

Abstract
microRNA-155 is an oncogenic microRNA that has been shown to be critical for B-cell maturation and immunoglobulin production in response to antigen. In line with its function in B-cell activation, miR-155, and its primary transcript, B-cell integration cluster (BIC), is induced by B-cell receptor (BCR) cross-linking. Using pharmacological inhibitors in the human B-cell line, Ramos, we show that activation of BIC and miR-155 expression by BCR signaling occurs through the extracellular signaling-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) pathways but not the p38 pathway. BCR activation results in the induction of c-Fos, FosB, and JunB, and expression of these are suppressed by ERK and JNK inhibitors. Reporter analysis established a key role for a conserved AP-1 site ∼40 bp upstream from the site of initiation but not an upstream NF-κB site or a putative c-Ets located at the site of initiation. Lastly, chromatin immunoprecipitation analysis demonstrated the recruitment of FosB and JunB to the miR-155 promoter following BCR activation. These results identify key determinants of BCR-mediated signaling that lead to the induction of BIC/miR-155.
MicroRNAs (miRNAs)2 have been shown to be key mediators of cell regulatory processes such as those controlling cell growth, differentiation, and development (1–3). The ability of at least some miRNAs to significantly alter cell processes and cell fate is attributable to their capacity to influence the expression of a large number of target mRNA species. miRNAs function to inhibit translation of mRNAs through specific but imperfect base pairing with their 3′-untranslated regions. The binding of miRNA protein complexes to mRNAs results in localization of the miRNA-protein-mRNA complex to a perinuclear compartment referred to as GW or P bodies, thereby preventing access to ribosomes, and in some cases, leading to the degradation of the respective mRNA (4, 5).
The microRNA, miR-155, is processed from a primary transcript, referred to as B-cell integration cluster (BIC), whose upstream region was originally identified as a common site of integration of the avian leukosis virus in lymphomas (6). Transgenic mouse studies demonstrated that B-cell targeted expression of BIC leads to the development of B-cell malignancies (7). Further, a number of miRNA profiling studies have shown elevation of miR-155 in a wide array of cancers including lymphomas (7–14). To date, miR-155 is one of the most highly implicated microRNAs in cancers.
miR-155 has been shown to play a critical role in lymphocyte activation in vivo (15, 16) and is induced by a number of immune cell stimuli including Toll-like receptor (TLR) ligands, tumor necrosis factor-α (TNF-α), interferon-β, and antigen (B-cell receptor (BCR) engagement) (9, 17, 18). The mechanisms through which miR-155 is regulated following TLR and interferon signaling in macrophages has recently come under study (17). In this study, TLR ligand-mediated activation of miR-155 was shown to occur through myeloid differentiation factor 88 (MyD88) and Toll/IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF)-dependent pathways (17). Interferon signaling was found to require an autocrine pathway involving TNF-α. Lastly, induction of miR-155 by poly(I-C) and TNF-α were shown to be inhibited by a Jun-activated kinase (JNK) inhibitor, suggesting that this pathway plays a role in induction of miR-155 in these systems. Here we have begun to analyze signaling pathways involved in activating miR-155 following B-cell receptor (BCR) engagement. This analysis identified critical pathways required for BCR-mediated miR-155 activation, some of which likely overlap with pathways activated by TLR and TNF-α signaling in macrophages. Together these studies define some of the fundamental miR-155 regulatory processes following immune cell activation and lays the groundwork for understanding some of the mechanisms through which BIC/miR-155 is overexpressed in tumors.
Experimental Procedures
Cell Culture and Treatments
The EBV-negative human Burkitt lymphoma cell line, Ramos, was cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and penicillin/streptomycin (Invitrogen). B-cell receptor cross-linking experiments were carried out by exposure to anti-human-IgM (Sigma, catalog number I 0759). An equal volume of fresh medium was added to Ramos cells (which were at densities of ∼1–2 × 106/ml) 1 day before treatment. On the day of treatment, cells were counted, and 2 × 107 cells were added to 10 ml of fresh complete RPMI medium containing 10 μm anti-IgM. Inhibitor experiments were carried out as above except that the inhibitors (all purchased from Calbiochem®) were added to cells resuspended in fresh medium 30 min prior to the addition of anti-IgM (to a final concentration of 10 μm). For RT-PCR analysis, cells were harvested 24 h following the addition of anti-IgM. For Western blot analysis, cells were treated as above except that cells were harvested at the indicated times (in the figures and/or figure legends for Figs. 4 and 5).
FIGURE 4. The AP-1 family members, JunB, c-Fos, and FosB are induced by BCR cross-linking.
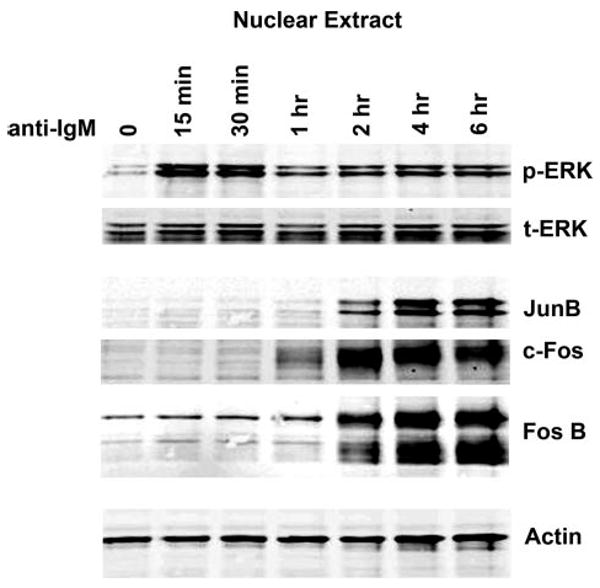
Time course analysis shows early activation of phospho-ERK (p-ERK) followed by induction of JunB, c-Fos, and FosB between 1 and 2 h after induction in Ramos cells. t-ERK, total ERK.
FIGURE 5. Induction of JunB, c-Fos, and FosB is inhibited by ERK and JNK inhibitors.

Ramos cells were pretreated with the indicated inhibitors prior to the addition of anti-IgM. Cells were lysed and subjected to Western blot analysis as described under “Experimental Procedures.” MEK1/2 Inh., MEK1/2 inhibition; JNK Inh., JNK inhibition; p38 Inh., p38 inhibition; DMSO, Me2SO; p-ERK, phospho-ERK.
RNA Preparation and Real-time RT-PCR
Total RNA was prepared using a Qiagen miRNeasy mini prep kit (catalog number 217004) according to the vendor's protocol. For primary BIC transcript analysis, 2 μg of total RNA was reverse-transcribed to make cDNA using SuperScript™ III first-strand synthesis system (Invitrogen, catalog number 18080-051). PCR reactions were carried out using the following primers and conditions: BIC forward (BIC-b), 5′-CTCTAATGGTGGCACAAA-3′; BIC reverse (BIC-c), 5′-TGATAAAAACAAACATGGGCTTGAC-3′ (14); G3PDH forward, 5′-GCCAAGGTCATCCATGACAACTTTGG-3′, and G3PDH reverse, 5′-GCCTGCTTCACCACCTTCTTGATGTC-3′ (19). PCR reactions were performed using the following parameters: 95 °C for 3 min followed by 40 cycles of denaturation (95 °C 30 s), annealing (62 °C 40 s), and extension (72 °C 40 s). Real-time PCR was conducted with 2 μl of 10-fold diluted cDNA using iQ™SYBR® Green Supermix (Bio-Rad catalog number 170-8882) and an iQ5 multicolor real-time PCR detection system (Bio-Rad).
Mature miR-155 expression was assessed using a mirVanaTM qRT-PCR miRNA detection kit (Ambion, catalog number 1558) kit with the mirVana qRT-PCR miR-155 primer set (Ambion, catalog number 30302) according to the manufacturer's protocol. PCR was carried out using the following conditions: 95 °C for 3 min followed by 40 cycles of 95 °C, 15 s and 60 °C, 30 s. The expression of BIC and miR-155 in experimental samples was determined by the comparative Ct method (2-(Δ)(Δ)Ct method), in which Ct is the threshold cycle and ΔΔCt = (ΔCt BIC) − (ΔCt reference RNA (G3PDH)).
BLAST Analysis of miR-155 Promoter versus Mouse Genome
1939 bases of sequence upstream and 562 bases of sequence downstream from the miR-155 start site in humans were used to blast the mouse genome using the National Center for Biotechnology Information (NCBI) Blastn algorithm with default settings. A single homology shown in Fig. 3 was identified as the only hit and corresponds to 8,703,897–8,703,794 and 8,703,618–8,703,488 of the mouse chromosome 11 (NCBI, reference assembly).
FIGURE 3. The conserved AP-1 element is important for mediating activation of BIC transcription following BCR activation.
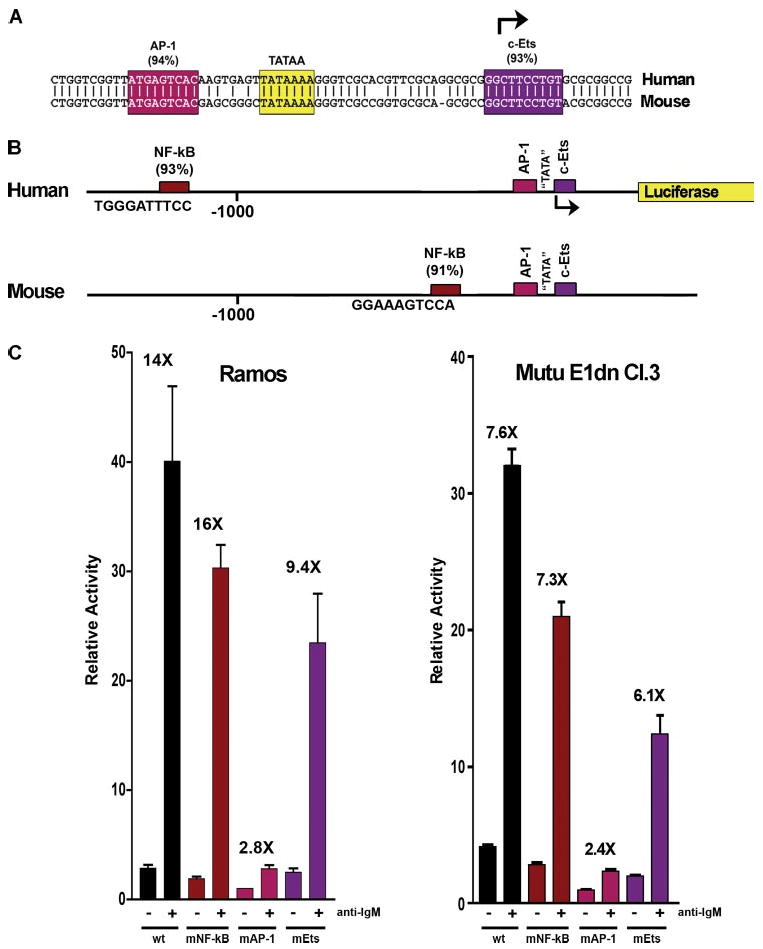
A shows the core homology region of the BIC promoter. B shows schematic representation of human versus mouse promoter. C, analysis of wild type (wt) versus mutant (mNF-κB, mAP-1, and mEts) reporters in response to BCR signaling in Ramos cells and Mutu E1dn Cl.3 (an EBV-negative derivative of the cell line, Mutu (20)).
5′ RACE Analysis
5′ RACE was carried out using total RNA isolated from the human B-cell line, Ramos, following exposure to anti-IgM, using a SMART RACE cDNA amplification kit (Clontech) according to the manufacturer's protocol. The gene-specific primer, 5′-CAGCCTACAGCAAGCCTTCAGCACTC-3′, which is complementary to the third exon of the human BIC/miR-155 primary transcript was used. One major PCR product with a size of 350 bp was obtained and cloned into the PCR4-TOPO cloning vector by TA cloning. Seven positive clones were sequenced to determine the 5′ most end in each case. The major start site of the human BIC transcript was found to be located at nucleotide 12,596,314 of the human chromosome 21 (NCBI, reference assembly).
Cloning and Mutagenesis of BIC/miR-155 Promoter
The human BIC/miR-155 promoter extending from −1494 to +228 relative to the start site was isolated from Mutu genomic DNA by PCR using the primers 5′-GCAGCTAGCCCAGGGTTGGAACTGAGTTTGA-3′ (forward primer) and 5′-GCAAAGCTTCAGTTAACCCGGCGGTGA-3′ (reverse primer). The isolated fragment was digested with NheI and HindIII and cloned into NheI and HindIII cut pGL3basic (Promega). The entire promoter region was then sequenced, and no discrepancies were identified relative to the GenBank™ genomic sequence.
Mutagenesis of the miR-155 reporter plasmid was carried out using a QuikChange II site-directed mutagenesis kit (Stratagene) using the following oligonucleotides: 5′-GTAAATTAAGTACTATGCTCGAGCCAGCTCTGACATG-3′ and 5′-CATGTCAGAGCTGGCTCGAGCATAGTACTTAATTTAC-3′ (NF-κB), 5′-CTGGTCGGTTATCTCGAGCAAGTGAGTTAT-3′ and 5′-ATAACTCACTTGCTCGAGATAACCGACCAG-3′ (AP-1), and 5′-CGCAGGCGCGGCTCGAGTGTGCGCGGCC-3′ and 5′-GGCCGCGCACACTCGAGCCGCGCCTGCG-3′ (c-Ets). In each case, the core transcription factor binding site was replaced with an XhoI restriction site. Mutations were initially screened by digesting with XhoI and then verified by sequence analysis.
Reporter Analysis
For each reporter plasmid, 5 × 106 Ramos cells were distributed into each of two T25 tissue culture flasks containing 10 ml of RPMI 1640 (+10% fetal bovine serum, + penicillin/streptomycin). 5 μg of the respective reporter vector plus 35 μg of the carrier plasmid, pUHD10, were mixed with 2.5 ml of Opti-MEM. 2.5 ml of diluted Lipofectamine 2000 (100 μl of Lipofectamine plus 2.5 ml of opti-MEM) was then added to each set of DNA/opti-MEM mixtures, and the tubes were mixed and incubated at room temperature for 20 min. For each transfection, 2.5 ml of DNA/Lipofectamine mixture was then added to each of two separate T25 flasks containing cells, and flasks were incubated at 37 °C, 5% CO2 for 24 h. At this time, affinity-isolated goat anti human IgM (catalog number I0759, Sigma) was added to a final concentration of 10 μm to the second flask for each reporter. Cells were harvested 24 h later and assayed for luciferase activity. Results are presented as the average of three independent experiments.
The reporter analyses shown in Fig. 7C were carried out as above except that cells were not treated with anti-IgM and cells were co-transfected with reporters plus either 1 μg of a control, JunB, or FosB expression vector or 0.5 μg of JunB plus 0.5 μg of FosB expression vectors. The reporter analyses of anti-IgM treated Mutu E1dn Cl.3 cells (20) were carried out as described for the anti-IgM analysis in Ramos cells except that cells were transfected by electroporation.
FIGURE 7. JunB and FosB bind and activate the BIC promoter through the AP-1 element.

A and B, electrophoretic mobility shift analysis was carried out using the BIC AP-1 promoter element and extracts from uninduced and anti-IgM induced Ramos cells. Competitor oligonucleotides sequences and antibodies used are described under “Experimental Procedures.” control Ab, control antibody. C, Ramos cells were transfected with either the wild type BIC reporter (wt) or the AP-1 site mutant BIC reporter (mAP-1) along either with 1 μg of control, JunB, or FosB expression vectors or with 0.5 μg of JunB plus 0.5 μg of FosB expression vector. All values are relative to wild type promoter activity plus control expression vectors. CNTL, control.
Western Blot Analysis
Ramos cells were treated as discussed above prior to harvesting for Western blot analysis. For whole cell lysates, cells were suspended in 1× SDS loading buffer and heated for 10 min at 90 °C to shear genomic DNA. To generate nuclear extracts, 7 × 107 Ramos cells were suspended in 300 μl of hypotonic buffer (HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 1 mm dithiothreitol, plus protease and phosphatase inhibitor cocktails), 18 μl of 10% Nonidet P-40 was added, and the mixture was vortexed on high for 10 s. The tubes were transferred to a microcentrifuge and spun for 2 min on high. The supernatant was taken off, and 100 μl of nuclear extraction buffer (20 mm HEPES (pH 7.9), 0.4 m NaCl, 1 mm EDTA, plus 1 mm dithiothreitol and protease and phosphatase inhibitor cocktails) was added, and the pellet was suspended immediately by vigorous pipetting. The tubes were put on a rotator for 15 min at 4 °C and spun for 5 min in a microcentrifuge (14,000 rpm). The supernatant was transferred to new tubes and quantitated for use in Western blot analysis. Antibodies recognizing phospho-ERK (Cell Signaling (catalog number 9106)), total ERK (Cell Signaling (catalog number 9102)), c-Jun (Cell Signaling (catalog number 2315 and catalog number 9165)), c-Fos (Cell Signaling (catalog number 2250)) and FosB (Cell Signaling catalog number 2251)), JunB (Santa Cruz Biotechnology (sc-8051)), and actin (Sigma (catalog number A4700)) were used for Western blot analysis. Western blotting was carried out using 25 μg of total cell lysate of each cell lysate. Signal detection was carried out using an Odyssey infrared imaging system (LI-COR Biosciences).
Immunoprecipitation
For immunoprecipitation, 2 × 107 cells were suspended in 500 μl of RIPA buffer (50 mm Tris-HCl, pH7.4, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 0.1% SDS) and incubated for 1 h at 4 °C. Lysates were then centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatants were transferred to new tubes. Cell lysates were precleared with 25 μl of protein A (for polyclonal antibody) or protein G (for monoclonal antibody) Sepharose (GE Healthcare) for 1 h. At the same time, 5 μl of the FosB antibody (Cell Signaling, catalog number 2251) and 20 μl of the JunB antibody (Santa Cruz, catalog number sc-8051) were incubated with 20 μl of protein A- or G-Sepharose in 400 μl of RIPA buffer for 1 h at 4 °C and then washed three times with RIPA buffer. The lysates were then centrifuged briefly, and the supernatants were transferred to the antibody-Sepharose complexes and incubated overnight on a rotator at 4 °C. The beads were washed three times with RIPA buffer with 5-min incubations (at 4 °C) for each wash. 25 μl of 2× SDS page loading buffer was then added to Sepharose bead complexes, and samples were heated for 5 min at 95 °C and loaded onto an SDS-PAGE gel for Western blot analysis.
Chromatin Immunoprecipitation
Ramos cells were cultured for 2 h in the absence or presence of 10 μm anti-IgM (Sigma, catalog number I 0759) (1 × 107 cells were used for each immunoprecipitation). Cells were then fixed with 1% formaldehyde for 30 min. Glycine was added to a final concentration of 0.125 m, and cells were spun down. Cells were washed 2× with 1× phosphate-buffered saline. Cell pellets were then suspended in ice-cold RIPA buffer and incubated on ice for 1 h Cell lysates were then sonicated to obtain DNA fragments averaging 600 bp. Lysates were then spun on high speed in a microcentrifuge at 4°C for 15 min, and supernatant was used for immunoprecipitations. For each immunoprecipitation, 350 μl of samples was precleared with 50 μl of protein A agarose beads with overnight incubation on a rotator at 4 °C. Beads were spun out, and supernatant was transferred to new tubes containing 50 μl of protein A agarose beads for a 4-h second preclearing step. Samples were then spun, and supernatant was transferred to a new tube containing the following antibodies: FosB (20 μl, Santa Cruz (catalog number sc-48X)), JunB (20 μl, Santa Cruz (catalog number sc-73X)), histone H4 (5 μl of H4, Abcam (catalog number ab31827)), and acetyl-histone H4 (lys8) (5 μl of Ac-H4, Upstate Biochemicals (catalog number 07-328)). Tubes were rotated at 4 °C overnight, and 50 μl of protein A agarose beads was added for a 2-h incubation on rotator at 4 °C. Samples were spun briefly, and supernatant was aspirated off. Beads were washed three times with 1× phosphate-buffered saline. Beads were incubated with 100 μl of Tris-EDTA containing 50 μg/ml RNase A at 37 °C for 1 h. 5 μl of 10% SDS plus 2 μl of 1 mg/ml proteinase K were added, and beads were incubated for 4 h at 42 °C with occasional mixing. Samples were then incubated overnight at 65 °C, phenol/chloroform-extracted, ethanol-precipitated, and resuspended in 20 μl of H2O. 1 μl was used for PCR reactions using the following conditions: 95 °C for 30 s, 30× 60 °C for 30 s, 72 °C for 30 s, 95 °C for 30 s. Primers used were as follows: BIC promoter primers, BICps, 5′-CCTGGTCGGTTATGAGTCAC-3′ and BICpas, 5′-GAGACTGAAGTCGGCGTACC-3′; and BIC intron 1 primers, BICintron1S, 5′-CATGGAAGGGTGACAAAACA-3′ and BICintron1AS, 5′-CGTTTTCCATTTGCCTAACA-3′.
Electrophoretic Mobility Shift Analysis
For each binding reaction, 10 μg of nuclear extract was combined with 0.1 pmol of IRdye700-labeled probe (LI-COR Biosciences) using standard binding conditions (LI-COR Biosciences (part number 829-07910) standard protocol). Labeled AP-1 probe oligonucleotides were purchased from LI-COR Biosciences: AP-1 sense, 5′-TCGGTTATGAGTCACAAGTGA-3′ and AP-1 antisense 5′-TCACTTGTGACTCATAACCGA-3′. Unlabeled competitor oligonucleotides were purchased from Integrated DNA Technologies: AP-1wt sense, 5′-TCGGTTATGAGTCACAAGTGA-3′, AP-1wt antisense, 5′-TCACTTGTGACTCATAACCGA-3′, AP-1mutant sense, 5′-TCGGTTATCTCGAGCAAGTGA-3′, and AP-1 mutant antisense, 5′-TCACTTGCTCGAGATAACCGA-3′. Supershift experiments were carried out using the following antibodies: JunB (Santa Cruz Biotechnology (sc-8051X)), FosB (Santa Cruz Biotechnology (sc-48X)) and normal rabbit IgG (sc-2027). Competition and supershift experiments were performed by preincubation with extract in binding buffer for 10 min, after which labeled probe was added for a further 20-min incubation at room temperature. Reactions were then loaded onto a pre-run 4% polyacrylamide retardation gel and run for 3 h at 300 V. Gels were scanned using an odyssey infrared imaging system (Li-Cor Biosciences).
Results
Induction of BIC/miR-155 Transcription following BCR Activation Requires the ERK and JNK Signaling Pathways
To identify pathways involved in mediating BIC promoter activation following BCR activation, the human B-cell line, Ramos, was treated with anti-IgM in the presence of ERK, JNK, or p38 pathway inhibitors. Cells were treated with either Me2SO or inhibitors of the upstream ERK effector, mitogen-activated protein kinase kinase (MEK (1/2)), PD98059 or U0126, the JNK inhibitor, SP600125, or the p38 inhibitor, SB203580, for 0.5 h prior to the addition of anti-IgM. 24 h later, cells were harvested, RNA was prepared, and the RNA was subjected to real-time RT-PCR to assess the levels of BIC and miR-155 transcripts. As shown in Fig. 1, the MEK inhibitors and the JNK inhibitor, SP600125 suppressed induction of BIC (Fig. 1A) as well as mature miR-155 (Fig. 1B), whereas the p38 inhibitor, SB203580, has little influence. These data demonstrate that activation of BIC transcription is mediated through the ERK and JNK pathways.
FIGURE 1. Inhibition of BIC and miR-155 expression by MEK and JNK inhibitors following activation of BCR signaling.
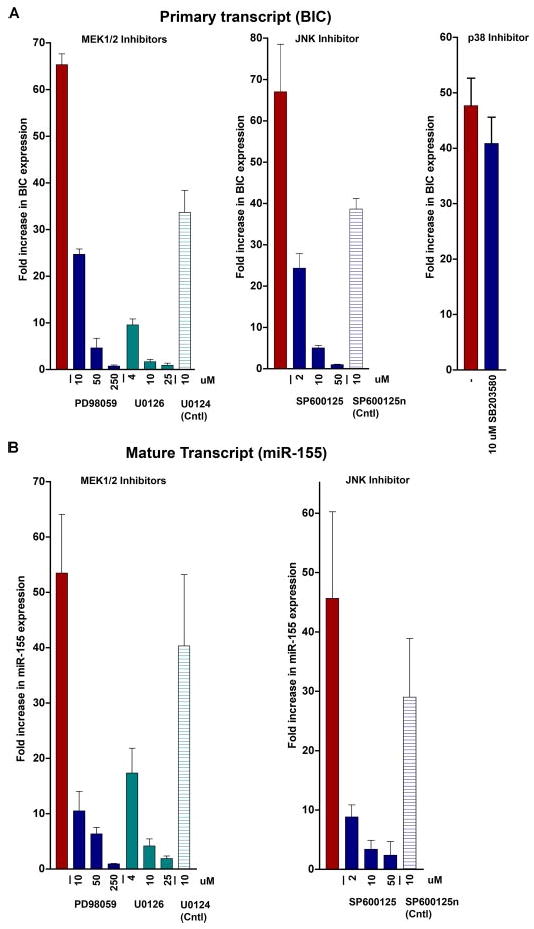
Ramos cells were pre-treated with the indicated inhibitors prior to the addition of anti-IgM. Total RNAs were then analyzed for BIC (A) and miR-155 (B) expression by real-time reverse transcription-PCR. All values are relative to G3PDH expression.
Identification of the BIC Transcription Initiation Site in BCR-activated Ramos Cells
BIC/miR-155 expression is highly regulated at the transcription level. Based on reported cDNA sequences, BIC has been previously identified as a three-exon gene with the miR-155 sequence located in exon 3 (21). Nevertheless, an analysis of the EST data base unexpectedly identified an EST (BI821816) that contained all three exons plus an additional exon located 134 bp upstream from the 5′ most extending EST, CR99368 (Fig. 2). Moreover, the sequences for the splice donor and acceptor of this transcript match well with consensus donor/acceptor consensus sequences. This raised concerns that BIC may be composed of four exons and/or that there may be differential promoter usage in different tissues. We therefore carried out 5′ RACE using RNA from Ramos cells following exposure to anti-IgM. Seven independent clones were sequenced, and none of these contained the upstream exon sequences (Fig. 2). This analysis revealed that at least in activated B-cells, the BIC transcript is composed of three exons with the major initiation site located at nucleotide 12,596,314 of chromosome 21 (NCBI reference assembly), inline with most of the ESTs identified as well as the previously reported cDNA structure (21). Further, a classical “TATA” box sequence is located 24 bp upstream from the start site (Fig. 2A), which is within the 20–30-bp spacing that generally separates these two transcriptional features in TATA-containing promoters.
FIGURE 2. Transcript analysis of BIC.

The indicated ESTs were identified by BLAST analysis of the EST data base using the miR-155 encoding exon of BIC. The start site in BCR-activated Ramos cells was derived by 5′ RACE analysis.
Evolutionary Conservation of BIC Promoter Sequences
To gain insights into possible conserved regulatory elements in the BIC promoter, a BLAST search against the mouse genomic data base was carried out using sequences surrounding the human BIC start site (1939 bp upstream to 562 bp downstream). A single 75 nucleotide homology was identified that is roughly centered around the TATA box (Fig. 3A). Importantly, the homologous sequences are located on mouse chromosome 16 ∼ 11 kb upstream from the mouse miR-155. This location is positionally similar to the 11,835- bp spacing between the human miR-155 sequence and the human sequences indicated in the promoter alignment.
The human BIC promoter was then analyzed for potential transcription factor binding sites using the program TFSEARCH (22). This analysis identified an AP-1 site and a c-Ets site with high probability scores within the human/mouse homology region (Fig. 3A). Also notable is a high scoring NF-κB site located 1150 bp upstream from the human BIC transcription start site (Fig. 3B). Although the AP-1 and c-Ets sites are conserved in mouse, the NF-κB site is not positionally conserved. Instead, there is a putative NF-κB site located more proximal to the human/mouse homology region (264 bases upstream from the c-Ets site (Fig. 3B)).
The AP-1 Site Is Required for Activation of BIC Transcription following BCR Engagement
The putative NF-κB, AP-1, and c-Ets sites were considered to be good candidates for mediating or contributing to the activation of BIC following activation of the BCR. To assess the possible contribution of these elements in mediating BIC regulation, the human BIC promoter was cloned upstream from a luciferase reporter gene, and mutants were generated at each of these promoter elements. Wild type or mutant reporters were transfected into Ramos cells or an EBV-negative derivative of the cell line, Mutu (20), and were either left untreated or stimulated with an anti-IgM antibody. As shown in Fig. 3C, mutation of the NF-κB site had little influence on promoter activation, and mutation of the c-Ets site had only a marginal influence on promoter activity. In contrast, mutation of the AP-1 site decreased basal promoter activity and substantially impaired response to BCR activation. These results indicate that the AP-1 site plays a central role in BIC promoter activity following activation of B-cell receptor signaling.
Induction of the AP-1 Family Members, Jun B, c-Fos, and FosB, Requires the ERK and JNK Signaling Pathways
We first analyzed members of the AP-1 family of transcription factors that may be responsible for induction of the BIC promoter through the AP-1 site by assessing c-Jun, JunB, c-Fos, and FosB expression by Western blot analysis. Using either whole cell lysates or nuclear extracts, we were consistently unable to detect c-Jun using two antibodies that recognize total c-Jun (Cell Signaling (L70B11 and 60A8)) or the phospho-specific antibodies recognizing phospho-c-Jun (Ser-63) or phospho-c-Jun (Ser-73). In contrast, Jun B levels were readily detected starting at 2 h after induction. Robust induction of both c-Fos and FosB were observed at 2 h and persisted through 6 h (Fig. 4).
The expression levels of Jun B, c-Fos, and FosB were then analyzed in the presence of MEK, JNK, and p38 inhibitors. As shown in Fig. 5, c-Fos induction is significantly inhibited by the MEK inhibitor, U0126, and slightly less well inhibited by the MEK inhibitor, PD98059, and the JNK inhibitor, SP600125. FosB expression was also lower in the presence of each of these inhibitors. The slowest migrating form of JunB was decreased by the MEK and JNK inhibitors, whereas the faster migrating form was only moderately affected. In contrast, the p38 inhibitor, SB202190, had little detectable influence on the expression of any of these AP-1 family members. These results show that the MEK/ERK and JNK pathways mediate induced expression of the AP-1 family members, JunB, c-Fos, and/or FosB, suggesting that these factors may play a role in the induction of BIC transcription.
JunB and FosB Are Recruited to the BIC Promoter following B-cell Receptor Engagement
As expected, co-immunoprecipitation experiments readily demonstrated interactions between Jun B and either FosB or c-Fos (Fig. 6, A–C), indicating that heterodimerization between these family members likely occurs following BCR activation in this system. The binding of JunB and FosB to the BIC promoter following B-cell receptor engagement was then assessed by chromatin immunoprecipitation analysis. As positive controls, antibodies that recognize histone H4 or the acetylated form of histone H4 were used. Although the level of overall histone H4 precipitated was not effected by BCR activation, elevated levels of the acetylated form were precipitated from cells exposed to anti-IgM (Fig. 6D). This provided confidence in the chromatin immunoprecipitation assay and provided evidence that the local chromatin environment at the BIC promoter is activated following BCR activation. Using the JunB- and FosB-specific antibodies, enhanced precipitation of the BIC promoter was similarly observed, indicating that JunB and FosB bind to the BIC promoter following activation of the B-cell receptor. In contrast, no signal was detected in FosB or JunB immunoprecipitates using primers that amplify a 162-bp fragment located in intron 1, 6 kb downstream from the site of transcription initiation (Fig. 6D). Therefore, FosB and JunB bind specifically to the BIC promoter following BCR activation and likely play a role in the activation of BIC expression.
FIGURE 6.

A–C, JunB co-precipitates with c-Fos and FosB following BCR activation. H.C. refers to the immunoglobulin heavy chain signal. Cntl Ab, control antibody; IP Ab, immunoprecipitation antibody. D, chromatin immunoprecipitation analysis shows that FosB and JunB bind to the BIC promoter (BICp) but not the first intron of the BIC gene following BCR activation. Quantitation was carried out using ImageJ software.
To further establish that JunB and FosB bind directly to the AP-1 site of the BIC promoter, electrophoretic mobility shift analysis was carried out using a 20-mer oligonucleotide probe spanning the AP-1 site. An intense shifted band is observed using extracts from Ramos cells treated with anti-Ig that is not observed in extracts from uninduced Ramos cells (Fig. 7A). This band is competed by excess unlabeled wild type competitor oligonucleotide but not a competitor oligonucleotide containing the AP-1 site mutation used in the reporter assay shown in Fig. 3C. Nearly 100% of this band is super-shifted using a JunB antibody, and this band is partially supershifted using an antibody against FosB (Fig. 7B). These data indicate that this induced band is composed of JunB, which is likely heterodimerized with FosB. Whether the lack of a complete supershift of this band with the FosB antibody is due to incomplete binding of the antibody or due to a portion of this band representing JunB heterodimerized to another AP-1 family member such as c-Fos is not clear at this time. Nevertheless, these data indicate that JunB is a major component of this complex and that FosB is a contributor.
Lastly, co-transfection experiments using either a wild type BIC reporter or the AP-1 mutant with control, JunB, FosB, or JunB plus FosB expression vectors were carried out to establish that the binding of these factors to the AP-1 site is capable of mediating promoter activation. Transfection of JunB or FosB expression vectors alone moderately activates the wild type promoter, whereas co-transfection of the JunB and FosB expression vectors together results in a more robust induction (Fig. 7C). No induction was observed using the AP-1 site mutant, indicating that activation likely occurs through the direct binding to the conserved AP-1 element.
Discussion
The 5′ RACE analysis described here identified the major start sites and a three-exon structure for BIC in a BCR-activated human B-cell system. Nevertheless, the splice junctions of the first intron of the BI821816 transcript match reasonably well to consensus splice donor acceptor sites, providing some evidence that this EST represents a bona fide transcript. BI821816 was derived from a pool of brain, lung, and testis RNAs, raising the possibility that there may be differential promoter usage in different tissues, suggesting added complexity to BIC regulation. Nevertheless, usage of the promoter mapped here in the BCR-activated B-cell line, Ramos, is not specific to this particular cell line. 5′ RACE analysis of another B-cell line that expresses high constitutive levels of BIC (due to the presence of EBV) revealed the same start site.3 It is therefore likely that this promoter is commonly used in B-cells and possibly other lymphoid tissues.
As shown in Fig. 5, inhibition of either the ERK or the JNK pathways inhibits the level of c-Fos and FosB induction. This likely occurs through ERK- and JNK-mediated phosphorylation of the T-cell factor family member, Elk-1, which is a critical determinant of c-Fos and FosB promoter activation (through the binding to the Ets motif of serum-response elements) (23–25). In addition, there is evidence for ERK-dependent phosphorylation of FosB (26), which may also contribute to the activation of FosB function. Inhibition of the ERK or JNK pathways primarily had an effect on the levels of the slower migrating JunB band, suggesting a possible role in influencing a modified form of JunB. It is possible that in addition to the lower overall levels of c-Fos and/or FosB, this change in the slower migrating form of JunB may also contribute to the inhibition of overall AP-1 activity by ERK and JNK inhibitors. Together, these results provide evidence for a key role in ERK- and JNK-mediated activation of FosB (and probably c-Fos) and JunB in the induction of BIC expression following BCR activation.
A number of previous studies have demonstrated robust expression of BIC in EBV-infected cells expressing the full repertoire of EBV latency genes (27, 28). This implicates (but does not prove) that EBV latency genes are responsible for the induction of BIC. JunB is a well established downstream target of EBV latency genes that is induced by EBV infection (29). It is therefore possible that EBV induces high level BIC expression at least in part through the induction of JunB.
The data shown in Fig. 3 indicate that the AP-1 site is important for both basal transcription and activated transcription, indicating that it is a crucial element in facilitating BIC transcription. Nevertheless, we still see a measurable response to BCR activation with the AP-1 site mutant (2.8-fold induction), suggesting that other promoter elements may contribute and/or cooperate with AP-1 family members to activate BIC transcription. This is unlikely to be due to incomplete ablation of the AP-1 site since the six core nucleotides of this element were mutated (wild type = ATGAGTCAC, mutant = ATctc-gagC). A small but measurable decrease in response was observed when the c-Ets site located at the start site was mutated, suggesting the possibility that this site may contribute to activation, possibly through the binding of the Ets family member, Elk-1. In addition, there is a consensus Elk-1 site (25) located at −310 to −300 (GTTTCCTTTT), which could play a role in facilitating activation in response to BCR and perhaps TLR activation. We anticipate that although the AP-1 site is an important determinant of promoter activity, other elements are also likely to contribute to promoter function and play a role in BIC promoter regulation under these or other activating conditions. O'Connell et al. (17) have shown that the JNK pathway is required for activation of BIC expression following the activation of certain TLR signaling pathways; however, the involvement of ERK or the FosB, c-Fos, or JunB AP-1 family members is not yet clear. Moreover, although it is likely that the BIC AP-1 site is important for TLR-mediated induction of BIC transcription, this has not to our knowledge been tested. Further studies will reveal how extensive the overlap is between these pathways in the activation of BIC/miR-155 expression.
AP-1 signaling plays a key role in mediating inflammatory responses in the immune system and is important for the induction of regulatory cytokines. In addition, AP-1 activation plays a role in immune cell activation and differentiation. For example, Carrozza et al. (30) showed that transgenic mice expressing the naturally occurring FosB dominant negative, Δ-FosB, under the direction of a T-cell-specific promoter resulted in impaired T-cell development. Interestingly, BIC/miR-155 knock-out mice showed a defect in immune cell activation (15, 16). It is therefore likely that the induction of BIC/miR-155 plays a key role in facilitating at least some of the phenotypic effects of AP-1 activation. Similarly, the association between AP-1 proteins and cancer is well established, and AP-1 family members support tumor growth, survival, and metastasis through a wide array of downstream genes. The highly implicated nature of miR-155 in tumorigenesis suggests that induction of BIC/miR-155 may be a critical component of AP-1 signaling that contributes to AP-1 oncogenic signaling.
Acknowledgments
We thank Dr. Jennifer Cameron for helpful insights throughout the course of this project. We also thank Dr. Matt Burow for the advice and Dr. Dexing Fang and Dr. Huichen Wang for excellent technical advice on immunoprecipitation and chromatin immunoprecipitation assays.
Footnotes
This work was supported by the National Institutes of Health research Grants GM48045 (to E. K. F.), DE017008 (to E. K. F.), and R01CA124311 (to E. K. F.), a grant from the Lymphoma Research Foundation (to Q. Y.), and a Mentoring a Program in Cancer Genetics National Institutes of Health Center of Biomedical Research Excellence award (to Prescott Deininger–P20 RR020152).
The abbreviations used are: miRNA, microRNA; BIC, B-cell integration cluster; BCR, B-cell receptor; TLR, Toll-like receptor; ERK, extracellular signaling-regulated kinase; JNK, c-Jun NH2-terminal kinase; MEK, mitogen-activated protein kinase/ERK kinase; TNF, tumor necrosis factor; RT, reverse transcription; qRT, quantitative RT; G3PDH, glyceraldehyde-3-phosphate dehydrogenase; RACE, rapid amplification of cDNA ends; RIPA, radioimmune precipitation buffer; EBV, Epstein-Barr virus; EST, expressed sequence tag; IL, interleukin.
X. Wang and E. Flemington, unpublished data.
References
- 1.Hwang HW, Mendell JT. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miska EA. Curr Opin Genet Dev. 2005;15:563–568. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Wienholds E, Plasterk RH. FEBS Lett. 2005;579:5911–5922. doi: 10.1016/j.febslet.2005.07.070. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Rivas FV, Wohlschlegel J, Yates JR, III, Parker R, Hannon GJ. Nat Cell Biol. 2005;7:1261–1266. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. Nat Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clurman BE, Hayward WS. Mol Cell Biol. 1989;9:2657–2664. doi: 10.1128/mcb.9.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A. J Pathol. 2005;207:243–249. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 9.van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, Jacobs S, Kluiver J, Diepstra A, Maggio E, Poppema S. Genes Chromosomes Cancer. 2003;37:20–28. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- 10.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tam W, Hughes SH, Hayward WS, Besmer P. J Virol. 2002;76:4275–4286. doi: 10.1128/JVI.76.9.4275-4286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 13.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 14.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taganov KD, Boldin MP, Chang KJ, Baltimore D. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph AM, Babcock GJ, Thorley-Lawson DA. J Virol. 2000;74:9964–9971. doi: 10.1128/jvi.74.21.9964-9971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron J, Yin Q, Fewell C, Lacey M, McBride J, Wang X, Lin Z, Schaefer B, Flemington E. J Virol. 2007 in press. [Google Scholar]
- 21.Tam W, Ben-Yehuda D, Hayward WS. Mol Cell Biol. 1997;17:1490–1502. doi: 10.1128/mcb.17.3.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazo PS, Dorfman K, Noguchi T, Mattei MG, Bravo R. Nucleic Acids Res. 1992;20:343–350. doi: 10.1093/nar/20.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yordy JS, Muise-Helmericks RC. Oncogene. 2000;19:6503–6513. doi: 10.1038/sj.onc.1204036. [DOI] [PubMed] [Google Scholar]
- 25.Treisman R, Marais R, Wynne J. EMBO J. 1992;11:4631–4640. doi: 10.1002/j.1460-2075.1992.tb05565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberger SF, Finch JS, Gupta A, Bowden GT. J Biol Chem. 1999;274:1124–1130. doi: 10.1074/jbc.274.2.1124. [DOI] [PubMed] [Google Scholar]
- 27.Jiang J, Lee EJ, Schmittgen TD. Genes Chromosomes Cancer. 2006;45:103–106. doi: 10.1002/gcc.20264. [DOI] [PubMed] [Google Scholar]
- 28.Kluiver J, Haralambieva E, de Jong D, Blokzijl T, Jacobs S, Kroesen BJ, Poppema S, van den Berg A. Genes Chromosomes Cancer. 2006;45:147–153. doi: 10.1002/gcc.20273. [DOI] [PubMed] [Google Scholar]
- 29.Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. J Virol. 2004;78:4108–4119. doi: 10.1128/JVI.78.8.4108-4119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrozza ML, Jacobs H, Acton D, Verma I, Berns A. Oncogene. 1997;14:1083–1091. doi: 10.1038/sj.onc.1200921. [DOI] [PubMed] [Google Scholar]