) of these samples indicate that, in the major form of this enzyme, the two chains are joined by a disulfide bond. As expected, on the basis of our previous results and those of Pudota et al. (34), a small amount of the two-chain carboxylase migrates as if it were not cross-linked.
Figure 4.

Western blot analyses of carboxylase and the mutant carboxylases G125L, G128L, and G132L. Freshly prepared microsomes from cells expressing two-chain carboxylase (lanes 1 and 6, panels B and C), G125L two-chain carboxylase (lanes 2 and 7, panels B and C), G128L two-chain carboxylase (lanes 3 and 8, panels B and C), and G132L two-chain carboxylase (lanes 4 and 9, panels B and C) were labeled by NEM and analyzed by SDS–PAGE under reduced (R) and nonreduced conditions (NR). Proteins were visualized by Western blot using the antibody to the C-terminal peptide (B) and the antibody to the N-terminal peptide (C) as the primary antibodies. The percentage of disulfide bond formed in the mutant carboxylases (A) relative to wild-type carboxylase was determined by scanning the gel in panel B. The N-terminal and C-terminal peptides are indicated by arrows. Disulfide-linked G128L mutant two-chain carboxylase (B) is indicated by an asterisk.
These latter results indicated to us that the two-chain molecule is functionally intact and, therefore, that it must have a structure similar to that of the one-chain molecule and will be a useful model for studying carboxylase.
Proline 378 Is Important for Disulfide Formation
Proline residues at the membrane interface of transmembrane helices often play important roles in orienting essential residues in directions appropriate for protein function through the formation of a helix kink and/or swivel angles (35, 36). In carboxylase, there is a proline near the lumenal helix surface of both TMD1 (P80) and TMD5 (P378). We thought these two prolines might contribute to formation of the disulfide bond between cysteines 99 and 450. To test this hypothesis, we mutated P80 and P378 to leucine individually in the two-chain carboxylase.
Mutation of residue P80, which has activity similar to that of wild-type carboxylase (results not shown), has a minor effect on disulfide formation. On the other hand, P378L significantly decreased the disulfide formation in carboxylase (Figure 2A). These results were derived from scanning the blot (Figure 2C) of the affinity-purified two-chain carboxylase and its P80L and P378L mutants under nonreducing conditions.
Figure 2.

Determination of disulfide bond formation in purified two-chain carboxylase and P80L and P378L two-chain carboxylases by SDS gel electrophoretic and Western blot analyses. Affinity-purified NEM-labeled two-chain carboxylase (in both panels B and C, lanes 1 and 5) and P80L two-chain (panels B and C, lanes 2 and 6) and P378L two-chain carboxylases (panels B and C, lanes 3 and 7) were analyzed by SDS–PAGE under reduced (R) and nonreduced (NR) conditions. Protein was visualized by silver stain (B) or Western blot using the antibody to the N-terminal peptide as the primary antibody (C). The percentage of disulfide bond formed in the mutants relative to that in carboxylase shown in panel A was determined by scanning the blot in panel C. The N-terminal and C-terminal peptides are indicated by arrows. Disulfide-linked P378L mutant two-chain carboxylase (B) and the dimer of the N-terminal peptide (C) are indicated by an asterisk.
The P80L two-chain carboxylase (lanes 2 and 6) behaves similarly to the wild-type two-chain carboxylase (lanes 1 and 5) under both reducing and nonreducing conditions. This indicates that the majority of the two peptides of the P80L mutant two-chain carboxylase are linked by a disulfide bond. On the other hand, P378L has dramatically less disulfide-linked two-chain carboxylase (lane 3). It also has other protein bands which might represent degradation products caused by incorrect folding of the carboxylase. This interpretation is consistent with our previous observations (13) in which mutation of C99 and/or C450, the two cysteines we identified as forming the disulfide bond, caused degradation of the purified carboxylase. These results suggest that proline 378, but not proline 80, is important for disulfide bond formation in carboxylase. This is consistent with the fact that P378, but not P80, is conserved across species as diverse as Drosophila and humans.
Interestingly, in the P378L sample (Figure 2B, lane 3), there is a protein band that migrates the same as the N-terminal peptide. Since, for the most part, there is no disulfide bond holding the two chains together, and we purify the protein using the C-terminal tag, if the disulfide bond were the only interaction between the two chains, one would expect the N-terminal peptide to be lost during purification.
To determine if this band represented the N-terminal peptide, we probed the gel with the N-terminal peptide antibody (anti-FLAG) for Western blot analysis. The band at 30000 apparent molecular weight is recognized by the N-terminal peptide antibody, indicating that it is the N-terminal peptide of the two-chain carboxylase (Figure 2C, lanes 3 and 7). Therefore, the N-terminal peptide copurified with the noncovalently linked C-terminal peptide, suggesting that, in addition to the disulfide, there must be other noncovalent linkages between the two peptides in the two-chain carboxylase. A likely source would be interaction between the TMDs.
We observed SDS-resistant oligomerization of carboxylase or its peptides (Figures 2–5). Oligomerization was especially prevalent in the TMD-rich N-terminal peptide. We previously observed high molecular weight aggregates when we heated samples to ensure protein denaturation in SDS/DTT before gel analyses. For that reason we now routinely treat our samples with SDS and DTT at room temperature for 15 min. These higher molecular weight forms appear to be more predominant in carboxylase without the disulfide bond. For example, among the nonreduced samples (Figure 2C) only that of the P378L (lane 3) two-chain carboxylase, in which most of the disulfide bond is disrupted, shows significant oligomers. In addition, the wild-type two-chain carboxylase only exhibits significant oligomer formation under reducing conditions (Figure 2C, lane 5). In all cases, however, the predominant form of the N-terminal peptide was the monomer. To investigate whether these bands, as we expected, were the result of sample preparation, we tested the effect of temperature, DTT, and SDS on the extent of oligomerization. The oligomer bands increased with incubation at 80 °C, especially with DTT but no SDS (results not shown). This indicates that the reduced form is more susceptible to association. The oligomers may be due to hydrophobic interactions between the TMDs as found in other membrane proteins (37–40). As we reported, the functional one-chain carboxylase is a monomer (15, 16). Therefore, we conclude that oligomerization of the N-terminal peptide is not likely to have any biological significance and, thus, should not affect the interpretation of our results.
Figure 5.

Western blot analyses of purified G128L and G132L two-chain carboxylases. Affinity-purified NEM-labeled G128L (lanes 1 and 4, both panels) and G132L two-chain carboxylases (lanes 2 and 5, both panels) were analyzed by SDS–PAGE under reduced (R) and nonreduced (NR) conditions. Protein bands were detected by Western blot using the C-terminal peptide antibody (A) or the N-terminal peptide antibody (B). The arrow in panel A indicates the C-terminal peptide, and the arrow in panel B indicates the N-terminal peptide.
TMD5 Is the Only TMD in the C-Terminal Peptide of the Two-Chain Carboxylase
According to our membrane topology of carboxylase, there are five TMDs in carboxylase, and TMD5 is the only TMD in the C-terminal peptide of the two-chain carboxylase (12). TMD5 must therefore direct the large hydrophilic carboxyl terminus of carboxylase to the lumen of the ER. We previously showed that all of the carboxylase glycosylation sites are in the C-terminal peptide and, thus, must be in the lumen, which adds further support for our model (30).
If our topology model is right, and TMD5 is the only TMD in the C-terminal peptide, then two things must be true: first, if TMD5 is disrupted in the one-chain carboxylase, the carboxyl terminus will be in the cytoplasm, and it will not be glycosylated, and second, TMD5 must be involved in any interaction that involves the C-terminal peptide and another TMD. If the first criterion is true, then this form of carboxylase will migrate faster in SDS–PAGE analysis than does glycosylated carboxylase (30). If the second is true, disruption of TMD5 in the two-chain carboxylase will result in a non-membrane-associated C-terminal peptide and the peptide will not appear in Western blots of microsomes. If, on the other hand, there is another TMD in the C-terminal peptide, or a membrane-associated domain, then glycosylation may be normal and the peptide should still be associated with the microsomal membrane.
To test this hypothesis, we simultaneously mutated residues L368 and L372 in TMD5 to proline (L368/372P) to disrupt the transmembrane helix. We made both the one-chain and two-chain forms of the mutant carboxylases. We prepared microsomes from insect cells expressing wild-type and these mutant carboxylases and analyzed them by SDS–PAGE under nonreducing conditions. Protein bands were visualized on the Western blot using the C-terminal peptide antibody (Figure 3A) and the N-terminal peptide antibody (Figure 3B). As expected if our model is correct, the one-chain L368P/L372P carboxylase (Figure 3A, lane 2) migrates faster than the wild-type carboxylase (lane 1). It migrates the same as the deglycosylated wild-type carboxylase (lane 4) when all carbohydrates were removed from wild-type carboxylase by endoglycosidase, PNGase F. In addition, migration of the L368P/L372P one-chain carboxylase is not affected by PNGase F treatment (lane 5). These results suggest that when TMD5 was disrupted in the one-chain carboxylase, the large hydrophilic carboxyl terminus of carboxylase is not translocated into the ER lumen for glycosylation.
Figure 3.

Western blot analyses of one-chain carboxylase and one- and two-chain L368/372P carboxylase and PNGase F-treated forms of these carboxylases. Microsomes from cells expressing either wild-type carboxylase or the carboxylase L368/372P were analyzed by SDS–PAGE under nonreduced conditions and transferred to a polyvinylidene difluoride membrane. Protein bands were visualized using the antibody to the C-terminal carboxylase peptide (A) or the antibody to the N-terminal peptide (B) employing ECL reagents. One-chain wild-type carboxylase (lane 1 in panels A and B), one-chain L368/372P (lane 2 in both panels), two-chain L368/372P (lane 3 in both panels), PNGase F-treated wild-type carboxylase (lane 4 in both panels), and PNGase F-treated one-chain L368/372P (lane 5 in both panels). One-chain carboxylase (A) and the N-terminal peptide (B) are indicated by arrows. The dimer of the N-terminal peptide (B) is indicated by an asterisk.
The result with the one-chain mutant suggests that disruption of TMD5 in the two-chain mutant will cause the C-terminal peptide to dissociate from microsomal membranes unless there is a membrane-associated domain in that peptide. In microsomes prepared from cells expressing the two-chain L368P/L372P carboxylase, we did not detect the C-terminal peptide. This result indicates that it is no longer associated with the membrane (Figure 3A, lane 3). In sum, this indicates that TMD5 is the last TMD in carboxylase, and the only one in the C-terminal peptide, and there is no membrane-associated domain in the C-terminal peptide.
To ensure that the result described above for the two-chain mutant was not because the C-terminal peptide was not expressed, we analyzed whole cell lysate of cells putatively expressing two-chain L368P/L372P. A Western blot analysis of the lysate from these cells using the C-terminal peptide antibody revealed a peptide migrating at approximately 16 kDa (data not shown). This suggests that the C-terminal peptide is expressed, but due to improper folding, it is more susceptible to proteolysis and is degraded.
Figure 3B shows the same blot as in Figure 3A but probed by the N-terminal peptide antibody. As expected, the one-chain form is detected as in Figure 3A. The two-chain L368P/L372P carboxylase (lane 3) N-terminal peptide is expressed properly even when its C-terminal partner is not associated with the membrane. These results suggest that TMD5 is the only TMD in the C-terminal peptide of the two-chain carboxylase.
An Interaction between TMD2 and TMD5 Is Important for Carboxylase Assembly
The obvious question at this point in our investigation was what is the other TMD interacting with TMD5? Recently, three motifs that are important for TMD helix–helix interactions have been identified: the GxxxG motif and its like, polar and hydrogen-bonding motifs, and proline motifs (for review, see ref 23). Glycine motifs have been shown to support a hydrogen bond network that stabilizes domain–domain interactions (41). Among the five TMDs in carboxylase, there is a GxxxG (residues 128 and 132) motif in TMD2. In addition, in human carboxylase TMD5, there is a GxxxT (residues 363 and 367) sequence that also has been identified as supporting TMD interactions. In both cases, since they are separated by three amino acids, the residues of interest will be on the same face of the TMD helix. The presence of the GxxxG sequence in TMD2 suggested to us that TMD2 was a likely candidate for TMD–TMD interactions, perhaps with TMD5. The GxxxT sequence in TMD5 may be involved in the interactions in which TMD5 participates. In order to investigate this potential TMD interaction, we made five mutant carboxylases. We changed the glycines in the TMD2 motif (128 and 132), as well as the nearby G125, to leucine. In addition we created two carboxylases with either G363L/T367L or E373L/Q374L mutations, all in TMD5. The E373L/Q374L was to test whether these polar and/or charged residues are important for a TMD interaction. We chose leucine for substitution since it tends to support helix formation and would therefore be less likely to disrupt the TMD structure. Our logic was that if these residues do support TMD association, then they may be important for ensuring the proximity necessary for disulfide formation, and thus mutation may result in less disulfide formation between the carboxylase’s two chains.
All three glycine mutations in the two-chain carboxylase caused decreased disulfide bond formation, and in addition, both peptides of G125L were degraded (Figure 4). This suggests that the glycines in TMD2 are important for carboxylase structure but does not tell us directly whether TMD2 interacts with another TMD, especially TMD5.
To further explore this question, we purified the two-chain carboxylases that were the least degraded, G128L and G132L. Our rationale for this experiment was that if the N-terminal and C-terminal peptides of either G128L or G132L two-chain carboxylase associate through interactions other than the disulfide bond or a TMD2-TMD5 interaction, then we would expect both peptides of the two-chain molecules to be purified using our methodology which, as described, employs an antibody tag on the C-terminus. The N-terminal peptide should be present in both reduced and unreduced samples. If, on the other hand, the disulfide bond and a TMD interaction between TMDs 2 and 5 are the only significant association between the N- and C-terminal peptides, then the N-terminal peptide should only be present in disulfide-bonded purified G128L and G132L two-chain mutants.
In the purified G128L and G132L two-chain carboxylases the N-terminal peptide is visible only in the reduced sample (Figure 5B, lanes 4 and 5). This result is consistent with our hypothesis that the TMD interaction is between TMD2 and, since it is the only TMD in the C-terminal peptide, TMD5. Additionally, this indicates that the only interactions between these peptides are the TMD interaction and the disulfide bond.
When we mutated TMD residues in the C-terminal peptide to test for polar/charge residues (G363L/T367L and E373L/Q374L) that might be involved in the TMD interaction, the proteins were highly degraded (results not shown). This result suggests the residues are important for structure formation but does not offer further information about the TMD interaction and the participating residues in TMD5. However, our model described below does suggest that these results are consistent with participation in a TMD interaction.
Analysis of the Homology Model in the Context of Our Experimental Results
In order to offer possible structural explanations for our experimental results, we constructed a model (Figure 6) of the TMD2 and TMD5 interaction. The residues that we changed in the carboxylase are highlighted using a space-filling representation. Residues that we mutated, G363, T367, E373, and Q374, along with F366, Y370, and L377 comprise the interacting surface of TMD5 (Figure 6A). G363L/T367L and E373L/Q374L two-chain carboxylases were heavily degraded, which, while not offering direct evidence as to structural interactions, indicates that these residues are important and is consistent with the model.
Figure 6.
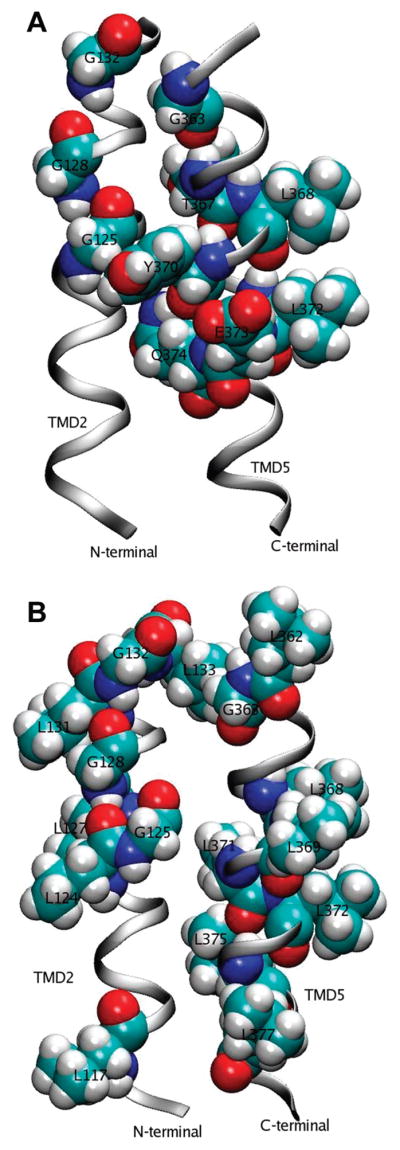
Homology model of TMD2 and TMD5. Two views of the homology model showing residues of interest in transmembrane domains 2 and 5 with space-filling representations.
In the model structure, G125, G128, and G132 in TMD2 are stacked along the helix, but only G125 appears to be part of the TMD5–TMD2 interface. In addition, all four methionines in TMD2 and Y119, F123, and A126 interact with TMD5. Since G125L is on the TMD2–TMD5 interface, our results showing degradation of G125L are consistent with the model. Apparently changing glycine to a bulkier leucine residue leads to a complete disruption of this interacting surface since the side chain would be competing for the space occupied in the wild-type enzyme by Y370 and F366 of TMD5. These latter residues have a π electron interaction.
If G125 forms part of the TMD2–TMD5 interface, as seems likely, then G128 and, therefore, G132 cannot. This is because G128 and G125 are separated by only two residues. Three intervening residues are necessary to complete the helix turn and position the residues on the same side.
The model suggests it is more likely that both G128 and G132 are on the helix face not associated with TMD5. This means that the two residues, which are on a face of TMD2 with four leucine residues (L117, L124, L127, and L131), might be interacting with another TM domain in addition to TMD5 (Figure 6B). According to our model, if G128 and G132 are not interacting with another TMD (besides TMD5), the helix should readily accommodate the leucine mutations because the substituted side chains would be oriented away from the TMD5 interface. However, our results indicate that G128 and G132 mutations do cause structural disruption, evidenced by decreased disulfide formation. If such an interaction surface exists, and the glycine motif supports the interaction, then, as we observe, substitution of the bulky leucine residues would disrupt this network.
If TMD2 forms a complex with TMD5 and one or more other TMDs, it may be that mutations disrupting either of the TMD2’s interactions might disrupt the complex as a whole. In other words, TMD2 may be acting as an anchor for the TMD complex. This would be consistent with our results that show the disulfide bond is disrupted in both G128L and G132L and that direct interaction between TMD2 and TMD5 is disrupted in those mutant carboxylases. A likely candidate for the other TMD involved would be TMD4, since it has five serine residues which are common in sequences identified as supporting TMD associations (42). However, since we only modeled TMDs 2 and 5, our homology construct does not constitute the entire neighborhood of G128 and G132; therefore, it is somewhat difficult to predict the outcome of G128L and/or G132L mutations with the current model. Our results do, however, indicate that further study of TMD interactions in the carboxylase will provide new structural information and perhaps insight into structure–function relationships in this enzyme.
It is too early to describe a detailed model for the carboxylase structure. However, we are tempted to speculate about how our results and previously published information fit together.
From what we know about carboxylase, purified carboxylase is a monomer; the propeptide binding site, glutamate binding site, and active site must be in close spatial proximity. In almost all vitamin K-dependent proteins the propeptide and Gla domain are adjacent in sequence. The propeptide anchors the substrate for multiple carboxylations without the propeptide dissociating, thus allowing the substrate to be repositioned, with respect to the active/binding site. Whether the carboxylase or the Gla domain moves is not known, but the propeptide remains bound. In addition, the glutamate site must be near the putative K218 active site residue for two reasons. First, the lysine presumably activates vitamin K (11), leading to removal of the γ proton of glutamate. Second, the positive charge of either K218 or K217 is likely to bind to the negatively charged glutamate. According to quantum chemical calculations, this second step is apparently necessary to facilitate the γ proton removal (43).
These notions fit with the idea that TMD interactions may be necessary for correct carboxylase conformation. The combination of the 99–450 disulfide bond and the proposed TMD2 and TMD5 interaction would bring the glutamate binding region around L394 (10) into proximity of the N-terminal sequence. The L394 sequence is near the exit of TMD5 from the membrane so its position is dependent on the location of TMD5. This is consistent with results implicating certain lumenal carboxylase N-terminal sequences in glutamate and propeptide binding, as well as K218’s apparent role in catalysis (6, 9, 11).
If the serines in TMD4 are interacting with TMD2 through the glycine motif or otherwise, then it would tend to limit the location of the long lumenal sequence containing K218 and make it more likely to be in proximity of the important sequences in the C-terminal region. Moreover, the disulfide bond would also tend to bring the C-terminal sequence involved in propeptide binding (438 to ~515) closer to the above regions.
In conclusion, we have provided further evidence supporting the existence of a disulfide bond in carboxylase. We have shown that the fifth transmembrane domain of carboxylase is the last and only such domain in the C-terminal peptide of our two-chain carboxylase. In addition, our results reveal the importance of proline 378 in the structure necessary for formation of the enzyme’s disulfide bond. Lastly, we found that it is likely the second and fifth transmembrane domains of carboxylase interact through previously described motifs commonly supporting these types of interactions and describe a structural model consistent with our results.
Footnotes
Abbreviations: ER, endoplasmic reticulum; TMD, transmembrane domain; NEM, N-ethylmaleimide; CHAPS, 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate; DTT, dithiothreitol; MOPS, 3-(N-morpholino)propanesulfonic acid (4-morpholinepropanesulfonic acid); MES, 2-(N-morpholino)ethanesulfonic acid (4-morpholineethanesulfonic acid); vitamin K1(20), 2-methyl-3-phytyl-1,4-naphthoquinone; PNGase F, peptide:N-glycosidase F; proFIX, 19 amino acid peptide comprising residues TVFLDHENANKILNRPKRY of human profactor IX; SRII, sensory rhodopsin II; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; PVDF, polyvinylidene fluoride.
This work was supported by National Institutes of Health (NIH) Grant HL48318 (to D.W.S.) and by the Intramural Research Program of the NIH and NIEHS (to L.P.).
References
- 1.Berkner KL. The vitamin K-dependent carboxylase. Annu Rev Nutr. 2005;25:127–149. doi: 10.1146/annurev.nutr.25.050304.092713. [DOI] [PubMed] [Google Scholar]
- 2.Furie B, Bouchard BA, Furie BC. Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood. 1999;93:1798–1808. [PubMed] [Google Scholar]
- 3.Stafford DW. The vitamin K cycle. J Thromb Haemostasis. 2005;3:1873–1878. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 4.Furie B, Furie BC. Molecular basis of gamma-carboxylation. Role of the propeptide in the vitamin K-dependent proteins. Ann NY Acad Sci. 1991;614:1–10. doi: 10.1111/j.1749-6632.1991.tb43687.x. [DOI] [PubMed] [Google Scholar]
- 5.Morris DP, Stevens RD, Wright DJ, Stafford DW. Processive post-translational modification. Vitamin K-dependent carboxylation of a peptide substrate. J Biol Chem. 1995;270:30491–30498. doi: 10.1074/jbc.270.51.30491. [DOI] [PubMed] [Google Scholar]
- 6.Yamada M, Kuliopulos A, Nelson NP, Roth DA, Furie B, Furie BC, Walsh CT. Localization of the factor IX propeptide binding site on recombinant vitamin K dependent carboxylase using benzoylphenylalanine photoaffinity peptide inactivators. Biochemistry. 1995;34:481–489. doi: 10.1021/bi00002a012. [DOI] [PubMed] [Google Scholar]
- 7.Wu SM, Mutucumarana VP, Geromanos S, Stafford DW. The propeptide binding site of the bovine gamma-glutamyl carboxylase. J Biol Chem. 1997;272:11718–11722. doi: 10.1074/jbc.272.18.11718. [DOI] [PubMed] [Google Scholar]
- 8.Lin PJ, Jin DY, Tie JK, Presnell SR, Straight DL, Stafford DW. The putative vitamin K-dependent gamma-glutamyl carboxylase internal propeptide appears to be the propeptide binding site. J Biol Chem. 2002;277:28584–28591. doi: 10.1074/jbc.M202292200. [DOI] [PubMed] [Google Scholar]
- 9.Kuliopulos A, Nelson NP, Yamada M, Walsh CT, Furie B, Furie BC, Roth DA. Localization of the affinity peptide-substrate inactivator site on recombinant vitamin K-dependent carboxylase. J Biol Chem. 1994;269:21364–21370. [PubMed] [Google Scholar]
- 10.Mutucumarana VP, Acher F, Straight DL, Jin DY, Stafford DW. A conserved region of human vitamin K-dependent carboxylase between residues 393 and 404 is important for its interaction with the glutamate substrate. J Biol Chem. 2003;278:46488–46493. doi: 10.1074/jbc.M307707200. [DOI] [PubMed] [Google Scholar]
- 11.Rishavy MA, Hallgren KW, Yakubenko AV, Shtofman RL, Runge KW, Berkner KL. Bronsted analysis reveals Lys218 as the carboxylase active site base that deprotonates vitamin K hydroquinone to initiate vitamin K-dependent protein carboxylation. Biochemistry. 2006;45:13239–13248. doi: 10.1021/bi0609523. [DOI] [PubMed] [Google Scholar]
- 12.Tie J, Wu SM, Jin D, Nicchitta CV, Stafford DW. A topological study of the human gamma-glutamyl carboxylase. Blood. 2000;96:973–978. [PubMed] [Google Scholar]
- 13.Tie JK, Mutucumarana VP, Straight DL, Carrick KL, Pope RM, Stafford DW. Determination of disulfide bond assignment of human vitamin K-dependent gamma-glutamyl carboxylase by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Biol Chem. 2003;278:45468–45475. doi: 10.1074/jbc.M309164200. [DOI] [PubMed] [Google Scholar]
- 14.Rishavy MA, Pudota BN, Hallgren KW, Qian W, Yakubenko AV, Song JH, Runge KW, Berkner KL. A new model for vitamin K-dependent carboxylation: the catalytic base that deprotonates vitamin K hydroquinone is not Cys but an activated amine. Proc Natl Acad Sci USA. 2004;101:13732–13737. doi: 10.1073/pnas.0404989101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt-Krey I, Haase W, Mutucumarana V, Stafford DW, Kuhlbrandt W. Two-dimensional crystallization of human vitamin K-dependent gamma-glutamyl carboxylase. J Struct Biol. 2007;157:437–442. doi: 10.1016/j.jsb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Presnell SR, Tripathy A, Lentz BR, Jin DY, Stafford DW. A novel fluorescence assay to study propeptide interaction with gamma-glutamyl carboxylase. Biochemistry. 2001;40:11723–11733. doi: 10.1021/bi010332w. [DOI] [PubMed] [Google Scholar]
- 17.Ottolia M, John S, Qiu Z, Philipson KD. Split Na+-Ca2+ exchangers. Implications for function and expression. J Biol Chem. 2001;276:19603–19609. doi: 10.1074/jbc.M101489200. [DOI] [PubMed] [Google Scholar]
- 18.Althage M, Karlsson J, Gourdon P, Levin M, Bill RM, Tigerstrom A, Rydstrom J. Functional split and crosslinking of the membrane domain of the beta subunit of proton-translocating transhydrogenase from Escherichia coli. Biochemistry. 2003;42:10998–11003. doi: 10.1021/bi034560x. [DOI] [PubMed] [Google Scholar]
- 19.Ehnes C, Forster IC, Kohler K, Biber J, Murer H. Functional studies on a split type II Na/P(i)-cotransporter. J Membr Biol. 2002;188:227–236. doi: 10.1007/s00232-001-0186-y. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Zhang XQ, Ahlers BA, Carl LL, Song J, Rothblum LI, Stahl RC, Carey DJ, Cheung JY. Cytoplasmic tail of phospholemman interacts with the intracellular loop of the cardiac Na+/Ca2+ exchanger. J Biol Chem. 2006;281:32004–14. doi: 10.1074/jbc.M606876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adler J, Bibi E. Promiscuity in the geometry of electrostatic interactions between the Escherichia coli multidrug resistance transporter MdfA and cationic substrates. J Biol Chem. 2005;280:2721–2729. doi: 10.1074/jbc.M412332200. [DOI] [PubMed] [Google Scholar]
- 22.Mo L, Xiong W, Qian T, Sun H, Wills NK. Coexpression of complementary fragments of ClC-5 and restoration of chloride channel function in a Dent’s disease mutation. Am J Physiol Cell Physiol. 2004;286:C79–C89. doi: 10.1152/ajpcell.00009.2003. [DOI] [PubMed] [Google Scholar]
- 23.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Curran AR, Engelman DM. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 25.DeGrado WF, Gratkowski H, Lear JD. How do helix-helix interactions help determine the folds of membrane proteins? Perspectives from the study of homo-oligomeric helical bundles. Protein Sci. 2003;12:647–665. doi: 10.1110/ps.0236503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanley TB, Jin DY, Lin PJ, Stafford DW. The propeptides of the vitamin K-dependent proteins possess different affinities for the vitamin K-dependent carboxylase. J Biol Chem. 1999;274:16940–16944. doi: 10.1074/jbc.274.24.16940. [DOI] [PubMed] [Google Scholar]
- 27.Gafvelin G, Sakaguchi M, Andersson H, von Heijne G. Topological rules for membrane protein assembly in eukaryotic cells. J Biol Chem. 1997;272:6119–6127. doi: 10.1074/jbc.272.10.6119. [DOI] [PubMed] [Google Scholar]
- 28.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 29.Mutucumarana VP, Stafford DW, Stanley TB, Jin DY, Solera J, Brenner B, Azerad R, Wu SM. Expression and characterization of the naturally occurring mutation L394R in human gamma-glutamyl carboxylase. J Biol Chem. 2000;275:32572–32577. doi: 10.1074/jbc.M006808200. [DOI] [PubMed] [Google Scholar]
- 30.Tie JK, Zheng MY, Pope RM, Straight DL, Stafford DW. Identification of the N-linked glycosylation sites of vitamin K-dependent carboxylase and effect of glycosylation on carboxylase function. Biochemistry. 2006;45:14755–14763. doi: 10.1021/bi0618518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9.0. University of California; San Francisco: 2006. [Google Scholar]
- 32.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DMJ, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 33.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pudota BN, Miyagi M, Hallgren KW, West KA, Crabb JW, Misono KS, Berkner KL. Identification of the vitamin K-dependent carboxylase active site: Cys-99 and Cys-450 are required for both epoxidation and carboxylation. Proc Natl Acad Sci USA. 2000;97:13033–13038. doi: 10.1073/pnas.97.24.13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordes FS, Bright JN, Sansom MS. Proline-induced distortions of transmembrane helices. J Mol Biol. 2002;323:951–960. doi: 10.1016/s0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 36.Orzaez M, Salgado J, Gimenez-Giner A, Perez-Paya E, Mingarro I. Influence of proline residues in transmembrane helix packing. J Mol Biol. 2004;335:631–640. doi: 10.1016/j.jmb.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 37.Choi S, Lee E, Kwon S, Park H, Yi JY, Kim S, Han IO, Yun Y, Oh ES. Transmembrane domain-induced oligomerization is crucial for the functions of syndecan-2 and syndecan-4. J Biol Chem. 2005;280:42573–42579. doi: 10.1074/jbc.M509238200. [DOI] [PubMed] [Google Scholar]
- 38.Favreau C, Bastos R, Cartaud J, Courvalin JC, Mustonen P. Biochemical characterization of nuclear pore complex protein gp210 oligomers. Eur J Biochem. 2001;268:3883–3889. doi: 10.1046/j.1432-1327.2001.02290.x. [DOI] [PubMed] [Google Scholar]
- 39.Johnson RM, Heslop CL, Deber CM. Hydrophobic helical hairpins: design and packing interactions in membrane environments. Biochemistry. 2004;43:14361–14369. doi: 10.1021/bi0492760. [DOI] [PubMed] [Google Scholar]
- 40.Kubista H, Edelbauer H, Boehm S. Evidence for structural and functional diversity among SDS-resistant SNARE complexes in neuroendocrine cells. J Cell Sci. 2004;117:955–966. doi: 10.1242/jcs.00941. [DOI] [PubMed] [Google Scholar]
- 41.Senes A, Ubarretxena-Belandia I, Engelman DM. The Cα–H···O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawson JP, Weinger JS, Engelman DM. Motifs of serine and threonine can drive association of transmembrane helices. J Mol Biol. 2002;316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- 43.Davis CH, Deerfield D, II, Stafford DW, Pedersen LG. Quantum chemical study of the mechanism of action of vitamin K carboxylase (VKC). IV Intermediates and transition states. J Phys Chem A. 2007;111:7257–7261. doi: 10.1021/jp068564y. [DOI] [PubMed] [Google Scholar]