

Abstract
Tandem mass spectrometry-based shotgun proteomics has become a widespread technology for analyzing complex protein mixtures. A number of database searching algorithms have been developed to assign peptide sequences to tandem mass spectra. Assembling the peptide identifications to proteins, however, is a challenging issue because many peptides are shared among multiple proteins. IDPicker is an open-source protein assembly tool that derives a minimum protein list from peptide identifications filtered to a specified False Discovery Rate. Here, we update IDPicker to increase confident peptide identifications by combining multiple scores produced by database search tools. By segregating peptide identifications for thresholding using both the precursor charge state and the number of tryptic termini, IDPicker retrieves more peptides for protein assembly. The new version is more robust against false positive proteins, especially in searches using multispecies databases, by requiring additional novel peptides in the parsimony process. IDPicker has been designed for incorporation in many identification workflows by the addition of a graphical user interface and the ability to read identifications from the pepXML format. These advances position IDPicker for high peptide discrimination and reliable protein assembly in large-scale proteomics studies. The source code and binaries for the latest version of IDPicker are available from http://fenchurch.mc.vanderbilt.edu/.
Keywords: bioinformatics, parsimony, protein assembly, protein inference, false discovery rate
Introduction
Liquid chromatography based tandem mass spectrometry (LC-MS/MS) has become the method of choice for large-scale identification of proteins present in complex biological samples.1,2 A typical LC-MS/MS experiment routinely generates tens of thousands of tandem mass spectra (MS/MS) due to recent advances in instrumentation. The generated MS/MS spectra are matched to a protein database using search engines like MyriMatch,3 Sequest,4 or Mascot.5 These search engines enumerate peptides from the database, predict their fragment ions, and match them to the MS/MS spectra, resulting in thousands of peptide identifications per LC-MS/MS experiment. Because a large proportion of MS/MS spectra cannot be matched successfully to peptide sequences, raw identifications must be filtered to retain the most accurate results.2,6
The size and the complexity of contemporary data sets require automated handling. Several methods that convert arbitrary search scores of raw identifications into statistical measures have been developed.7-11 A common technique, exemplified by IDPicker,7 employs decoy database searches to compute the False Discovery Rate (FDR) of raw identifications. The FDR-based methods generally use only one scoring metric reported by a search engine, making them easily portable to new search engines. Database search engines, however, generally report multiple scoring metrics to assess the quality of a spectrum match. If all these metrics are used in combination, higher discrimination is possible than any single score.12–17
The DTASelect16 v2.0 and PeptideProphet17 tools can combine multiple scores for each identification into a single discriminant score through statistical analysis. Because search engine scores differ in type and distribution, however, these complex systems must be tuned separately for each search engine. The Percolator15 tool introduced an alternative method that can combine multiple scores from any search engine via FDRs and machine learning. Here, we introduce a generic score combination method that does not require any statistical distribution inference or machine learning.
Peptides present in trypsin-digested samples can be identified using a semitryptic or unconstrained database search. These searches have an added advantage of identifying nonenzymatic and semitryptic peptides that are generated due to chymotryptic activity and in-source fragmentation. Peptides with basic residues at the C-terminus tend to produce higher scoring fragment ion spectra than other peptides.18 Choosing a single threshold for a mix of tryptic, semitryptic, and other peptides preferentially retains peptides that have basic residues at the C-terminus. This scenario is comparable to the effect seen in identifications for peptides of different charges; if scores for +3 peptides are higher than for +2 peptides, it makes little sense to apply the same threshold score to both charge states.17,19,20
Researchers routinely check for contaminating proteins present in their LC-MS/MS experiments by identifying spectra against a multispecies protein database such as Swiss-Prot. Other researchers make use of databases such as the human subset of “NCBI NR” in order to recognize mutant or isoform variants of proteins. Such databases often contain highly homologous protein sequences due to natural sequence diversity. As a result, the search results contain large numbers of peptide identifications that map to multiple proteins, yielding spurious peptide identifications that confuse the process of protein assembly.
In this report, we describe recent improvements to IDPicker.7 In summary, IDPicker filters peptide identifications to a desired FDR using decoy database matches, builds a bipartite graph of peptide–protein relationships, and assembles a list of protein identifications through a parsimony reduction of the bipartite graph. The new version of IDPicker improves peptide identification by automatically combining multiple scores reported by database search engines. The software partitions peptides based on their number of tryptic termini and observed charge state, and it estimates their error rates separately, improving identification sensitivity in semitryptic and unconstrained database searches. The new IDPicker features a novel filter to remove spurious protein identifications from multispecies or NCBI NR database search results. We evaluated the new software using multiple data sets obtained from several MS platforms with different sample complexities. We also compared the performance of IDPicker to the most commonly used program, PeptideProphet, in three data sets. The new IDPicker is now accessible by a graphical user interface (GUI) for easy incorporation into the current workflow of any proteomics laboratory.
Materials and Methods
Improvement of IDPicker
IDPicker7 is an analysis pipeline for assembling confident and parsimonious protein identification lists from raw spectral identifications. The software consists of three separate modules: idpQonvert, idpAssemble, and idpReport. The first module, idpQonvert, reads peptide identifications from pepXML21 files, estimates their FDR values using decoy matches, and records this information to an XML file. The second module, idpAssemble, categorizes the generated XML reports into appropriate groups (e.g., technical replicates or 2-D gel LC fractions combined as a single group), filters the peptides to a user-chosen FDR value (typically 5%), and generates a unified XML file with all the information. The final module, idpReport, reads the unified XML file, generates a parsimonious list of proteins, and generates HTML reports of protein, peptide, and spectral identifications. Several algorithmic changes were made to IDPicker in order to improve the sensitivity of its peptide identification and accuracy of its protein assembly. A new graphical user interface (GUI) was also developed to make it easier for new users of the tool to make use of the IDPicker modules.
Multiple Score Combination and Peptide Separation
IDPicker can combine complementary scoring metrics of a search engine using decoy sequences and FDRs without making any assumptions about the underlying distribution of search engine scores. Users can specify which scoring metrics are to be included from their search results. To accommodate scores of different distributions, IDPicker can normalize scoring metrics to a quantile scale prior to score combination. The combined score for a search engine is defined as a weighted summation of its subscores: S = w1s1 + w2s2 + wnsn, where each subscore si is associated with a particular weight wi. Weights may either be user-defined (static) or automatically determined using a Monte Carlo simulation method (dynamic). In the dynamic mode, IDPicker tests random score weights to determine which maximizes the total number of confident identifications for the specified FDR.
A database search may be constrained to consider only tryptic peptides or it may be left unconstrained, to consider any peptide that can be generated from protein sequences. These unconstrained searches yield peptides with different numbers of tryptic termini (NTT): fully tryptic (NTT = 2), semitryptic (NTT = 1), and nonspecific (NTT = 0). A “tryptic terminus” is one that conforms to a cut after arginine, to a cut after lysine, or to a protein terminus. A peptide that was bounded by two standard trypsin cutting sites yields an NTT of 2. A peptide for which only one of the termini corresponds to a standard trypsin cutting site, however, is semitryptic, with NTT = 1. A peptide that lacks a standard trypsin cutting site on both ends yields an NTT of 0.
IDPicker was modified to partition spectral matches from a database search into nine separate peptide classes based on the NTT (0, 1, or 2) and Z state (charge state 1+, 2+, or 3+) of the observed peptides. For each peptide class, a combined search score is generated and its threshold corresponding to the user-specified FDR is computed using the above method. Peptide identifications passing the score thresholds of each class are pooled for protein assembly.7
Robust Protein Assembly for Multispecies and NCBI NR Database Searches
When databases that contain many homologous sequences are used for identification, spectra may be erroneously matched to sequences that differ from the true peptide by minor sequence changes. These peptides can greatly complicate the process of protein sequence assembly (see Figure 1). A new filter “minimum additional peptides per protein group” was introduced in IDPicker to counteract these near-miss peptides. The parsimony analysis adds a protein to a cluster only if it explains a minimum number of peptides that were not previously explained by the proteins already present in the candidate cluster. Setting this filter to >1 can greatly reduce the number of orthologous protein identifications in multispecies database searches.
Figure 1.

Robust protein assembly for high sequence homology database searches. In this diagram, seven peptides observed in human serum are associated with the ceruplasmin sequence from three different species. Most protein assembly tools would include all three proteins because each is associated with at least two peptides, with at least one peptide being unique to each protein sequence. IDPicker, however, is able to screen out the mouse and rat sequences by requiring proteins to explain more than one new peptide for inclusion in the final list. The two peptides starting with “MYYS” differ at the fifth amino acid; this sequence difference probably reflects that the serum used in this study was a pool, reflecting the variant sequences of a population of blood donors. The two peptides starting with “MFTT”, on the other hand, are isobaric; the differing sequences “DQ” and “EN” are exactly the same mass. Of all the y ions generated by the two sequences starting with “MFTT”, only y4 could distinguish the peptides.
Graphical User Interface (GUI)
A new GUI was developed to make it easier for new users to work with IDPicker. Supplemental file 1 (SF1) in the Supporting Information contains screenshots of the GUI during analysis of search engine results from an example data set. The GUI guides users through the process of selecting pepXML files, organizing these files in a multilayer experimental hierarchy, and configuring the thresholding that the software will use for filtering raw identifications and assembled proteins. The GUI then launches the analysis and tracks its progress in creating the HTML reports. IDPicker reports can be evaluated via a Web browser or spreadsheet. The reports are intended to satisfy the Paris guidelines (http://www.mcponline.org/misc/ParisReport_Final.dtl), showing explicitly which peptides support particular proteins. The GUI can also export the identification reports as ZIP or CSV files for easy sharing. If IDPicker reports are viewed via the GUI, users can employ an integrated spectrum viewer for manual validation of spectral matches. A screenshot of this viewer can be seen in Figure 2.
Figure 2.

A Screenshot of IDPicker GUI report. Three samples from cancer subjects and three samples from control subjects were arranged in a tree hierarchy to reflect their biological meaning. Each sample has three replicate LC-MS/MS experiments that were grouped together. The final protein identification report arranges the protein, peptide, and spectral identifications in the above-described hierarchy. The numbers of identifications at each node are reported by summarizing the identifications of its child nodes. For example, the above report starts with the “root” level of hierarchy, designated by the “/” label, that summarizes all identifications present in the analysis. Following the root node, the numbers of identifications for next lower level hierarchies (cancer and control groups) are summarized, followed by each sample and individual technical replicate. The report also contains a navigation frame (shown on the left side) that allows the user to browse the protein identifications using different indices. Users can also manually validate the spectral matches using a built-in spectrum viewer. For example, the bottom window highlights the fragment ion matches of a tandem mass spectrum that was mapped to the peptide “IAQWQSFQLEGGLK”.
Data Sets
Three shotgun proteomics data sets of different complexities were used to demonstrate the utility of improved IDPicker. The data sets are available for download from Vanderbilt University Mass Spectrometry Research Center's Web site (http://www.mc.vanderbilt.edu/msrc/bioinformatics/data.php).
Data Set I: Serum Orbi
The 12 most abundant proteins were depleted from 50 μL of human serum using an IgY-12 LC2 (Beckman Coulter, Fullerton, CA) column following the manufacturer's instructions. Proteins present in the flow-through fraction were reduced with 50 mM dithiothreitol (DTT), alkylated with 100 mM iodoacetamide (IAM), and digested by incubating overnight at 37 °C with trypsin added at 1:50 enzyme/substrate ratio. Digestion was quenched by adding formic acid (FA) at pH 2.0 to a final concentration of 5%. The resulting peptide mixture was lyophilized and reconstituted with 0.1% FA to a final concentration of 0.2 μg/μL. Two microliter portions of peptide mixture were analyzed using an LTQ-Orbitrap hydrid mass spectrometer (Thermo, San Jose, CA) equipped with an Eksigent 1D Plus NanoLC (Eskigent, Dublin, CA) system. Peptides were solid-phase extracted using an inline column (100 μm × 6 cm) packed with Jupiter C18 resin (5 μm, 300 Å, Phenomenex, Torrence, CA) and separated on a capillary tip (100 μm × 11 cm, Polymicro Technologies, Phoenix, AZ) packed with the same C18 resin as previously described.22,23 Following the injection, peptides were solid-phase extracted by washing with 0.1% FA (mobile phase A) for 15 min at a flow rate of 1.5 μL/min. Mobile phase B consisted of acetonitrile (ACN) with 0.1% FA. Peptides were separated using a gradient of 2–25% B for 35 min, followed by a rapid increase of B from 25–90% in 15 min, and held at 90% B for 9 min before returning to initial conditions of 98% A. Survey scans were collected in the Orbitrap at a resolution of 60 000 within a mass range of 300–2000 Da. Following each survey scan, the eight most intense ions were selected for MS/MS fragmentation in the LTQ portion of the instrument using the dynamic exclusion feature (exclusion mass width of ±0.6 Da, exclusion duration of 60 s, and repeat count of 1). A total of five replicate LC-MS/MS experiments were performed and 32 740 MS/MS spectra were collected. Binary spectral data present in the raw files were converted to mzML format using msConvert tool of the ProteoWizard24 library. DTAs or MGFs were extracted from the mzML files using mzxml2search program of Trans-Proteomic Pipeline21 (Institute of Systems Biology, Seattle, WA).
Data Set II: DLD1 LTQ
Human colon adenocarcinoma cells (DLD-1 cell line, American Type Culture Collection, Manassas, VA) were grown in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin antibiotics at 37 °C and 5% CO2. Cells were harvested (at >90% confluence), washed in 1× phosphate-buffered saline (PBS), and spun down at 1000g for 5 min. Cell pellets were collected and stored at –80 °C until further use. Protein was extracted from the cell pellets by subjecting them to 1 h vigorous shaking (1000 rpm) followed by repeated sonication (3 × 20 s) in 200 μL of ammonium bicarbonate buffer (NH4HCO3; pH 8.0) with 50% tetrafluoroethylene (TFE). Concentration of extracted protein was estimated using BCA protein assay (Pierce, Rockford, IL). Protein samples were reduced using 100 mM DTT and 40 mM tris(2-carboxymethyl)-phosphine (TCEP) at either pH 8.0 or pH 4.0. Reduced samples were pH adjusted by adding 600 μL of 50 mM NH4HCO3 buffer (pH 8.0), alkylated with IAM, and digested by incubating overnight at 37 °C with trypsin added at 1:50 enzyme/substrate ratio. Peptides were solid phase extracted using C18 column and resuspended in 200 μL of 0.1% FA for reverse-phase separations using an Eskigent nanoLC (Eskigent, Dublin, CA) system and an LTQ (Thermo, San Jose, CA) mass spectrometer. Two microliter portions of peptide mixture were applied at 0.7 μL/min to a trap cartridge (Phenomenex, Torrance, CA), and then shifted to a 100 mm × 11 cm Jupiter C18 capillary column (Phenomenex, Torrance, CA) using a mobile phase containing 0.1% of FA. The peptide mixture was resolved using a 2–90% ACN gradient over 60 min at a flow rate of 0.7 μL/min. The five most abundant ions following each survey scan were selected for MS/MS fragmentation using dynamic exclusion (mass width –1.0 to 2.0 m/z; repeat count 1; and duration of 1). A total of four LC-MS/MS experiments were performed (2 replicates of the sample reduced at pH 8.0 and 2 replicates of the sample reduced at pH 4.0), and 51 652 tandem mass spectra were collected. Binary spectral data were converted to mzML, DTA, and MGF formats as for the “Serum Orbi” set.
Data Set III: Plasma QSTAR
Human blood collection and plasma processing were performed using the protocol developed by the Clinical Proteomics Technologies Assessment for Cancer (CPTAC) Biospecimen Working Group (http://proteomics.cancer.gov/). The 14 most abundant proteins were depleted from batches of 40 μL of human plasma using a MARS-Hu-14 (4.6 × 100 mm) column from Agilent (Santa Clara, CA) on a Michrom Paradigm MG4 HPLC system (Auburn, CA) following the manufacturer's instructions. The protein flow-through fraction was collected and readjusted to the original volume using a 5 kDa MWCO centrifugal concentrator (Sartorius AG, Goettingen, Germany). Depleted plasma samples were reduced with DTT, alkylated with IAM, and digested using trypsin as previously described.25 One microliter aliquots of peptide mixture (approximately 1 μg/μL) were analyzed using an Eksigent nano-LC 2D HPLC system (Eksigent, Dublin, CA) connected to a quadrupole time-of-flight (QqTOF) QSTAR Elite mass spectrometer (MDS SCIEX, Concorde, Canada). Peptides were loaded on a guard column (C18 Acclaim PepMap100, 300 μm i.d. × 5 mm, 5 μm particle size, 100 Å pore size, Dionex, Sunnyvale, CA), washed for 10 min using mobile phase A containing 0.1% FA at a flow rate of 20 μL/min, and transferred to an analytical C18-nanocapillary HPLC column (C18 Acclaim PepMap100, 300 μm i.d. × 15 cm, 3 μm particle size, 100 Å pore size, Dionex, Sunnyvale, CA). Mobile phase B consisted of ACN with 0.1% FA. Peptides were separated using a gradient of 2–40% B for 120 min, followed by a rapid increase of B from 40–90% in 15 min, and held at 90% B for 9 min before returning to initial conditions of 98% A (flow rate 300 nL/min). All mass spectra were recorded at a resolution of 12 000–15 000. Following each survey scan, the six most abundant ions were selected and fragmented using the advanced information dependent acquisition (IDA) feature along with QSTAR Elite specific features such as “Smart Collision” and “Smart Exit” (fragment intensity multiplier set to 2.0 and maximum accumulation time of 1.5 s). MS/MS spectra were acquired using the dynamic exclusion feature (exclusion mass width 50 mDa m/z and exclusion duration of 60 s) of the mass spectrometer. A total of five replicate LC-MS/MS experiments were performed and 35 290 MS/MS spectra were collected. WIFF files were processed using Protein Pilot software (version 2.0.1, Applied Biosystems, Carlsbad, CA) and tandem mass spectra were exported in MGF file format. Generated MGF files were transcoded to mzXML format using LibMSR software, a precursor of the ProteoWizard24 library.
Database Searching and Peptide Validation
Table 1 summarizes the search engines, protein sequence databases, and parent/fragment ion mass tolerances used to process the data sets. All protein databases contained sequences in both forward and reverse orientations for estimation of protein and peptide identification error rates. All search engines were configured to use a static mass shift of 57.0215 Da for alkylated cysteines and allow the variable modification of oxidation of methionine (+15.9949 Da). MyriMatch and Mascot were also configured to allow formation of N-terminal pyroglutamate (–17.0265 Da) as a variable modification. Detailed configuration of all search engines is given in Supplemental File 2.
Table 1.
Experimental Data Sets Summary
| data set namea | replicates | average no. of MS/MS scans | database search tools | precursor/fragment m/z tolerance | databases used for searchb |
|---|---|---|---|---|---|
| Score Combination Evaluation Data Sets | |||||
| Serum Orbi | 5 | 6548 | MyriMatch, Sequest, Mascot | 0.1/0.5 | ipi.HUMAN.v3.47 |
| Plasma QSTAR | 5 | 7058 | MyriMatch, Sequest, Mascot | 0.25/0.25 | ipi.HUMAN.v3.47 |
| DLD1 LTQ | 4 | 12913 | MyriMatch, Sequest, Mascot | 1.25/0.5 | ipi.HUMAN.v3.47 |
| Peptide Segregation Evaluation Data Sets | |||||
| DLD1 LTQ | 4 | 12913 | MyriMatch | 1.25/0.5 | ipi.HUMAN.v3.47 |
| Serum Orbi | 5 | 6548 | MyriMatch | 0.1/0.5 | ipi.HUMAN.v3.47 |
| Parsimony Performance Evaluation Data Sets | |||||
| DLD1 LTQ | 4 | 12913 | MyriMatch | 1.25/0.5 | uniprot_sprot-rel56.2 |
| Serum Orbi | 5 | 6548 | MyriMatch | 0.1/0.5 | uniprot_sprot-rel56.2 |
Data set names represent sample type and the mass spectrometer used in the analysis (see Materials and Methods for additional details).
All protein databases contained reversed sequence entries (decoys) for estimation of false discovery rates. Exhaustive database search configurations were made available in Supplemental File 2.
All identifications were processed from pepXML format. Identifications in Sequest OUT files were transcoded into pepXML format using out2xml program of the Trans-Proteomic Pipeline21 (Institute of Systems Biology, Seattle, WA). The RefreshParser tool (also from ISB) corrected the peptide/protein associations in the produced pepXML file. Mascot (Matrix Science, London, U.K.) DAT files were translated to pepXML format using the Export Search Results option from the Select Summary Report. Throughout this study, IDPicker was configured to derive score thresholds to yield a 5% False Discovery Rate (FDR). Peptides passing these thresholds were considered as legitimate identifications. IDPicker assembled protein identifications from the legitimate peptide identifications using parsimony rules.7
Alternative Peptide Validation Using PeptideProphet
Sequest search engine results from all three data sets were loaded into PeptideProphet (Trans-Proteomic Pipeline version 4.2), and peptide-spectrum match (PSM) probabilities were computed using default parameters. For each MS/MS, only the top scoring PSM was retained for further analysis. A simple Perl script was used to extract the peptide probability and decoy status from the retained PSMs. The script was configured to assign a decoy-status of F for forward hits and R for reverse hits. PSMs matching both forward and reversed sequences were omitted from both counts, as in IDPicker. The PSMs were ordered based on the decreasing probability score, and the probability threshold that corresponds to 5% FDR was computed using the high scoring reverse hits. This FDR computation method mimics the method used by IDPicker. The PSMs that pass the computed probability threshold were considered as confident identifications by PeptideProphet.
Results and Discussion
Combining Multiple Scores Increases Confident Identifications
Most search engines report multiple scoring metrics for peptide-spectrum matches. These scores can be combined using different weights to generate a single score for the search engine. The combined score needs to be optimized for each sample due to the technical and biological variability of the samples. We have implemented a new Monte Carlo method in IDPicker that efficiently combines multiple scores, on a per sample basis, using decoy sequences and false discovery rates (see Materials and Methods).
In this study, the effect of score combination on peptide identifications was evaluated using three different search engines: MyriMatch,3 Sequest,4 and Mascot.5 The following primary and secondary scores from each search engine were considered for score combination: MVH and mzFidelity from MyriMatch; XCorr and DeltaCN from Sequest; IonScore and IdentityScoreThreshold from Mascot. The MyriMatch MVH (“Multivariate Hypergeometric”) score evaluates the probability that a random peptide would match to fragments as intense in the observed spectrum as a particular candidate sequence. The MyriMatch mzFidelity score, currently under development, is a metric to evaluate how closely observed fragment m/z values match their expected locations. MyriMatch and Sequest scores were converted to quantiles and combined using the Monte Carlo method. Mascot scores were combined as “IonScore-IdentityScoreThreshold” using static weights following the Matrix Science recommendation.
Peptides present in the “Serum Orbi” data set were identified using all three search engines (see Materials and Methods). For each search engine result, IDPicker was configured to use either its primary score or a combination of its scores while deriving score thresholds at an FDR of ≤5%. Figure 3A–C shows the percent overlap of confident peptide identifications passing the score thresholds when using either single or multiple scores. Similar analysis was also performed for “Plasma QSTAR” and “DLD1 LTQ” samples and the results are shown in Figure 3D–I.
Figure 3.

Combining multiple scores from a search engine improves peptide identification rate. Tandem mass spectra from three different samples were matched to IPI human protein database (version 3.47) using MyriMatch, Sequest, and Mascot search engines (see Materials and Methods for additional details). Peptide identifications from all search engines were loaded into IDPicker. For each search engine, IDPicker was configured to use either its primary score or a combination of its scores to identify peptides at an FDR ≤5%. Panels A–I show the percent overlap between valid peptide identifications when IDPicker was using either a single score or multiple scores from respective search engines. Combining multiple scores from a search engine yielded more peptide identifications from all samples. There were few peptide identifications that were identified only when using the primary score of a search engine but not the score combination.
Irrespective of the sample type, combining multiple scores from a search engine consistently identified more peptides than using a single score (Figure 3). Sequest benefited significantly more from score combination than MyriMatch and Mascot. One possible interpretation is that XCorr is very good for ranking peptides for a particular spectrum but is more comparable among multiple spectra when DeltaCN is included. Subtracting away the Identity Score Threshold in Mascot or combining the mzFidelity score in MyriMatch produced far smaller gains in identification. Figure 3 also highlights that some identifications may be lost when multiple scores are combined.
Comparison of PeptideProphet versus IDPicker Score Combination Methods
Probabilistic frameworks like PeptideProphet17 can combine multiple search engine scores into a single discriminant score and compute peptide probabilities based on mixture modeling of the score distributions. These frameworks have also been extended to use decoy database entries to accurately model the incorrect and correct score distributions.11,20 However, algorithms implementing the mixture model techniques are very complex and are not easily extensible to accommodate new search engines or different scoring metrics of an existing search engine.
In contrast, the score combination implemented in IDPicker is based on a simple nonparametric Monte Carlo simulation method. This approach uses decoy sequences and FDRs to combine multiple scoring metrics of a search engine. Because IDPicker is computing aggregate identification FDRs rather than individual identification probabilities, its approach can be configured to combine multiple scoring metrics from arbitrary search engines as long as decoy sequences are included in the database.
In this study, we compared the score combination methods implemented in IDPicker with PeptideProphet using Sequest search engine results from three different types of data sets (see Table 1). The search results from each data set were separately loaded into PeptideProphet and IDPicker. Both algorithms were configured to filter the peptide-spectrum matches (PSMs) using 5% FDR (see Materials and Methods). Figure 4 compares the total number of confident PSMs identified by PeptideProphet and IDPicker in each replicate of the “Serum Orbi”, “Plasma QSTAR”, and “DLD1 LTQ” data sets. IDPicker consistently produced more confident PSMs than PeptideProphet from the “Serum Orbi” data set, while the opposite was true for the “Plasma QSTAR” data set (Figure 4). Both algorithms produced similar number of confident PSMs from the “DLD1 LTQ” data set (Figure 4). In short, neither algorithm consistently out-performed the other. These data demonstrate that the performance of the simple IDPicker score combination model is on par with the far more complex statistical model in PeptideProphet.
Figure 4.

Comparison of IDPicker and PeptideProphet score combination methods. Tandem mass spectra from three different samples were matched to the IPI human protein database (version 3.47) using the Sequest search engine (see Materials and Methods). The search results were separately processed by IDPicker and PeptideProphet. Both algorithms were configured to filter PSMs using a 5% FDR threshold. The total number of confident PSMs identified by both algorithms in each replicate of all three data sets is shown above. IDPicker produced more confident PSMs than PeptideProphet in some data sets and vice versa, but the algorithms performed similarly in all data sets, with a maximum difference of 5.7%. The simple nonparametric score combination method implemented in IDPicker performs as well as the complex probabilistic frameworks implemented in PeptideProphet, but the IDPicker score combination method can be more easily extended to combine multiple search scores from new search engines.
The Percolator15 tool can also combine multiple scores from arbitrary search engines by generating a linear classifier and retraining it for each individual data set using target and decoy matches (machine learning). This tool is designed to work with separated target-decoy database searches. These types of searches may be less sensitive than the concatenated target-decoy database searches used by IDPicker.10 Several attempts to include Percolator in the above comparison were unsuccessful because the software is designed for searches in which forward and reversed sequences are separated.
Partitioning Peptides Based on Number of Tryptic Termini (NTT) and Z State Improves Peptide Identification Rate
Search engines may be configured to identify peptides that do not conform to trypsin cutting sites on both termini. Unconstrained searches yield identifications with different NTT, allowing the algorithms to identify alternative cutting sites for other proteases, chymotryptic activity for trypsin, and in-source fragmentation of peptides. Of all candidate peptides considered in an “unconstrained” search, only about 1% correspond to peptides with trypsin cutting sites on both ends (NTT = 2). Approximately an order of magnitude more peptides are “semitryptic”, with a trypsin cutting site on only one end of the peptide (NTT = 1). The great majority of candidate peptides compared to spectra in an unconstrained search do not match trypsin cutting sites on either end (NTT = 0). Peptides of different NTT values are likely to produce scores in different ranges due to the positions of basic residues and the relative concentrations of peptide ions produced in digestion. This phenomenon is more widely appreciated with respect to peptide charge. For quite some time, the identifications of triply charged peptides have been required to meet different scoring requirements than identifications of doubly charged peptides. The new IDPicker compensates for these scoring differences by explicitly segregating peptide identifications on the basis of both peptide charge and NTT value, improving the numbers of identifications produced from each RPLC separation.
The effect of peptide partitioning was determined for three different search strategies: fully tryptic, semitryptic, and unconstrained. The following four different peptide partition styles were tested for each database search strategy: (A) no partitioning, (B) Z state (1+, 2+, or 3+) only, (C) NTT (0, 1, or 2) only, (D) both Z state and NTT. Tandem mass spectra from a whole cell lysate data set (“DLD1 LTQ”) were searched against an IPI Human protein database with MyriMatch using all three search strategies. Peptide identifications from each database search were loaded into IDPicker and peptides were partitioned using all four partition styles separately. For each partition style, IDPicker determined the number of identifications that met the 5% FDR threshold. The results from the four technical replicates were averaged in Figure 5A. A comparable analysis was also performed using a human serum data set (“Serum Orbi”) and results were shown in Figure 5B.
Figure 5.

Partitioning peptides based on charge state (Z) and number of tryptic termini (NTT) improves peptide identification. “DLD1 LTQ” and “Serum Orbi” samples were matched to the IPI human protein database (version 3.47) using three search strategies: fully tryptic, semitryptic and unconstrained. Peptide identifications from each search were loaded into IDPicker and partitioned into and separate classes using four different methods shown in the figure. The average numbers of peptide identifications that have an FDR ≤ 5% when using a particular partition method are computed using reverse sequences present in the database and plotted for “DLD1 LTQ” (A) and “Serum Orbi” (B) data sets separately. The error bars in A and B represent the standard deviations from the replicates. Separating peptide identifications based on NTT and Z state improved the number of identified peptides in semitryptic and unconstrained searches. Improvement of peptide identification rate in the fully tryptic search (NTT = 2) is due to Z state only.
As expected, partitioning peptide identifications according to charge state (Z) improved the peptide identification rate regardless of database search strategy and the type of sample. When the search included semitryptic or nonspecific peptides, partitioning peptides on both NTT and Z state outperformed the other partitioning methods (Figure 5). Since falsely identified spectra are more likely to be nonspecific or semitryptic, partitioning the identifications by their NTT values has the effect of separating most false identifications from most of the true identifications.
The concept of using both NTT and Z state values of peptides for improved validation is not novel to this work. PeptideProphet first implemented this concept by generating separate discriminant scores for each charge state and conditioning the identification probabilities on peptide NTT values. The same concept was implemented in the new IDPicker by segregating the identifications into separate classes on the basis of peptide NTT and Z state values and calculating their FDRs separately. This approach is simple and easily configurable to handle peptides with higher charge states (>3+) that are typically seen in high resolution data sets (approximately 10%–15% of MS/MS in LTQ-Orbitrap data sets are from higher charge state peptides).
Does an Unconstrained Search Always Yield More Peptide Identifications?
The white bars in panels A and B of Figure 5 show the number of peptide identifications from a fully tryptic search, semitryptic search, and an unconstrained search of “DLD1 LTQ” and “Serum Orbi” data sets, respectively. Similar trypsin digestion protocols were used to perform proteolysis of both “DLD1 LTQ” and “Serum Orbi” samples. However, more semitryptic peptides were identified in the “Serum Orbi” sample than the “DLD1 LTQ” sample (compare the difference between fully tryptic and semitryptic searches in Figure 5, panels B and A). Both samples are complex, but the depleted serum sample is disproportionately dominated by a few major proteins. These major proteins often contribute large numbers of semitryptic peptides. Although semitryptic peptides might be generated at lower probability than the tryptic peptides for a given protein, the semitryptic peptides for a high concentration protein may compete successfully with the tryptic peptides of low concentration proteins in being selected for fragmentation. “DLD1 LTQ” is less dominated by a small number of major proteins, and thus shows less advantage for the semitryptic search.
Reducing Spurious Subsumable Protein Identifications in Multispecies and NCBI NR Database Searches
Multispecies and NCBI NR protein databases contain numerous homologous sequences due to natural sequence diversity. Matching tandem mass spectra against these databases often generates protein identifications that have overlapping peptide identifications (see Figure 1). A new “minimum additional peptides per protein group” filter was added to the protein assembly process to resist adding spurious homologous proteins. This filter adds a new protein to the minimal list of protein identifications only if it contributes a specified number of distinct peptide identifications that are not already explained by other proteins. The new “minimum additional peptides per protein group” filter may at first be confused with the existing “minimum distinct peptides per protein” filter of IDPicker. The latter filter is applied to all proteins in the report, typically requiring each protein to be supported by at least two different peptide sequences for inclusion. The new filter, though, is used to control how a cluster of overlapping peptide/protein associations is decomposed into a minimal protein list (aka parsimony process). If zero additional peptides are specified, even subset and subsumable proteins will be reported; this is equivalent to disabling parsimony. If one additional peptide is required, then the parsimony behavior described in the prior IDPicker publication results. When the minimum additional peptide count is set to 2, IDPicker can discard homologous proteins from the list. We emphasize that none of the existing protein assembly tools provide as much user control over the parsimony process as does the new IDPicker.
The effect of this new filter in reducing spurious orthologous protein identifications from a multispecies database search was tested using human “DLD1 LTQ” and “Serum Orbi” data sets. Both these data sets were matched to a multispecies Swiss-Prot database using MyriMatch. Peptide identifications were loaded into IDPicker and filtered at FDR of 5%. Valid peptide identifications were used to assemble a minimal list of protein identifications. Proteins that shared degenerate set of peptides were grouped together as a single entity. Three different settings (1, 2 or 3) were used for the “Minimum additional peptides per protein group” filter in the assembly process. At each setting, the number of human and nonhuman protein groups that passed the filter were shown for “DLD1 LTQ” and “Serum Orbi” samples in Figure 6, panels A and B, respectively. The new IDPicker filter reduces the number of orthologous protein identifications from Swiss-Prot, with minimal effect on the human proteins reported.
Figure 6.
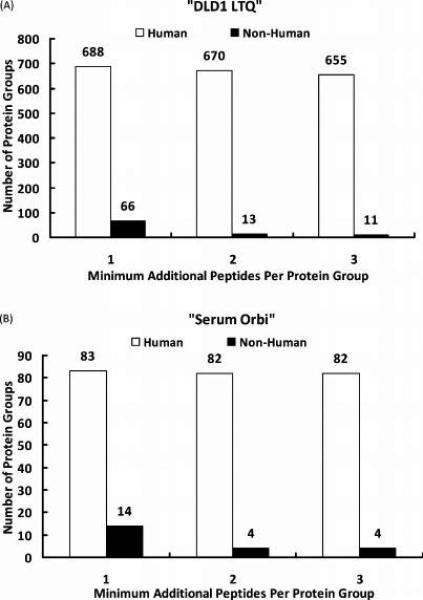
Reduction of orthologous protein identifications in a multispecies database search. Two different human samples (“DLD1 LTQ” and “Serum Orbi”) were matched to the Swiss-Prot multispecies database (version 56.2) using MyriMatch. Protein groups (containing indistinguishable proteins) were assembled from peptide identifications using IDPicker. Three different settings were used for the “Minimum additional peptides per protein group” filter in the assembly process. At each setting, the total numbers of human and nonhuman protein groups were enumerated and plotted for both “DLD1 LTQ” (A) and “Serum Orbi” (B) samples. Setting the filter to 2 dramatically reduced the number of nonhuman (orthologous) protein identifications from a multispecies database search without significantly affecting the number of human (paralogous) protein identifications.
Conclusions
The new version of IDPicker improves the sensitivity and reliability of peptide and protein identification. By combining multiple scores from a search engine, the new software made particular gains in processing Sequest results. Its new peptide partitioning strategy improved sensitivity for semitryptic and unconstrained searches. The new GUI should make the advanced features of the software more accessible to new users.
IDPicker reports can be shared among laboratories because they are simple directories of HTML files. Support for reading identifications from pepXML format enables compatibility with any workflow that can produce them. The current version of IDPicker was developed as stand-alone Microsoft Windows desktop software. The latest version of IDPicker is available under the Mozilla Public License at http://fenchurch.mc-.vanderbilt.edu/.
Supplementary Material
Acknowledgment
D. L. Tabb, Z.-Q. Ma, and M. C. Chambers were supported by NIH grants R01 CA126218 and U24 CA126479. S. Dasari was supported by NIH grant R01 CA126218. M. D. Litton and S. M. Sobecki were supported by NIH grant U24 CA126479. The “DLD1 LTQ” and “Serum Orbi” data sets were collected by P. J. Halvey and L. J. Zimmerman, respectively, under NIH grant U24 CA126479 at Vanderbilt University. B. Schilling at the Buck Institute for Age Research and P. M. Drake at UCSF provided the “Plasma QSTAR” data, which was supported by an NCRR shared instrumentation grant S10 RR024615 (to B. W. Gibson; Buck Institute for Age Research) and by NIH Grants U24 CA0126477 (to S. J. Fisher; UCSF) and a U24 Subcontract to B. W. Gibson as part of the NCI Clinical Proteomic Technologies for Cancer (http://proteomics.cancer.gov) initiative.
Footnotes
Supporting Information Available: IDPicker GUI instruction for a demo project; MyriMatch configurations; Sequest configurations; Mascot configurations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 3.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007;6(2):654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eng JKM,AL, Yates JR. An approach to correlate tandem mass-spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;(5):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 5.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods. 2007;4(10):787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 7.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007;6(9):3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X, Dondeti V, Dezube R, Maynard DM, Geer LY, Epstein J, Chen X, Markey SP, Kowalak JA. DBParser: web-based software for shotgun proteomic data analyses. J. Proteome Res. 2004;3(5):1002–1008. doi: 10.1021/pr049920x. [DOI] [PubMed] [Google Scholar]
- 9.Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 2008;7(1):29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 10.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4(3):207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 11.Choi H, Ghosh D, Nesvizhskii AI. Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res. 2008;7(1):286–292. doi: 10.1021/pr7006818. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Li J, Xie H, Zhu Y, He F. A new strategy to filter out false positive identifications of peptides in SEQUEST database search results. Proteomics. 2007;7(22):4036–4044. doi: 10.1002/pmic.200600929. [DOI] [PubMed] [Google Scholar]
- 13.Du X, Yang F, Manes NP, Stenoien DL, Monroe ME, Adkins JN, States DJ, Purvine SO, Camp DG, II, Smith RD. Linear discriminant analysis-based estimation of the false discovery rate for phosphopeptide identifications. J. Proteome Res. 2008;7(6):2195–2203. doi: 10.1021/pr070510t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore RE, Young MK, Lee TD. Qscore: an algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass Spectrom. 2002;13(4):378–386. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 15.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 16.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002;1(1):21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 18.Tabb DL, Huang Y, Wysocki VH, Yates JR., III Influence of basic residue content on fragment ion peak intensities in low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 2004;76(5):1243–1248. doi: 10.1021/ac0351163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resing KA, Meyer-Arendt K, Mendoza AM, Aveline-Wolf LD, Jonscher KR, Pierce KG, Old WM, Cheung HT, Russell S, Wattawa JL, Goehle GR, Knight RD, Ahn NG. Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Anal. Chem. 2004;76(13):3556–3568. doi: 10.1021/ac035229m. [DOI] [PubMed] [Google Scholar]
- 20.Choi H, Nesvizhskii AI. Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. J. Proteome Res. 2008;7(1):254–265. doi: 10.1021/pr070542g. [DOI] [PubMed] [Google Scholar]
- 21.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005;1 doi: 10.1038/msb4100024. 2005 0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortes HJ, Pfeiffer CD, Richter BE, Stevens T. Porous ceraminc bed supports for fused silica packed capillary columns used in liquid chromatography. J. High Resolut. Chromatogr. Chromatogr. Commun. 1987;10:446–448. [Google Scholar]
- 23.Licklider LJ, Thoreen CC, Peng J, Gygi SP. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal. Chem. 2002;74(13):3076–3083. doi: 10.1021/ac025529o. [DOI] [PubMed] [Google Scholar]
- 24.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics. 2007;6(12):2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.