

Abstract
Autophagy/Macroautophagy is known for its role in cellular homeostasis, development, cell survival, aging, immunity, cancer and neurodegeneration. However, until recently, the mechanisms by which autophagy contributes to these important processes were largely unknown. The demonstration of a direct cross-talk between autophagy and NF-κB opens up new frontiers for deciphering the role of autophagy in diverse biological processes. Here, we review our current understandings of autophagy, with a focus on its role in tumor suppression, NF-κB inactivation and selective protein degradation in mammals. We also list some most intriguing and outstanding questions that are likely to engage researchers in the near future.
Keywords: Autophagy, Cancer, Hsp90, IKK, NF-κB, Proteasome, Selective Degradation, Therapy
1. Introduction
Loss of balance between protein synthesis and degradation leads to various pathogenic conditions, particularly cancers and neurodegenerative disorders (1, 2). While protein degradation is mediated primarily by the proteasome and autophagy (1, 2), protein synthesis involves a series of processes, including mRNA transcription, protein translation, protein folding/maturation and subsequent conformation maintenance by the Hsp90 (heat shock protein of 90 kDa) chaperone complex (3, 4). Not surprisingly, targeting the proteasome and Hsp90 has become one of the most attractive therapeutic strategies for combating these diseases. As a matter of fact, several inhibitors of the proteasome and Hsp90 have being used for cancer treatment in clinics or clinic trials (1, 4, 5). In addition, the signaling pathways that are regulated by the proteasome and Hsp90 have also become very attractive targets for drug development. Due to this significance, the mechanisms of how the proteasome and Hsp90 are regulated as well as how they regulate their downstream signaling pathways have been well documented.
Compared to the proteasome pathway, autophagy and its downstream signaling pathways are much less defined. Recent studies have demonstrated a key role of autophagy in tumor suppression and neurodegeneration prevention. Interestingly, autophagy can selectively degrade signaling regulatory proteins, thereby controlling the signaling pathways such as NF-κB that are important for cell functions. Furthermore, autophagy forms a net with the proteasome and Hsp90 in protein regulation. These new findings raise important questions and open new, exciting research avenues.
2. The proteasomal degradation pathway
The proteasome is a large multi-catalytic protease complex (26S) composed of two 19S regulatory caps and a core proteolytic 20S cylinder, which are responsible for substrate recognition and degradation, respectively (1). By and large, the proteasome degrades proteins that are covalently attached with poly-ubiquitin chains. Thus, ubiquitination serves as a molecular trigger for degradation of specific proteins in the proteasome. Protein ubiquitination involves the sequential action of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). This reaction starts with formation of a thiolester linkage between E1 and ubiquitin, followed by transfer of ubiquitin to an E2. Finally, E3 recruits a specific protein substrate to the E2-ubiquitin, where the ubiquitin is conjugated to a specific lysine in the protein substrate. Since the E3 is required for substrate recognition, it is this enzyme that controls the specificity of the ubiquitination reaction. Accordingly, cells contain a large number of different E3s but only one E1 and a few of E2s.
3. Regulation of NF-κB by the ubiquitin-proteasome system
The ubiquitin-proteasome system (UPS) regulates a broad array of basic cellular processes, one of which is the activation of the NF-κB transcription factors. Mammalian cells express five NF-κB members, RelA (p65), RelB, c-Rel, NF-κB1 p50 and NF-κB2 p52, which usually form p50/Rel or p52/Rel heterodimers, though they may also function as various homo- and hetero-dimers (6). The NF-κB dimers are normally sequestered as latent complexes in the cytoplasm by the IκB inhibitors such as IκBα or IκB-like inhibitors such as p100. Accordingly, NF-κB induction is mainly based on inducible IκB degradation (canonical/classical pathway) or p100 processing (non-canonical/alternative pathway) (Fig. 1).
Figure 1. Differential regulation of NF-κB by autophagy, Hsp90 and proteasome.
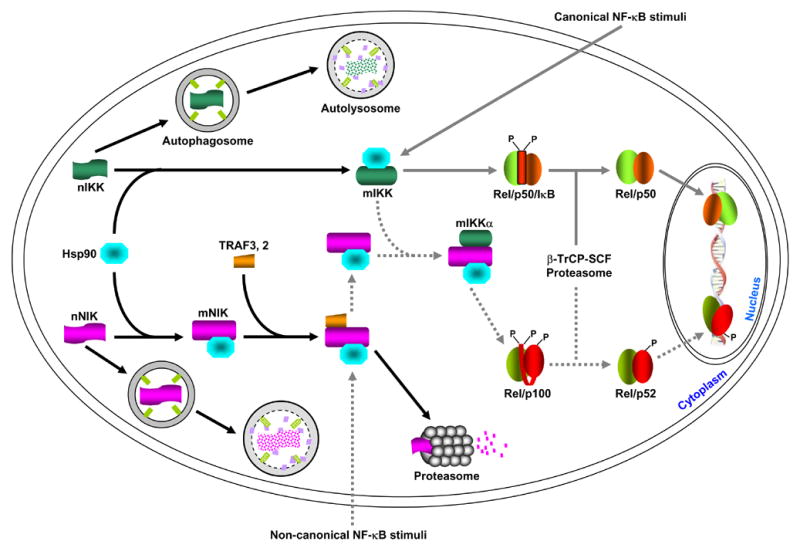
Newly synthesized IKK and NIK proteins (labeled as nIKK and nNIK, respectively) are rapidly captured by the Hsp90 chaperone complex (for simplicity, the three IKK subunits, IKKα, IKKβ and IKKγ, are just indicated as IKK). Two possible roles of this association are: promote the maturation and/or maintain the correct conformation of the IKK and NIK proteins. When Hsp90 function is absence (e.g. inhibition by geldanamycin, GA), the nascent IKK and NIK proteins cannot maturated and/or the mature proteins (labeled as mIKK and mNIK) cannot maintain the correct conformation, resulting in degradation via the autophagy pathway. In response to the classical NF-κB stimuli, IKK is activated, therefore leading to IκB proteasomal degradation and NF-κB activation. Hsp90 seems also involved in the formation of a large IKK signalsome required for IKK activation, although it is not required for the association among the three IKK subunits themselves. Unlike the mature IKK proteins that are relatively stable, the mature NIK proteins are quickly degraded by the proteasome. This proteasomal degradation is mediated by TRAF3 and possibly also by other proteins such as TRAF2. The non-canonical NF-κB stimuli somehow lead to TRAF3 degradation and/or dissociation from NIK, thereby protecting them from the proteasomal degradation. The NIK proteins freed from TRAF3 then have a chance to activate and recruit the mature IKKα into p100 for its processing and subsequent NF-κB activation. Although it always binds to NIK, Hsp90 is not required for the formation of the NIK/IKKα/p100 complex. However, it remains to be investigated whether Hsp90 is involved in the NIK regulation by TRAF3 or by the upstream NF-κB stimuli. Of note, IKK and NIK proteins escaped from GA-mediated autophagic degradation by autophagy inhibition remain their abilities, at least partially, in NF-κB activation. These results suggested that nascent IKK and NIK proteins may have some activity or they can become mature, possibly less efficiently, without Hsp90. So, the major role of Hsp90 might be to suppress autophagic degradation of IKK and NIK but not to help their folding.
The canonical NF-κB pathway can be activated by a myriad of stimuli, such as proinflammatory cytokines, mitogens, and antigens (7). These stimuli lead to rapid and transient activation of the IκB kinase (IKK) signalsome, which consists of at least two catalytic subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit, IKKγ (NEMO). Once activated, IKK phosphorylates the IκB proteins to induce their ubiquitination by the β-TrCP E3 ligase complex and degradation by the 26S proteasome, therefore unleashing NF-κB dimers into the nucleus for gene expression (6).
Different from the canonical pathway, the non-canonical NF-κB pathway can be triggered only by limited stimuli and is largely involved in B-cell maturation and lymphoid development under physiological condition (6). The non-canonical NF-κB stimuli somehow prevent basally translated NF-κB-inducing kinase (NIK) protein from undergoing proteasomal degradation mediated by the tumor necrosis factor receptor-associated proteins (TRAF), such as TRAF3 and TRAF2 (8–10). Stabilized NIK in turn specifically activates and recruits the IKKα subunit of IKK into the p100 complex to phosphorylate p100, leading to its ubiquitination by the β-TrCP E3 ligase complex and processing by the 26S proteasome (11–16). The processed p52 together with its binding partner then translocates into the nucleus to regulate target gene expression.
The NF-κB target genes are involved in different aspects of immune responses, cell survival, proliferation, differentiation and development (7). The tight control of NF-κB is thus essential for normal cellular function: defects in its activation lead to animal developmental deficiency or even death, while persistent activation of either NF-κB pathway may result in various pathogenesis, particularly immune-related disorders and cancers (6, 17, 18).
4. Differential regulation of NF-κB by Hsp90 and autophagy
As described above, NIK is required for activation of the non-canonical NF-κB pathway and IKK is involved in activation of both canonical and non-canonical pathways. However, the mechanisms of how NIK and IKK are regulated are still obscure. Recent studies demonstrate that both NIK and IKK are clients of Hsp90. Inhibition of Hsp90 by its specific inhibitor geldanamycin (GA), a very promising anti-tumor drug in clinic trial II, leads to dissociation from Hsp90 and subsequent degradation of NIK and IKK (19, 20). These results are highly consistent with a key role of Hsp90 in folding of nascent proteins, refolding of stress-denatured proteins and conformational maintenance of mature proteins (5). Surprisingly, the degradation of NIK and IKK is not mediated by the UPS, because genetic or biochemical blockage of the UPS fails to block their degradation (19, 20). These studies provide the first line of examples that an Hsp90 client can be degraded, at least alternatively, by a mechanism distinct from the proteasome. Further mechanistic studies indicate that the GA-induced degradation of NIK and IKK is largely mediated by autophagy (19, 20). Thus, NF-κB is differentially controlled by the UPS, Hsp90 and autophagy (Fig. 1).
5. Autophagic degradation pathway
The word “autophagy” is derived from the Greek and means to eat (“phagy”) oneself (“auto”). Unlike the UPS only degrades protein on a molecule-by-molecule basis, autophagy is able to degrade not only a single protein but also protein aggregate and organelle. In mammals, three modes of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). These three modes differ with respect to the pathway by which cytoplasmic material is delivered to the lysosome but share in common the final steps of lysosomal degradation of the cargo with eventual recycling of the degraded material (21). In CMA, the substrate protein is specifically recognized by a chaperone complex containing hsc70 (heat shock cognate of 70 kDa) and then delivered into the lysosome (22). In microautophagy, the cargo is engulfed directly at the lysosomal surface by invagination, protusion, and/or septation of the lysosomal membrane. In contrast, macroautophagy is characterized by that the cargo is sequestered into a double membrane structure termed autophagosome before delivery to the lysosome (Fig. 2).
Figure 2. Schematic model of autophagy.
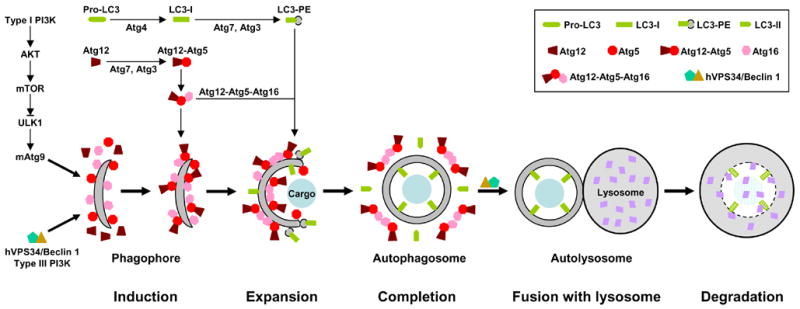
Autophagy involves at least five steps: 1. induction; 2. expansion; 3. completion; 4. docking and fusion; 5. degradation. For example, nutrient starvation, the prototypic stimulus of autophagy, leads to inhibition of the type I PI3K-AKT-mTOR signaling and activation of type III PI3K hVPS34/Beclin 1 (Atg6). Inhibition of mTOR unleashes ULK1 (Atg1), which in turn induces redistribution of mAtg9 from trans-Golgi to late endosome. Activation of hVPS34/Beclin 1, on the other hand, generates PIP3 [PtdIns(3,4,5)P3] on endomembrane. These events result in isolation and decoration with Atg5 and Atg16 of a small template membrane dubbed as phagophore. The expansion of this crescent structure requires conjugation of two ubiquitin-like proteins Atg12 and LC3 (Atg8) to Atg5 and phosphatidylethanolamine (PE), respectively. The Atg12-Atg5 conjugate binds to Atg16 and this trimeric complex polymerizes. The Atg12-Atg5-Atg16 oligomers on the surface, particularly at the tips, of phagophore initiate its elongation and curvature by recruiting cytosolic LC3-PE. The cargo may also contribute to the initiation and expansion of phagophore by serving as a scaffold. During the formation of autophagosome, the Atg12-Atg5-Atg16 complexes redistribute and concentrate mostly on the external lipid bilayers. Once the autophagosome is completed, the Atg12-Atg5-Atg16 complexes dissociate from it. The LC-II on the external lipid bilayer of the autophagosome is also released into the cytosol due to the cleavage mediated by Atg4. The uncoated autophagosome then fuses with the lysosome with the help of hVPS34/Beclin 1. The sequestered cargo together with the LC-II trapped in the lumen of the autophagosome is degraded within the autolysosome. During autophagic process LC3 is converted from LC-I (non-lipidated, long form) to LC3-II (PE-conjugated, short form) and translocated sequentially from the cytosol to phagophores, autophagosomes and autolysosome (punctate structures), it can be monitored easily by immunoblotting and immunofluresecnce/fluresecence, so it is the best and most used marker for autophagy. Currently, 3-MA (3-methyladenine) and AICAR (5-aminoimidazole-4-carboxaminde riboside) are two most widely used inhibitors for autophagy. The inhibitory function of 3-MA is attributed to its ability in suppressing type III PI3K, although it also suppresses type I PI3K. On the other hand, the mechanism by which AICAR inhibits autophagy is still unclear. One possibility is that AICAR activates p70S6K (70 kDa kinase for ribosomal protein S6) by functioning as an activator of AMPK (AMP-activated protein kinase), the original function of AICAR, although activated AMPK can inhibit mTOR. Other possibilities are that AICAR may inhibit type III PI3K (similar to 3-MA) or it may actually activate mTOR via an unidentified mechanism (35–37, also see Figure 3).
Macroautophagy is the main form of autophagy and usually directly referred to autophagy (same herein). In addition to its constitutive/basal activation in protein/organelle turnover for cellular housekeeping, autophagy is a highly regulated process and can be induced by various stimuli, such as stress, cytokines, pathogens, misfolded or aggregated proteins, damaged or surplus organelles, and even inhibition of protein synthesis, caspase, Hsp90, CMA or the proteasome. The center of autophagy activation is to form autophagosome, which involves a series of steps: isolation membrane to form phagophore (induction step), elongation of phagophore (expansion step) and maturation of autophagosome (completion step). This dynamic process is controlled by a set of proteins called Atg (autophagy related) proteins and requires two ubiquitin-like conjugation systems (21, 23).
In mammals, the two involved ubiquitin-like proteins are microtubule-associated protein 1 light chain 3 (MAP1LC3 or simply as LC3) (homologue of yeast Atg8) and Atg12. LC3/Atg8 is synthesized as a larger precursor called proLC3. The C-terminal portion of nascent proLC3 is removed by a cleavage of Atg4, a cysteine protease (24, 25). The result of this proteolytic event is to expose a glycine residue at C-terminus of mature LC3 (also called LC3-I), which is a prerequisite for the modification reaction mediated by Atg7, an E1 like protein, and Atg3, an E2 like protein specific for LC3. Notably, LC3/Atg8 is finally linked to phosphatidylethanolamine (PE), a lipid moiety, instead of a protein. During the lipidation reaction LC3-I is further processed to generate short lipidated LC3 called LC3-II or LC3-PE (26). Although the lipidated LC3 itself is able to associate with lipid bilayers, its recruitment to the phagophore depends on the other ubiquitin-like conjugation system involving Atg12 (27). Atg12, like the LC3, is also activated by Atg7. However, the E2 like protein for Atg12 is Atg10, and the final target is the Atg5 protein but not lipid. The covalent linkage of Atg12 to Atg5 leads to formation of an Atg12-Atg5-Atg16 trimeric complex, which further forms larger oligomers (28, 29). The Atg12-Atg5-Atg16 oligomers on phagophore surface initiate elongation and curvature of phagophore by recruiting cytosolic LC3-PE and Atg12-Atg5 conjugate as well as Atg16 to the phagophore, particularly to its tips (30). At the same time, the Atg12-Atg5-Atg16 complexes redistribute and concentrate mostly on the external lipid bilayers while LC3-II is located both outside and inside the completed autophagosome. Once or immediately before the autophagosome is completed, the Atg12-Atg5 conjugate and Atg16 dissociate from it. The LC-II on the external lipid bilayer of the autophagosome is also released into the cytosol by Atg4-mediated cleavage. Through this uncoating/retrieval event, these important components can be reutilized. Most importantly, only uncoated autophagosome is allowed to fuse with the lysosome. The LC-II trapped in the lumen of the autophagosome together with sequestered cargo is then broken down in the lysosome lumen.
The core pathway described above is regulated differentially by type I and type III PI3 kinases (PI3Ks). Activation of type I PI3K and its downstream signal transduction components, such as PDK1, Akt and mTOR, suppresses autophagy, while negative regulators of type I PI3K signaling, such as PTEN, act as activators of autophagy. In sharp contrast, type III PI3K hVPS34 is required for autophagy activation. Type I PI3K-Akt-mTOR signaling inhibits autophagy by indirect inhibition of ULK1/Atg1, a serine/threonine kinase required for autophgay (31, 32). One important function of ULK1/Atg1 is to induce redistribution of mAtg9, a multi-spanning transmembrane protein required for autophagy, from the trans-Golgi network to late endosome, a possible membrane source for phagopore (33). Another possible function of Atg1 is to bind to and regulate LC3, which might facilitate LC3 recruitment into the newly formed phagopore (34). Type III PI3K hVPS34 and its regulatory protein Beclin 1/Atg6 are involved in both autophagosome formation and transportation to the lysosome. One possible role of hVPS34/Beclin 1 is to facilitate the recruitment of some important proteins, such as Atg5 and Atg16, into phagophore for the induction and formation of autophagosome. Many stimuli, such as starvation and growth factors, target the PI3K pathways for autophagy induction or suppression. However, a variety of conditions that activate autophagy suggest the existence of different activating signals, although the intermediate signaling cascades have not yet been fully elucidated (Fig. 3).
Figure 3. Simple diagram of autophagy signaling.
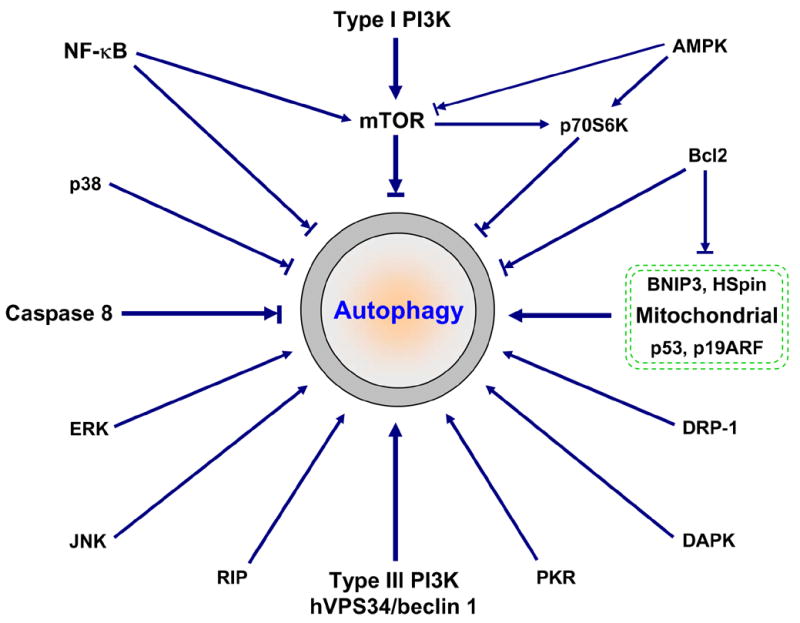
In addition to type I and type III PI3K signaling (Figure 2), many other signaling proteins have been linked to autophagy regulation. These signaling proteins often cross-talk and form a very complicated regulatory net. For example, NF-κB-mediated autophagy repression may involve mTOR activation, Bcl2/Bcl-xL upregualtion and RIP degradation (6, 38). Upregulated Bcl2/Bcl-xL can bind to and suppress the inhibitory function on autophagy of BNIP3 and HSpin, two BH-3-like domain containing protein located at mitochondrial (39, 40). Bcl2 may also bind to Beclin 1, thereby directly regulating autophagy (41, 42). Similar to NF-κB activation, activated caspase 8 cleaves RIP, leading to suppression of autophagy. There are at least two different but related mechanisms involved in RIP-mediated activation of autophagy: JNK activation and mitochondria stimuli (ROS accumulation, ATP depletion and Ca2+ release, etc.) (see Figure 5 for details). Tumor suppressors p53 (via DRAM) and p19ARF also target mitochondria for autophagy activation (43, 44). It seems that JNK, like PKR/elF2α (double-stranded RNA-activated protein kinase/eukaryotic initiation factor-2 alpha), positively regulates autophagy by inducing autophagy-related gene expression (45–47). Currently, very little is known about how p38 MAPK, ERK/GAIP (extracellular signal-regulated protein kinase/Gα interacting protein), DAPK (death-associated protein kinase) and DRP-1 (death-associated related protein kinase-1) integrate into autophagy (48–50). Stimuli to these signaling proteins may alter autophagy activity. However, the final outcome depends not only on these signaling regulators but also on cells and circumstances.
6. Selective degradation of cellular proteins by autophagy
It is generally believed that autophagy is a non-selective, bulk degradation system of long-lived proteins and organelles; while the proteasome and CMA selectively degrade cellular proteins. However, this general believe has been challenged recently. Emerging evidence indicates that the primary targets of autophagy are diffuse cytosolic proteins, but not protein aggregates (51–53). More importantly, autophagic degradation is highly selective, at least in certain cases. As mentioned above, IKK and NIK, two key regulators of NF-κB, are selectively degraded by autophagy when Hsp90 function is inhibited. It is worthy to note that the degradation of IKK and NIK is not mediated by CMA, because the GA treatment actually disrupts their association with Hsp90 (19, 20). As we already discussed, Hsp90 may be involved in substrate protein recognition and delivery to the lysosome in CMA (54). Moreover, the GA-induced degradation of NIK and IKK requires Beclin 1 and ATG5, and is associated with the conversion of LC3-I to LC3-II. On the other hand, two different autophagic inhibitors 3-MA and AICAR block their degradation (19, 20). Similarly, catalase is also selectively degraded by autophagy, but not by the proteasome or CMA, upon caspase inhibition (55). Interestingly, activation of autophagy by nutrient deprival fails to trigger autophagic degradation of catalase, suggesting a different level of autophagic selection and different mechanisms for autophagy activation. Furthermore, autophagic degradation of organelles is also highly selective, as evidenced by that autophagy can recognize and eliminate invading microbe or remove mitochondria in a highly specific and regulated manner without affecting any other organelles (56, 57).
Right now, how autophagy specifically selects cargo is still largely unknown. One immediate speculation is that certain autophagy genes may be involved in the cargo selection, directly or indirectly. A hint for this has already been provided by the study of Shigella flexneri, an invasive bacterium. VirG, a surface protein of this bacterium, can bind to Atg5 and this binding induces autophagy, leading to specific pathogen encapsulation into autophagosome and subsequent clearance (58). Given a wide range of cargoes and a large number of Atg proteins, it seems unlikely that Atg5 is the only Atg protein involved in cargo selection. Indeed, another study indicates that LC3/Atg8 is able to physically associate with the polyubiquitin-binding protein p62/SQSTM1 (sequestosome 1), resulting in selective recruitment and autophagic degradation of the p62-bound ubiquitinated proteins (59). Likewise, proteins other than p62 may be responsible for different cargo selections. It is also possible that the chaperone proteins may be involved in certain cargo selection for autophagy, although disruption association between Hsp90 and NIK or IKK leads to their autophagic degradation. In further support, autophagy has been found able to efficiently compensate CMA for its function in protein degradation (60). Since the selectivity of this pathway is conferred by means of the recognition of a KFERQ-like motif in the CMA substrates by the cytosolic chaperone complex (22), it is of interest to confirm whether the pentapeptide amino acid motif is also involved in autophagic degradation. Identify more selectively degraded cargoes will help determine the molecular basis of the selective action and functions of autophagy.
7. Cross-talk between CMA, the UPS and autophagy
The selectivity of autophagic degradation of cellular proteins is also supported by the cross-talk among different proteolytic systems. This cross-talk may occur at different levels: the same protein can be degraded by different proteolytic systems (61, 62); same components are shared by different proteolytic systems (Ubiquitin for all three of them, lysosome for autophagy and CMA); they may be mutual substrates for each other; blockage of one pathway is compensated, at least temporally or partially, by activation of others. For example, selective blockage of CMA by knocking down LAMP-2A, the lysosomal membrane receptor for the substrate-Hsc70 chaperone complex, upregulates autophagy. Interestingly, the increased autophagy is sufficient to compensate CMA in protein degradation (58). Similarly, inhibition of the proteasomal pathway has been found to increase autophagy in various cells (63–68). And the proteasome inhibition could also lead to activation of CMA (65). On the other hand, autophagy and CMA can also regulate the activity of the proteasome, since the intact proteasome is a well-known substrate of autophagy and CMA can selectively degrade some essential subunits of the proteasome (69).
Irrespective of the involved mechanisms, the cross-talk is also reflected by the observation that these proteolytic systems can be coordinately regulated by the same stimuli. For example, it is well-known that nutrient starvation induces autophagy and CMA but inhibits the proteasome. However, only autophagy is activated initially when nutrient is removed. It reaches maximal activation within several hours and then gradually decays (70–73). The time-course of activation of CMA overlaps only partially with autophagy, reaching maximal activity later on and last long time (74, 75). Although it is unclear how the sequential activation of autophagy and CMA is coordinated, inhibition of the proteasome by starvation seems due to the degradation of the proteasome by autophagy and CMA (69).
8. Mechanisms of autophagy-mediated tumor suppression
The cross-talk between these proteolytic systems, especially between the UPS and autophagy, is physiologically and pathogenically important. For instance, defect in proteasomal or autophagic pathway leads to ubiquitinated protein aggregate, inclusion body formation and neurondegenerative diseases, demonstrating an essential role of their synergy in cellular homeostasis (1, 2). On the other hand, autophagic degradation of the proteasome is also important for cellular homeostasis, in the light of that tumorigenesis is often accompanied by high proteasomal activity with a decrease in autophagic capacity. Although whether autophagic limitation of the proteasome contributes to tumor suppression remains to be established, a negative role of autophagy in tumorigenesis has been clearly suggested recently. Consistent with the observation that its capacity is decreased during progression of many tumors, autophagy can be induced in tumors by many anti-tumor agents including rapamycin, tamoxifen, arsenic trioxide and radiation, resulting in tumor cell death (76–80). Moreover, many oncoproteins, such as AKT, c-Myc, PI3K and Ras, are potent inhibitors of autophagy while many tumor suppressors, such as PTEN, p53 and p19ARF, are activators of autophagy (43, 44, 81, 82). The most important evidence driving to this conclusion is the demonstration of Beclin 1, a key regulator of autophagy, as a haploinsufficient tumor suppressor gene (83–85).
In addition to restricting proteasomal activity, several other potential mechanisms might also contribute to the tumor suppression function of autophagy: decreasing genotoxic stress; promoting cell death; limiting necrosis and inflammation; regulating of signaling pathways that contribute to tumorigenesis by selectively degrading and/or even modulating signaling proteins, etc (Fig. 4).
Figure 4. Roles of autophagy in tumor suppression.
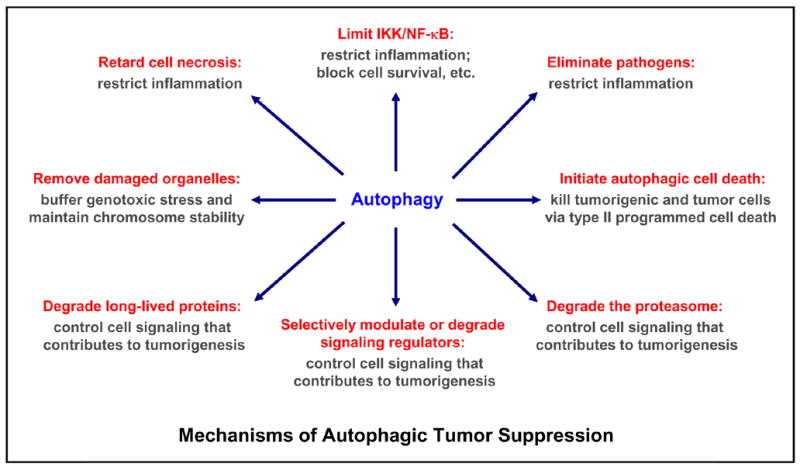
Multiple mechanisms are involved in autophagy-mediated tumor suppression: limit genotoxic damage and maintain chromosome stability by removing damaged or surplus organelles (e.g. mitochondria and endoplasmic reticulum); restrict inflammation by controlling NF-κB activation, blocking cell necrosis, eliminating pathogens; regulating signaling involved in tumorigenesis (particularly, the cell survival and proliferation signaling, e.g. mTOR, NF-κB) by modulating signaling regulatory proteins (e.g. mTOR) or by degrading signaling regulatory proteins (e.g. IKK, NIK), long-lived proteins, proteasome and other organelles; or direct kill harmful cells by type II programmed cell death/autophagic cell death. While autophagy plays an essential role in preventing tumor formation, it may also contribute to tumor progression by allowing tumor cells survival under metabolic stress (low-oxygen and low-nutrient, a common occurrence in tumors). In addition, it may protect some tumor cells against anticancer treatments by blocking the apoptotic pathway and lowering immune-mediated tumor surveillance (also see Figure 5).
Given its essential role in removing damaged and/or surplus organelles for cellular homeostasis, one can speculate that autophagy suppresses tumor formation via limiting the exposure of cellular DNA to various genotoxic stresses such as free radicals. For example, mitochondria are the major site of reactive oxygen species (ROS) production in the cell. The removal of damaged and excess mitochondria through autophagy would decrease the levels of cellular oxidants, therefore decreasing the basal DNA mutation rate and suppressing tumorigenesis.
Another speculated mechanism by which autophagy suppresses tumor progression is that it may contribute to cell death of tumorigenic or tumor cells. The autophagic cell death is now termed as type II programmed cell death while apoptosis is termed as type I programmed cell death. The third means of cell death is necrotic cell death (Fig. 5). However, the ability of autophagy in cell death seems paradoxical. Autophagy is an adaptive response to nutrient starvation and energy depletion, allowing cells to survival temporarily. On the other hand, tumor cells are often subjected to metabolic stresses. Indeed, autopahgy facilitates temporary survival of tumor cell under these stress conditions (86). Prolonged metabolic stresses and genotoxic stresses, however, may eventually lead to autophagic cell death of tumor cells, particularly when apoptosis is suppressed, which is usually the case in tumor cells. That may partially account for the tumor suppression function of Atg5 (87). During tumor development and progression, the three means of cell death coordinate together. Initially, apoptosis may be the predominate means to kill tumor potential cells. If apoptosis fails or it is suppressed, autophagy can maintain cell viable in a short term but possibly lead to cell death in a long term. Activated autophagy may also trigger tumor cell death by re-initiating apoptosis (87–89). Tumor cells that have escaped from both apoptosis and autophagy are then largely eliminated by necrotic cell death due to various stresses, especially nutrient and oxygen limitations. Through these three different death mechanisms, tumor cells are usually eradicated (Fig. 5).
Figure 5. Interplay among apoptosis, autophagy and necrosis.
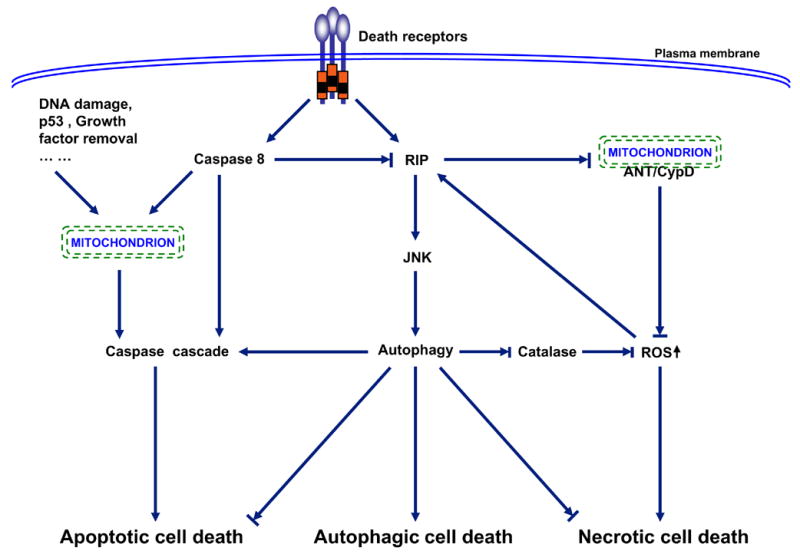
Three major patterns of cell death have been described: apoptosis or type I programmed cell death, autophagy or type II programmed cell death, and necrosis. Hallmarks of apoptotic cell death include activation of caspase cascade, DNA fragmentation, and membrane blebbing. Autophagic cell death is associated with the formation of autophagosomes, double membrane vacuoles that deliver the trapped targets into lysosomes for degradation and recycling. Necrotic cell death is characterized by cellular edema and eventually breakdown of the plasma membrane, leading to release of the cellular components and induction of inflammatory response (90, 91). The three death pathways are interconnected and often entangled. In general, apoptosis activation usually leads to inhibition of both autophagy and necrosis. This inhibition partially attributes to caspase 8-mediated cleavage of RIP1 (receptor interacting protein 1) (92–98). One of the important functions of RIP1 in cell autophagy and necrosis is to enhance ROS (reactive oxygen species) production and diminish ATP generation through prevention the interaction between ANT (adenosine nucleotide translocator) and CypD (cyclophilin D) within mitochondrial membrane (99–102). Another important function of RIP1 in ROS accumulation is to trigger degradation of catalase, the major enzymatic ROS scavenger, via JNK1-medaited autophagy (55). The activation of autophagy may first start as a survival attempt by cleaning up dysfunctional mitochondira, recycling proteins for energy and nutrient, and blocking necrosis and apoptosis (86, 103). However, when this attempt fails, autophagy may trigger apoptotic cell death, or autophagy itself even becomes cytotoxic, leading to autophagic cell death (87–89). This functional shift of autophagy may be due to up-regulation of autophagy-related genes (e.g. Atg4, 5, 7, 12 and beclin 1), leading to over-activation of autophagy (47, 104–105). However, an open mind should be kept that autophagy might only function as a pre-survival mechanism associated with or eventually contributing to cell death; but autophagy itself is not the direct death executor (87–89). In tumorigenic and tumor cells, the apoptosis and autophagy are often suppressed. In this case, necrotic cell death becomes dominant. Thus, which cell death ensues really depends on cells and circumstances. In addition, it is also possible that more than one type of cell death may occur simultaneously.
Although evidence proving a direct linking between autophagic cell death and tumor suppression is still lacking, a role of autophagy, in concert with apoptosis, in restricting cell necrosis and its mediated inflammation seems clear (86). Contrary to apoptosis and autophagic cell death, necrosis always leads to inflammatory responses. Inflammation may facilitate tumor regression via immune-mediated tumor surveillance, but chronic inflammation can lead to tumor formation even in the presence of competent immune system. As such, necrosis and inflammation are common features of tumors. Nevertheless, restricting inflammation by preventing cell necrosis may be one of the important mechanisms by which autophagy suppresses tumorigenesis.
The major mechanism for autophagy-mediated inflammation restriction and subsequent tumor suppression might be actually through targeting NF-κB (19, 20). NF-κB is the master mediator of inflammatory response and plays a causative role in tumorigenesis, particularly formation of the inflammation-associated tumors, and in resistance of malignant cells to apoptosis-based tumor surveillance (6, 17). Autophagy can selectively degrade NIK and IKK, resulting in restriction of both basal and inducible activation of NF-κB (19, 20). But more importantly, autophagy inhibition leads to IKK/NF-κB recovery from GA in malignant cells and significantly dampens the cytotoxicity of GA (data not shown). Thus, it seems plausible that NF-κB might provide a molecular link between autophagy and its function in suppressing both inflammation and tumor progression. Clearly, this hypothesis needs to be tested by in vivo animal model. Moreover, other unidentified targets of autophagy may be also involved (Fig. 4). Remarkably, NF-κB activation can also prevent autophagy activation induced by inflammatory cytokines (38). Given the general functions of NF-κB and autophagy in a plethora of cellular processes, this cross-inhibition provides some mechanistic insights into their actions.
9. Conclusions
During recent years much significant progress has already been made on autophagy. Like many important initial discoveries, these novel findings change some dogmas about autophagy and also raise more questions than they answered. Of the many unanswered questions, the most important, in author’s opinion, are listed here. 1. What is the detailed mechanisms regulating autophagy? These include defining the membrane source(s) of autophagosome, identifying the mammalian Atg homologues and determining their exact roles in autophagy. 2. How do different signals integrate into autophagy? 3. How does autophagy specifically select cargo? 4. What is the role of cargo in autophagosome formation and autophagy activation? 5. What is the exact role of autophagy in tumor suppression? An open mind needs to be kept that the role of Beclin 1 as a haploinsufficient tumor suppressor gene might not completely reflect its role in autophagy. It seems that Beclin 1 has functions beyond autophagy, as evidenced by that Beclin 1 deficient mice die much earlier than Atg 5 or Atg7 deficient mice (84, 85, 106, 107). Further, there is no report yet that autophagy defective mice other than Beclin 1 deficient mice display a pronounced increase in the incidences of tumor, although overexpression of Atg5 inhibits tumor growth (87). 6. How does autophagy coordinate with other proteolytic systems and cell death pathways under both physiological and pathogenic conditions? 7. In addition to NF-κB, are there other downstream targets of autophagy that may be involved in its functions? The answers for these questions will contribute significantly to our further understanding of autophagy and provide solid molecular basis for therapeutic strategies fighting against autophagy-associated diseases, particularly tumors and neurodegenerative diseases.
Acknowledgments
This study is supported in part by research grants from the New Jersey Commission on Cancer Research, Busch Biomedical Research Foundation, Fifth District AHEPA Cancer Research Foundation, American Cancer Society RSG-06-066-01-MGO and National Institutes of Health/National Cancer Institute R01 CA116616 to G. X.
Biography

Gutian Xiao, Ph. D. Dr. Gutian Xiao is currently an Assistant Professor in the Cell Biology and Neuroscience Department at Rutgers State University of New Jersey. His research centers on NF-κB signal transduction pathways. A novel signaling pathway leading to NF-κB activation termed as the non-canonical NF-κB pathway was first described by Xiao and colleagues. Presently, Dr. Xiao is focusing his efforts on elucidating the molecular mechanisms of how NF-κB and autophagy are cross-regulated and how this cross-regulation contributes to tumorigenesis for preventive and therapeutic purposes. Dr. Xiao has more than 36 scientific papers within the last eight years and is funded by awards from the National Institutes of Health (NIH), American Cancer Society (ACS), New Jersey Commission on Cancer Research (NJCCR), Fifth District AHEPA Cancer Research Foundation (ACRF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Exp Biol Med (Maywood) 2006;231:1197–211. doi: 10.1177/153537020623100705. [DOI] [PubMed] [Google Scholar]
- 2.Klionsky DJ. Good riddance to bad rubbish. Nature. 2006;441:819–20. doi: 10.1038/441819a. [DOI] [PubMed] [Google Scholar]
- 3.Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. Molecular Cell Biology. 5. 2004. pp. 59–72.pp. 108–30. [Google Scholar]
- 4.Dai C, Whitesell L. HSP90: a rising star on the horizon of anticancer targets. Future Oncol. 2005;1:529–40. doi: 10.2217/14796694.1.4.529. [DOI] [PubMed] [Google Scholar]
- 5.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 6.Xiao G, Rabson A, Young W, Qing G, Qu Z. Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–93. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 8.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-κB-inducing kinase by the tumor necrosis factor receptor-associated protein 3-induced degradation. J Biol Chem. 2004;279:26243–50. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 9.Qing G, Qu Z, Xiao G. Stabilization of basally translated NF-κB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-κB2 p100. J Biol Chem. 2005;280:40578–482. doi: 10.1074/jbc.M508776200. [DOI] [PubMed] [Google Scholar]
- 10.He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–8. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J Biol Chem. 2004;279:30099–105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 12.Xiao G, Harhaj EW, Sun SC. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001;8:401–9. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 13.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 14.Qing G, Qu Z, Xiao G. Regulation of NF-κB2 p100 processing by its cis-acting domain. J Biol Chem. 2005;280:18–27. doi: 10.1074/jbc.M406619200. [DOI] [PubMed] [Google Scholar]
- 15.Xiao G, Civijic ME, Fong A, Harhaj EW, Uhlik MT, Waterfield M, Sun SC. Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J. 2001;20:6805–15. doi: 10.1093/emboj/20.23.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu Z, Qing G, Rabson A, Xiao G. Tax deregulation of NF-κB2 p100 processing involves both β-TrCP-dependent and -independent mechanisms. J Biol Chem. 2004;279:44563–72. doi: 10.1074/jbc.M403689200. [DOI] [PubMed] [Google Scholar]
- 17.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 18.Sun SC, Xiao G. Deregulation of NF-κB and its upstream kinases in cancer. Cancer and Metastasis Rev. 2003;22:405–22. doi: 10.1023/a:1023733231406. [DOI] [PubMed] [Google Scholar]
- 19.Qing G, Yan P, Qu Z, Liu H, Li H, Xiao G. Hsp90 Regulates Processing of NF-κB2 p100 Involving Protection of NF-κB-inducing Kinase (NIK) from Autophagy-mediated Degradation. J Biol Chem. 2007 doi: 10.1038/cr.2007.47. In revision. [DOI] [PubMed] [Google Scholar]
- 20.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IκB kinase (IKK) Cell Res. 2006;16(11):895–901. doi: 10.1038/sj.cr.7310109. Comments in: Cell Res, 2006; 16(11): 855–56. [DOI] [PubMed] [Google Scholar]
- 21.Levine B, Klionsky DJ. Development by Self-Digestion Molecular Mechanisms and Biological Functions of Autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 22.Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–44. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 24.Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A Single Protease, Apg4B, Is Specific for the Autophagy-related Ubiquitin-like Proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L*. J Biol Chem. 2003;278:51841–50. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- 25.Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/Autophagin-1 Cleaves the Carboxyl Termini of Three Human Atg8 Homologues and Delipidates Microtubule-associated Protein Light Chain 3- and GABAA Receptor-associated Protein-Phospholipid Conjugates*. J Biol Chem. 2004;279:36268–76. doi: 10.1074/jbc.M401461200. [DOI] [PubMed] [Google Scholar]
- 26.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12–Apg5 conjugate. J Cell Sci. 2003;116:1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 30.Abeliovich H, Dunn WA, Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol. 2000;151:1025–34. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuroyanagi H, Yan J, Seki N, Yamanouchi Y, Suzuki Y, Takano T, Muramatsu M, Shirasawa T. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics. 1998;51:76–85. doi: 10.1006/geno.1998.5340. [DOI] [PubMed] [Google Scholar]
- 32.Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, Shirasawa T, Muramatsu M. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun. 1998;246:222–7. doi: 10.1006/bbrc.1998.8546. [DOI] [PubMed] [Google Scholar]
- 33.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006 doi: 10.1242/jcs.03172. in press. [DOI] [PubMed] [Google Scholar]
- 34.Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, Koga H, Muramatsu M. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res. 2000;85:1–12. doi: 10.1016/s0169-328x(00)00218-7. [DOI] [PubMed] [Google Scholar]
- 35.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–6. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 36.Moller MT, Samari HR, Seglen PO. Toxin-induced tail phosphorylation of hepatocellular S6 kinase: evidence for a dual involvement of the AMP-activated protein kinase in S6 kinase regulation. Toxicol Sci. 2004;82:628–37. doi: 10.1093/toxsci/kfh273. [DOI] [PubMed] [Google Scholar]
- 37.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;28:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 38.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-κB activation represses TNF α-induced autophagy. J Biol Chem. 2006;281:30373–82. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 39.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–68. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yanagisawa H, Miyashita T, Nakano Y, Yamamoto D. HSpin1, a transmembrane protein interacting with Bcl-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ. 2003;10:798–807. doi: 10.1038/sj.cdd.4401246. [DOI] [PubMed] [Google Scholar]
- 41.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 43.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–75. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 44.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001;21:4347–68. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Pro Natl Acad Sci USA. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–91. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 48.vom Dahl S, Schliess F, Reissmann R, Gorg B, Weiergraber O, Kocalkova M, Dombrowski F, Haussinger D. Involvement of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver. J Biol Chem. 2003;278:27088–95. doi: 10.1074/jbc.M210699200. [DOI] [PubMed] [Google Scholar]
- 49.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–5. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 50.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 52.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 53.Mizushima N, Hara T. Intracellular quality control by autophagy: how does autophagy prevent neurodegeneration? Autophagy. 2006;2:302–4. doi: 10.4161/auto.2945. [DOI] [PubMed] [Google Scholar]
- 54.Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001;114:2491–9. doi: 10.1242/jcs.114.13.2491. [DOI] [PubMed] [Google Scholar]
- 55.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 57.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–5. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 58.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–31. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 59.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Pro Natl Acad Sci USA. 2006;103:5805–10. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cuervo A, Hu W, Lim B, Dice J. IκB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell. 1998:91995–2010. doi: 10.1091/mbc.9.8.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenk SE, Susan PP, Hickson I, Jasionowski T, Dunn WA., Jr Ubiquitinated aldolase B accumulates during starvation-induced lysosomal proteolysis. J Cell Physiol. 1999;178:17–27. doi: 10.1002/(SICI)1097-4652(199901)178:1<17::AID-JCP3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 63.Wojcik C, Schroeter D, Wilk S, Lamprecht J, Paweletz N. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71:311–8. [PubMed] [Google Scholar]
- 64.Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, Cuervo AM, Keller JN. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 1003;86:489–97. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 65.Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–80. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harada M, Kumemura H, Omary MB, Kawaguchi T, Maeyama N, Hanada S, Taniguchi E, Koga H, Suganuma T, Ueno T, Sata M. Proteasome inhibition induces inclusion bodies associated with intermediate filaments and fragmentation of the Golgi apparatus. Exp Cell Res. 2003;288:60–9. doi: 10.1016/s0014-4827(03)00162-9. [DOI] [PubMed] [Google Scholar]
- 67.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–62. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell. 2006;17:2674–83. doi: 10.1091/mbc.E05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuervo A, Palmer A, Rivett A, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x. [DOI] [PubMed] [Google Scholar]
- 70.de Waal E, Vreeling-Sindelarova H, Schellens J, Houtkooper J, James J. Quantitative changes in the lysosomal vacuolar system of rat hepatocytes during short-term starvation. A morphometric analysis with special reference to macro- and microautophagy. Cell Tissue Res. 1986;243:641–8. doi: 10.1007/BF00218073. [DOI] [PubMed] [Google Scholar]
- 71.Ueno T, Muno D, Kominami E. Membrane markers of endoplasmic reticulum preserved in autophagic vacuolar membranes isolated from leupeptin-administered rat liver. J Biol Chem. 1991;266:18995–9. [PubMed] [Google Scholar]
- 72.Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–72. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- 73.Meijer AJ. Amino acids as regulators and components of nonproteinogenic pathways. J Nutr. 2003;133:2057S–62S. doi: 10.1093/jn/133.6.2057S. [DOI] [PubMed] [Google Scholar]
- 74.Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physi. 1995;269:C1200–8. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 75.Fuertes G, Martin De Llano J, Villarroya A, Rivett A, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J. 2003;375:75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 77.Bursch W, Ellinger A, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 78.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–8. [PubMed] [Google Scholar]
- 79.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 80.Yao KC, Komata T, Kondo Y, Kanzawa T, Kondo S, Germano IM. Molecular response of human glioblastoma multiforme cells to ionizing radiation: cell cycle arrest, modulation of the expression of cyclin-dependent kinase inhibitors, and autophagy. J Neurosurg. 2003;98:378–84. doi: 10.3171/jns.2003.98.2.0378. [DOI] [PubMed] [Google Scholar]
- 81.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 82.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 83.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 84.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sc USA. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 88.Furuya D, Tsuji N, Yagihashi A, Watanabe N. Beclin 1 augmented cis-diamminedichloroplatinum induced apoptosis via enhancing caspase-9 activity. Exp Cell Res. 2005;307:26–40. doi: 10.1016/j.yexcr.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 89.Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–63. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 91.Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;358:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- 92.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–26. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 94.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–21. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 95.Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA, Liu ZG. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822–8. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 96.Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L, Karin M, Zhang J, Liu ZG. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol. 2004;24:5914–22. doi: 10.1128/MCB.24.13.5914-5922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 98.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–82. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 100.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 101.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 102.Temkin V, Huang Q, Liu H, Osada H, Pope RM. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol. 2006;26:2215–25. doi: 10.1128/MCB.26.6.2215-2225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ, Coughlin SM, Zuyderduyn SD, Jones SJ, Marra MA. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol. 2003;13:358–63. doi: 10.1016/s0960-9822(03)00082-4. [DOI] [PubMed] [Google Scholar]
- 105.Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol. 2003;13:350–57. doi: 10.1016/s0960-9822(03)00085-x. [DOI] [PubMed] [Google Scholar]
- 106.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 107.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]