

Abstract
Background
Anthracyclines are the most effective drugs available in the treatment of neoplastic diseases. However, they have profound consequences on the structure and function of the heart causing with time a cardiomyopathy that leads to congestive heart failure.
Methods and Results
Administration of doxorubicin (DOXO) in rats led to a dilated myopathy, heart failure and death. To test whether DOXO effects on cardiac anatomy and function were mediated by alterations in cardiac progenitor cells (CPCs), these cells were exposed to the anthracycline which increased the formation of reactive oxygen species (ROS), DNA damage, expression of p53, telomere attrition and apoptosis. Additionally, DOXO resulted in cell cycle arrest at the G2/M transition leading to a significant decrease in CPC growth. DOXO elicited multiple molecular adaptations; the massive apoptotic death occurring in CPCs in the presence of the anthracycline imposed on the surviving CPC pool the activation of several pathways aiming at the preservation of the primitive state, cell division, lineage differentiation and repair of damaged DNA. To establish whether delivery of syngeneic progenitor cells opposed the progression of DOXO cardiotoxicity, EGFP-labeled CPCs were injected in the failing myocardium and this treatment promoted regeneration of cardiomyocytes and vascular structures, improving ventricular performance and animal survival.
Conclusions
Our results raise the possibility that autologous CPCs can be obtained before antineoplastic drugs are given to cancer patients and subsequently administrated to individuals who are particularly sensitive to the cardiotoxicity of these agents for prevention and/or management of heart failure.
Keywords: Heart failure, Cardiotoxicity, Antineoplastic drugs, Cardiac Stem Cells
Anthracyclines are some of the most effective drugs currently available in the treatment of neoplastic diseases.1 However, anthracyclines have profound consequences on the structure and function of the heart causing with time a cardiomyopathy that leads to intractable congestive heart failure.2 The cardiotoxicity of anthracyclines is dose-dependent and this limits its clinical implementation at optimal antitumor efficacy. Doxorubicin (DOXO) is the most powerful and widely used anthracycline and considerable effort has been made to elucidate the etiology of DOXO-induced cardiotoxicity to prevent the mechanisms implicated in the initiation and dramatic evolution of ventricular dysfunction.3 The generation of reactive oxygen species (ROS) is a critical mediator of myocardial damage4 but the target cell(s) actually responsible for the deterioration of cardiac performance remains to be determined.
The recognition that the adult heart in animals and humans contains a pool of resident primitive cells, which are self-renewing, clonogenic and multipotent in vitro and regenerate myocytes and coronary vessels in vivo5–8 raises the question whether the effects of DOXO on cardiac homeostasis and repair are primarily directed to the stem cell compartment partially ablating the reserve of functionally-competent cardiac progenitor cells (CPCs). CPCs are particularly sensitive to oxidative stress and rapidly die by apoptosis. Myocytes are more resistant to ROS formation than CPCs, strengthening the possibility that loss of CPCs together with the attenuated generation of a myocyte progeny may be critical in the development of DOXO-mediated cardiomyopathy. Theoretically, CPCs can be isolated from biopsy samples, and after their expansion in vitro, can be implanted locally within regions of damage where they reconstitute the injured myocardium.5–8 This strategy may allow aggressive chemotherapy followed by CPC repopulation of the depleted myocardium which may rescue the cardiomyopathic heart. These hypotheses have been tested in the current study to determine whether DOXO-induced cardiomyopathy can be viewed as a stem cell disease and whether CPC therapy reverses heart failure in an animal model. Here, we report that intramyocardial injection of syngeneic CPCs positively interferes with anthracycline cardiotoxicity largely restoring the structural and functional integrity of the diseased heart.
Methods
CPCs and DOXO
Clonogenic c-kit-positive CPCs were infected with a retrovirus carrying EGFP. CPCs were treated for 12, 24 and 48 h with 0.1, 0.5 and 1 μM DOXO concentrations. CPC apoptosis and proliferation were determined.
Telomere-Telomerase-System
Telomerase activity was measured by quantitative PCR and telomere length by Q-FISH.
RT-PCR Array
The transcriptional profile of CPCs in the absence and presence of DOXO was assessed by quantitative RT-PCR array.
Animal Studies
Fischer 344 rats with DOXO-induced cardiomyopathy were treated with CPCs. A total of 5 × 104 EGFP-labeled CPCs were injected at 4 sites in the left ventricular myocardium. This dose was selected based on previous results in which the delivery of progenitors varying from 10,000 to 100,000–200,000 produced similar positive effects on myocardial regeneration.
Data Analysis and Statistics
Results are presented as mean ± SD.
For additional information see supplementary Materials and Methods.
Results
Doxorubicin and CPC Death and Growth
To establish the effects of DOXO on clonogenic c-kit-positive CPCs,5 these cells were exposed to 0.1, 0.5 and 1 μM DOXO for 12, 24 and 48 hours. Cell viability was assessed by a colorimetric MTT assay. In the presence of 0.1 μM DOXO, CPC survival was not affected. However, DOXO at 0.5 and 1 μM reduced, respectively, CPC viability by 24% and 33% at 24 hours, and by 66% and 90% at 48 hours (Figure 1A). Additionally, apoptosis measured by TdT assay, DNA laddering and caspase-3 activity increased with time and the dose of DOXO. These three indicators of apoptosis peaked after 48 hours of treatment with 1 μM DOXO (Figure 1B–1D). TdT assay was restricted to adherent cells and, following 48 hours of exposure to 1 μM DOXO, the number of adherent CPCs was reduced by ~90%, indicating that this drug promoted apoptosis in almost all cells.
Figure 1.



CPC death and growth. A, The viability of CPCs is negatively affected by dose and time of exposure to DOXO. B–D, CPC apoptosis measured by TdT assay (B), DNA laddering (C) and caspase-3 activity (D) increases with the concentration of DOXO and from 12 to 48 h. The expression of active caspase-3 is shown as fold changes with respect to control (c). TdT labeling (white) of apoptotic CPCs (c-kit, green) is shown by immunolabeling in panel B. E and F, BrdU (E, red) and phospho-H3 (F, yellow) labeling of CPCs decreases progressively with DOXO. *,**,† P<0.05 vs. c, 0.1 μM and 0.5 μM DOXO, respectively.
The impact of DOXO on CPC division was determined by BrdU and phospho-H3 labeling. The number of BrdU-positive CPCs and the mitotic index decreased with increasing concentration of DOXO and time (Figure 1E and 1F). Moreover, the molecular regulators of G1, G1/S transition and G2/M transition were measured. Cyclin D1, which drives cells from G1 to S, is activated by the cyclin dependent kinase cdk4 and this complex phosphorylates Rb inhibiting its repressive function on cell cycle progression. During G2, the cyclin B1-cdc2 complex is inactivated by phosphorylation. At the end of G2, the cdc25 phosphatase dephosphorylates this complex and cells enter mitosis. Cyclin D1, cdk4 and phosphorylated Rb decreased in CPCs exposed to DOXO in a dose and time dependent manner. The increase in cyclin B1 and cdc2 phosphorylation may reflect the arrest of the cell cycle at the G2/M transition (Figure 2A; supplementary Figure IA). These data are consistent with the delay in decrease of BrdU labelling in CPCs with respect to phospho-H3 (Figure 1E and 1F).
Figure 2.



Cell cycle regulators and oxidative stress. A–D, DOXO affects the expression of cyclins, cyclin-dependent kinases, Rb (A) and cyclin-dependent kinase inhibitors (B), increases DNA damage (C: 8-OH-dG, red) and decreases the activity of SOD and catalase (D) in CPCs (C: c-kit, green).
Subsequently, the protein level of the cyclin dependent kinase inhibitors p21Cip, p27Kip1 and p16INK4a was determined in CPCs. DOXO resulted in a transient increase of p21Cip and a persistent increase in p16INK4a (Figure 2B; supplementary Figure IB). However, the expression of p27Kip1 in CPCs was not affected by DOXO. The early upregulation of p21Cip may represent an attempt of CPCs to repair DNA damage while the persistent high quantity of p16INK4a indicates irreversible growth arrest and cellular senescence.
Doxorubicin and oxidative stress in CPCs
There is general consensus that the generation of ROS plays a relevant role in the development of anthracycline-induced cardiomyopathy.2,4 To determine whether a similar process was operative in CPCs, the presence of 8-OH-deoxyguanosine (8-OHdG) was measured in nuclei by immunocytochemistry and confocal microscopy. DOXO treatment was characterized by a striking increase in the number of 8-OHdG-positive CPCs (Figure 2C). Moreover, the expression of the antioxidant enzymes manganese superoxide dismutase (Mn SOD), copper-zinc superoxide dismutase (Cu/Zn SOD) and catalase did not change while the activity of these enzymes decreased markedly at 48 hours failing to counteract ROS-mediated DNA damage (Figure 2D; supplementary Figure IIA). DOXO resulted in an average 30% shortening of telomeres in CPCs and a shift to the left in the distribution curve of telomere lengths. Additionally, the percentage of CPCs with telomeres less than 8 kbp increased 4-fold with DOXO (Figure 3A). Telomere attrition occurred in spite of the preservation of telomerase activity in DOXO-treated CPCs (Figure 3B).
Figure 3.



Telomere-telomerase and p53 function. A, Telomere shortening in DOXO-treated CPCs: nuclei (blue) were stained with a telomere probe (magenta). Lymphoma cells with long (L5178Y-R, 48 kbp) and short (L5178Y-S, 7 kbp) telomeres are shown for comparison. The average telomere length is indicated together with the degree of telomeric shortening and the fraction of cells with telomeres equal/shorter than 8 kbp. B, Telomerase activity was comparable in control and DOXO-treated CPCs. Samples treated with RNase were used as negative and HeLa cells as positive control. C and D, DOXO activates the DNA-damage response in CPCs. Immunoblotting for Bax and Bad are shown in the upper part of panel D. Protein expression is shown as fold changes with respect to c. See Figure 1 for symbols.
Dysfunctional telomeres trigger a DNA damage response in which the major determinant is the transcription factor p53. The ataxia-telangiectasia mutated (ATM) protein kinase is required for phosphorylation of p53 at serine 15; ATM kinase and phospho-p53 at serine 15 and 20 were upregulated in DOXO-treated CPCs (Figure 3C; supplementary Figure IIB). ATM kinase expression peaked at 12 hours while phospho-p53 at serine 15 and 20 increased mostly at 12 and 24 hours and remained elevated at 48 hours. Phosphorylation at serine 15 activates a cascade of post-translational modifications of p53 which result in transcription of p53 target genes followed by activation of apoptosis or cellular senescence.9 In the current study, p53 phosphorylation at serine 15 was accompanied by enhanced but transient expression of p21Cip1 (see above) possibly in an attempt to promote DNA repair. Also, the pro-apoptotic proteins Bax and Bad increased in DOXO-treated CPCs (Figure 1E and 3D). The prolonged upregulation of p16INK4a in CPCs is consistent with the role of this protein in the modulation of irreversible growth arrest and cellular senescence. P16INK4a rarely co-localizes with DNA double-strand breaks and represents a delayed response10 which follows the induction of p53 and p21Cip1.
Thus, anthracyclines promote oxidative stress and the activation of p53 which together inhibit the growth and survival of CPCs supporting the notion that defects in progenitor cell function may condition the development of the cardiac myopathy in vivo. Additionally, these in vitro observations raise the possibility that CPC death may represent the primary event responsible for impaired myocyte turnover, accumulation of senescent cells, apoptosis and the onset of ventricular dysfunction, unrecognized aspects of DOXO-mediated cardiotoxicity. The in vivo experiments discussed in the subsequent sections aim at the documentation that alterations at the level of the controlling cell, the CPC, dictate the dramatic outcome of DOXO treatment in patients with neoplastic diseases.
Doxorubicin and Cardiac Anatomy and Function
To evaluate the effects of anthracyclines in vivo, Fisher 344 rats were injected intraperitoneally over a period of 14 days with six doses of DOXO11 (supplementary Figure III). One week following the last administration, there was a significant impairment of left ventricular (LV) function characterized by a decrease in ejection fraction (EF) which decreased further at 6 weeks (Figure 4A). The question was then whether the abnormalities detected echocardiographically were due to the prolonged presence of DOXO in the organism or the anthracycline had an acute toxic effect which persisted with time depressing myocyte mechanical behavior. Since DOXO has a half-life of ~30 hours and its direct action on cells is no longer detectable after 1–2 days,12 myocyte contractility and Ca2+ transients were determined in LV myocytes isolated from animals at 3 weeks. Sarcomere shortening and Ca2+ transients in myocytes were decreased with DOXO (Figure 4B). The time constant (τ) of Ca2+ decay and the time to 90% relaxation of myocytes were longer in these cells.
Figure 4.



DOXO-induced cardiomyopathy. A, Echocardiography in control (C) and at 3 and 6 weeks (wks) after the first injection of DOXO. B, Defects in myocyte mechanics and calcium transient in cells isolated from DOXO-treated hearts at 3 weeks. C–E, DOXO administration enhances apoptosis (C: TdT, white) and senescence (D: p16INK4a, yellow) and decreases proliferation (E: Ki67, green) of cardiomyocytes (α-sarcomeric actin: α-SA, red) in vivo. F, Myocyte number and volume in control and DOXO-treated rats. *,** P <0.05 vs. C and 3 wks, respectively.
To establish whether DOXO activated cell death, cardiomyocyte apoptosis was determined. In comparison with control hearts, DOXO treatment resulted in a 7-fold and 4-fold increase in myocyte apoptosis at 3 and 6 weeks, respectively (Figure 4C). Importantly, corresponding increases in the fraction of cardiomyocytes expressing the senescence-associated protein p16INK4a were 2-fold and 3-fold (Figure 4D). More than 70% of LV myocytes were p16INK4a positive at 6 weeks. Conversely, myocyte formation measured by the expression of Ki67 decreased 95% and 65% at 3 and 6 weeks, respectively (Figure 4E). Therefore, myocyte loss was not counteracted by an adequate formation of new cells leading to a significant decrease in the aggregate number of parenchymal cells in the LV myocardium. This reduction in myocyte number was more pronounced at 6 than at 3 weeks. Additionally, myocyte cell volume increased with time reflecting the inadequate level of myocyte regeneration seen in the presence of DOXO (Figure 4F). Collectively, these observations suggest that DOXO led to a cardiac myopathy in which myocyte death predominates and contributes together with the depression in cell mechanics to the deterioration of ventricular function in this animal model.
Doxorubicin and CPC Transcriptional Profile
To establish whether DOXO treatment influences CPC fate, the molecular identity of these cells was defined by analyzing their transcriptional profile following exposure to the anthracycline. We have employed quantitative RT-PCR array and examined a restricted set of genes linked to the undifferentiated state of the cells and their specification to cardiovascular lineages. Additionally, genes involved in cell proliferation, survival, death and senescence were studied (supplementary Figure IV).
DOXO induced profound changes in global gene expression of CPCs: 103 and 21 genes were upregulated and downregulated, respectively. DOXO resulted in a 9-fold increase in the expression of the ATP-binding cassette ABC transporter Abcg2/Mdr1 which is implicated in drug efflux and cell protection from toxic agents.13 Although c-kit receptor mRNA was similar in untreated and treated CPCs, transcripts for the downstream effectors MITF and Snail-homolog 2 (Slug) increased in the presence of the anthracycline (Figure 5; supplementary Figure IV).
Figure 5.

Doxorubicin and gene expression in CPCs. Transcriptional profiling of CPCs in the absence and presence of DOXO by quantitative RT-PCR array. Transcript expression is shown as fold changes with respect to control.
Genes involved in self-renewal and progenitor cell expansion,14,15 including fibroblast growth factor 8 (FGF8) and 10 (FGF10), the catalytic subunit of telomerase (TERT) and the histone acetyltransferases Myst1 and Myst2 were more abundant in DOXO-treated than untreated-CPCs. Similarly, Numb and Prospero-related protein (Prox1) that modulate asymmetric division16 were higher with DOXO (Figure 5). Importantly, transcripts for Klf4, Klf5, Oct4 and c-myc were significantly increased in CPCs exposed to the anthracycline. Growth differentiation factor-3 (GDF3) and Nanog were enhanced with DOXO while Sox2 was decreased but these changes in gene expression were not significant (supplementary Figure IV). Klf4, Sox2, c-Myc and Oct4 are the four genes that promote reprogramming of fibroblasts into inducible pluripotent stem cells.17 The core Klf circuitry, composed of Klf2, Klf4 and Klf5, is critical for the preservation of the undifferentiated state of embryonic stem cells.17 Together with GDF3, these genes integrate into the Nanog transcriptional network that specifies the stemness of various progenitors.18 Additionally, several cell cycle regulators comprising cyclins D1, E and A2 and the cyclin-dependent kinase cdc2 were more abundant in DOXO treated CPCs.
The mechanisms that control cardiomyogenesis in the adult heart are largely unknown. However, the differentiation of CPCs into myocytes reiterates partly the molecular programs of cardiac development. The majority of cardiac regulatory transcription factors were upregulated in DOXO treated-CPCs. They included GATA4, GATA5, MEF2A, Tbx1, Tbx3, Tbx20 and Hand2. Consistently, the downstream targets BNP, α-sarcomeric actin, myosin-light-chain-4 and β-myosin-heavy-chain were more highly expressed in these cells. Notch1 receptor is a critical determinant of the transition of CPCs to amplifying myocytes.19 Although Notch1 expression was decreased, transcripts of the Notch pathway, including the Delta-like-3 and the Jagged1 ligands, the mastermind-like 1 co-factor and the Hes1 effector, were more abundant in DOXO treated-CPCs (Figure 5; supplementary Figure IV).
The positive effect of DOXO on CPC commitment was not restricted to the myocyte lineage. The expression of several vascular-specific genes increased in CPCs in response to DOXO. This molecular adaptation involved mostly EC-related genes including Vezf1, Flk1, Flt1, Tie2, PECAM, multimerin, selectin and von-Willebrand-factor. Together with the enhanced expression of Flk1, the upregulation of GATA1, CD34 and Tal1 indicated that the anthracycline triggered the activation of the molecular program controlling the formation of hemangioblasts.20 For the acquisition of SMC lineage, only TGF-β receptor 1 and SM myosin-heavy-chain were upregulated in DOXO treated-CPCs (Figure 5; supplementary Figure IV). Similarly, a group of p53-related genes implicated in cell death, DNA damage response and growth arrest were more expressed in these cells. They included ATM kinase, Rad50, Mre11, Bax, p21Cip1, Gadd45a and Mdm2 (Figure 5; supplementary Figure IV).
Collectively, these findings at the transcriptional level indicate that DOXO triggers multiple biological adaptations in CPCs. The massive apoptotic death occurring in CPCs in the presence of the anthracycline imposes that the surviving CPC pool activates several pathways aiming at the preservation of the primitive state, cell division, lineage differentiation and repair of damaged DNA.
Doxorubicin and CPC Death and Growth In Vivo
The data above raised the possibility that one of the major consequences of DOXO on cardiomyocyte death, hypertrophy and dysfunction in vivo was mediated by defects at the level of the progenitor cell compartment. Therefore, these variables of CPC function were evaluated quantitatively in the LV myocardium. In comparison with control hearts, DOXO produced a 5-fold and 8-fold increase in CPC apoptosis at 3 and 6 weeks, respectively (Figure 6A). Additionally, the fraction of p16INK4a positive CPCs which reached irreversible growth arrest10 was dramatically increased in these hearts (Figure 6B). In contrast, the percentage of Ki67-positive CPCs was severely reduced with DOXO treatment (Figure 6C). These findings were consonant with the enhanced oxidative stress and DNA damage promoted by DOXO, as documented by the generation of 8-OHdG in CPC nuclei (Figure 6D). Collectively, the impact of DOXO on CPC apoptosis and senescence decreased by 79% and 94% the compartment of functionally-competent CPCs in the LV myocardium at 3 and 6 weeks, respectively (Figure 6E). Thus, anthracyclines have negative effects on cell viability and growth, depleting the CPC pool available for cardiac homeostasis and repair.
Figure 6.

CPC death and growth. A–C, DOXO administration enhances apoptosis (A: TdT, white), senescence (B: p16INK4a, yellow) and decreases proliferation (C: Ki67, yellow) of CPCs (c-kit, green) in vivo at 3 and 6 weeks after the first injection of anthracycline. D, Oxidative damage (8-OH-dG, magenta) in CPCs increases after DOXO treatment in vivo. E, Number of functionally competent CPCs in control and DOXO-treated rats at 3 and 6 weeks. *,** P< 0.05 vs. C and 3 wks, respectively.
CPC Repopulation of the Myocardium
If the detrimental consequences of anthracyclines on the heart were dependent on the loss of CPCs, exogenously administered immunocompatible CPCs would be expected to restore partly cardiac function and structure improving the outcome of the dilated myopathy and animal survival. Therefore, DOXO-treated rats at 3 weeks were divided in two groups (supplementary Figure V). The first group received intramyocardial injections of syngeneic CPCs (DOXO-CPC) and the second vehicle only (DOXO-vehicle). CPCs were genetically tagged with EGFP for the identification of their progeny. All animals were sacrificed 3 weeks later, i.e., 6 weeks after the onset of DOXO and 3 weeks after CPCs or vehicle delivery.
Shortly after cell implantation, preliminary studies were performed to document by immunocytochemistry the presence of EGFP-positive CPCs within the myocardium. Additionally, the expression of Ki67 in EGFP-positive CPCs was demonstrated to prove that these cells, at least in part, successfully engrafted and continued to grow within the recipient myocardium (supplementary Figure VI). Following treatment, animals were exposed continuously to BrdU to label newly formed structures within the damaged decompensated heart. Therefore, regenerated myocytes and coronary vessels were expected to be both EGFP and BrdU positive in DOXO-CPC hearts. Previous results at 2 days after delivery of a comparable number of cells was ~20%. However, this value is the product of two variables: death of the non-engrafted cells and proliferation of engrafted cells.21
Three weeks after CPC therapy, there was an amelioration of the conditions of the animals; they were less lethargic and had modest or none abdominal enlargement. The amount of fluid in the abdomen was ~6-fold lower in DOXO-CPC (5.3±2.8 ml) than in DOXO-vehicle (30±14 ml; P<0.001) rats. Most importantly, mortality rate was dramatically reduced following CPC injection (Figure 7A). At 3 weeks, before treatment, mortality averaged 45%. However, from 3 to 6 weeks, animal mortality was decreased by 66% with CPC implantation. In the animals that survived, cardiac function was largely restored by CPC administration. With respect to DOXO-vehicle rats, LV developed pressure and +dP/dt and −dP/dt were markedly increased in DOXO-CPC hearts, reaching hemodynamic values similar to those in control animals (Figure 7B). Similarly, EF was essentially restored by CPC delivery (supplementary Figure VII). The decrease in ventricular mass and wall thickness, and the increase in chamber diameter and volume with the DOXO-myopathy were partly reversed by cell therapy (Figure 7C), suggesting that CPCs promoted myocardial regeneration contributing to the recovery of structure and function of the damaged heart.
Figure 7.


CPC treatment, ventricular function and animal survival. A–C, Mortality (A), ventricular hemodynamics (B) and cardiac anatomy (C) of untreated and CPC-treated animals at 6 weeks.
Large clusters of newly formed cardiomyocytes were detected throughout the LV wall. These cells were EGFP- and BrdU-positive, and expressed the contractile protein α-sarcomeric actin (Figure 8A–8C). Areas of myocardial regeneration were identified in all CPCs-treated animals and varied in size from 0.05 to 2.5 mm2. Connexin 43 and N-cadherin were detected between new myocytes, and preexisting and regenerated myocytes demonstrating that formed cells expressed the junctional proteins responsible for electrical and mechanical coupling (supplementary Figure VIII). Seeding of EGFP-positive cells was also shown by PCR that confirmed the morphological results (supplementary Figure IX).
Figure 8.



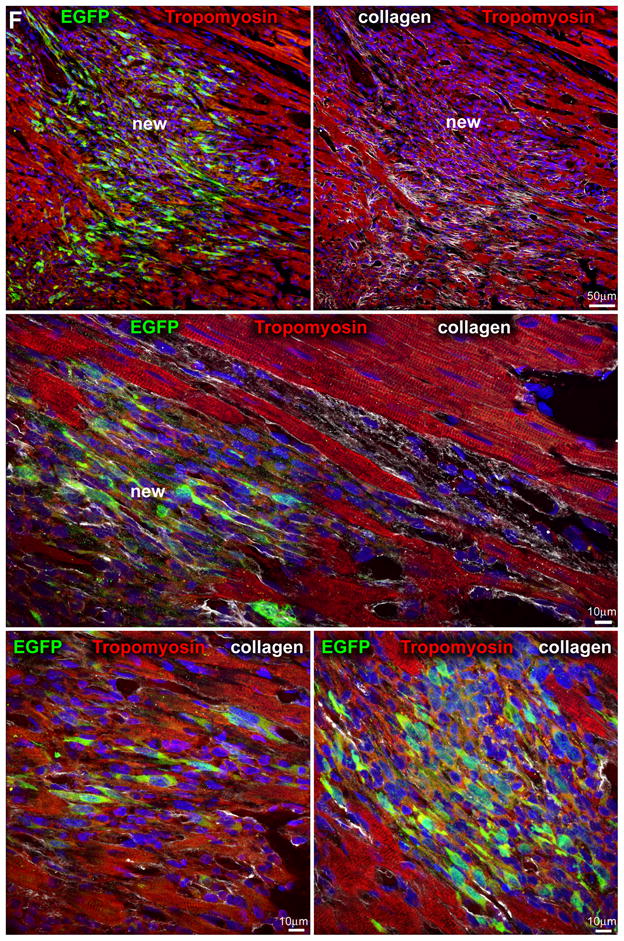
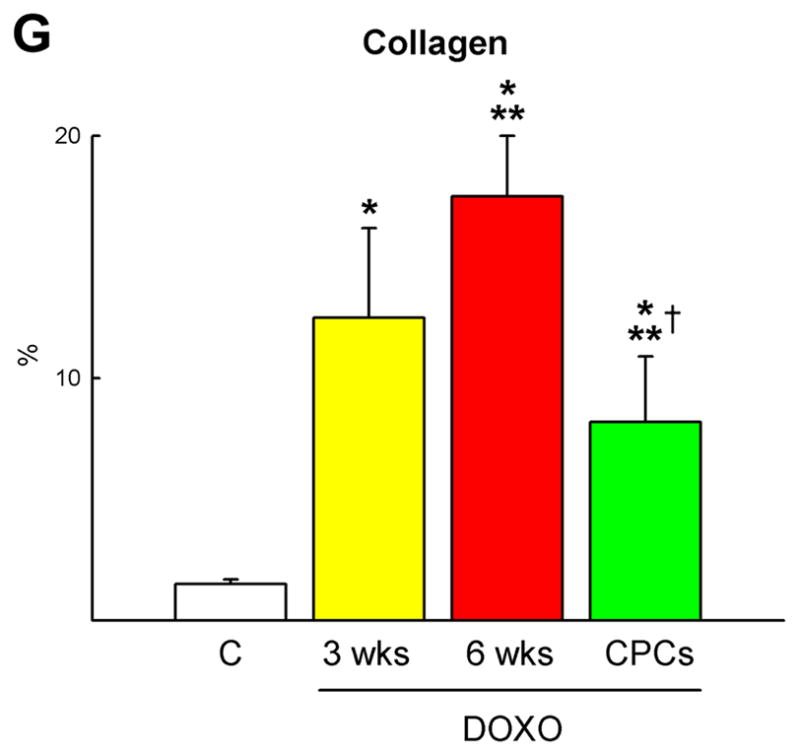
Myocardial regeneration and DOXO-induced cardiomyopathy. A, Low-power view of transverse sections of the LV wall of DOXO-treated rats 3 weeks after CPC injection. Arrows define areas of myocardial regeneration (green). Newly formed myocytes express EGFP (green) and α-SA (red). B, The area of myocardial regeneration included in the rectangle is shown at higher magnification in the adjacent panel. Small myocytes in the cluster express EGFP (green) and α-SA (red). C, Newly formed EGFP-positive (green) myocytes are labeled by BrdU (white) and express α-SA (red). D, Number of newly formed capillaries and arterioles. E, Foci of collagen (yellow) accumulation at times surrounding small vessels. F, Regenerated myocytes (new) are positive for EGFP (green) and tropomyosin (red) and replace areas of fibrosis (collagen, white). G, Fraction of collagen within the myocardium. *,**,† P <0.05 vs. c, 3 wks and 6 wks, respectively.
These foci of cardiac tissue consisted of cardiomyocytes and resistance arterioles and capillaries distributed throughout the regenerated myocardium (supplementary Figure X). New myocytes retained a fetal-neonatal phenotype varying in volume from 300 to 4,300 μm3 (supplementary Figure XI). Similarly, the number of capillaries and arterioles (Figure 8D) reflected those of a developing heart.8 Myocardial regeneration decreased by 52±12% the extent of DOXO-induced tissue damage. Following cell therapy, replacement fibrosis was 34% and 53% lower than in untreated animals at 3 and 6 weeks, respectively (Figure 8E–8G; supplementary Figure XII), indicating that CPC differentiation restored partly the structural integrity of the cardiomyopathic heart. However, activation of resident CPCs or the recruitment of circulating progenitors may have contributed to cardiac repair.
Discussion
The etiology of DOXO-induced cardiomyopathy remains unclear although several mechanisms including DNA damage, attenuation in protein synthesis, enhanced release of catecholamines, alterations in the adrenergic system and defects in Ca2+ homeostasis have been identified, together with the remarkable increase in oxidative stress and lipid peroxidation.1,2,4 These detrimental effects, however, result in a rather unspecific cardiac pathology1–3 in which cytoplasmic vacuolization, loss of myofibrils, and the relatively modest increase in interstitial and replacement fibrosis in the ventricular wall are not consistent with the severity of the disease and the dramatic manifestations of heart failure in animal models and humans.2,3 Understanding anthracycline-mediated cardiomyopathy is further complicated by the timing of appearance of cardiotoxicity. Ventricular dilation, wall thinning and depressed cardiac function may become evident during the course of therapy or later following the completion of treatment. The possibility to develop heart failure persists throughout life in patients who successfully survive cancer and its management with this antineoplastic agent.3
The results of the current study offer an alternative mechanism underlining the pathophysiology of DOXO-induced cardiomyopathy. Our in vitro and in vivo findings indicate that the determining event responsible for the initiation and evolution of the myopathy occurs at the level of the CPC compartment. Cardiotoxicity of the anthracycline is not restricted to cardiomyocytes but affects even more dramatically resident CPCs. Over a period of 6 weeks, DOXO led to an almost complete depletion of the CPC pool within the myocardium. Inhibition of CPC division in combination with the accumulation of oxidative DNA damage, growth arrest, cellular senescence and apoptosis decreased by 94% the number of functionally-competent progenitors in the failing heart. This devastating consequence of DOXO on CPCs interfered with the physiological turnover of cardiomyocytes and their regeneration in the presence of diffuse cell death. The number of ventricular myocytes was reduced by 48% and the majority of cells expressed the senescence-associated protein p16INK4a. Hypertrophied p16INK4a-positive myocytes show depressed mechanics and Ca2+ transients.22 Collectively, the lack of activation of CPCs and formation of a myocyte progeny, myocyte loss and impaired contractile behavior of spared, enlarged myocytes appear to be critical variables of the development of heart failure with DOXO administration. Thus, DOXO-cardiomyopathy is primarily a stem cell disease that conditions the pathophysiology of cardiomyocytes and ventricular hemodynamics.
Treatment of CPCs with DOXO induced dramatic changes of the cellular transcriptome, highlighting the molecular mechanisms implicated in the functional response of CPCs to anticancer drugs. DOXO evoked multiple and apparently contradictory pathways in CPCs by activating simultaneously transcriptional regulators that promote multipotency and trigger differentiation.15,18 By necessity, gene expression profiling was obtained in the subset of cells that survived DOXO toxicity and attempted to restore the homeostatic balance within the CPC compartment. To counteract ongoing apoptotic death, spared CPCs enhanced the expression of the complex network of transcription factors required for the maintenance of stemness. These genes integrate into the chromatin- regulatory-loop that involves the Nanog promoter and is hierarchically supervised by Oct4.18 Klf4, Klf5, Oct4, c-myc, GDF3 and Nanog were upregulated in CPCs. This cluster of genes was initially believed to be restricted to embryonic stem cells;18 however, the same transcriptional system appears to control the undifferentiated state of CPCs and other adult progenitor cells.23
Additionally, DOXO upregulated in CPCs Flk1, GATA1, CD34 and Tal1 which control the formation of embryonic and adult hemangioblasts.20 Hemangioblasts are common precursors for endothelial and hematopoietic cell lineages but, as shown above, the presence of several EC transcripts in the absence of bone marrow markers indicated that CPCs preferentially acquired the EC phenotype. The attempt of DOXO-CPCs to differentiate into vascular cells was coupled with the activation of transcription factors that modulate cell fate and expression of contractile proteins during cardiac morphogenesis. These regulatory genes were upregulated in DOXO-CPCs and comprised the cardiac-restricted members of the GATA family, the Mef2 transcription factors, the T-box-family of transcription factors and genes of the secondary heart field.
To strengthen the possibility that targeted therapies for cancer patients may be implemented in a manner that maximizes their antitumor effects24 without creating another equally devastating disease such as chronic heart failure,25 we have successfully documented that immunocompatible CPCs may be employed to repopulate the cardiomyopathic heart with new cardiomyocytes and coronary vessels. Myocardial regeneration mediated by delivery and differentiation of CPCs restored largely cardiac hemodynamics, and most importantly decreased dramatically animal mortality. Our findings have provided the first experimental documentation that negative ventricular remodeling characterized by cavitary dilation, wall thinning, severely impaired function and ascites, typically present in chronic heart failure,26 can be reversed by CPC-therapy. Although extreme caution has to be exercised in the translation of these animal studies to human beings, the possibility is raised that myocardial biopsies may be obtained before antineoplastic drugs are given to cancer patients. Autologous CPCs can be isolated and expanded from these myocardial samples8 for the treatment of heart failure in individuals who are particularly sensitive to the cardiotoxicity of these chemotherapeutic agents.
A difficult question to answer concerns the variability in the response to DOXO in patients and the rather common observation that anthracycline-induced-cardiomyopathy may develop years after the administration of the anti-cancer-drug. Two important factors may account for these human data which appear to be in contrast with our experimental results. The first may be related to the age of the patient which conditions the growth behavior and resistance to apoptosis of resident CPCs27 and the second may reflect the intrinsic properties of CPCs which are dictated by patient’s medical history. The latter may have influenced negatively telomere length and telomerase that together protect the pool size of CPCs, and their ability to self-renew, divide asymmetrically and survive. Finally, experimental findings cannot be easily translated to human beings.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by grants from the Italian Ministry of Education and the NIH.
Footnotes
Disclosures
None
Clinical Perspective
The mechanisms by which anthracyclines lead to the development of a dilated cardiomyopathy in cancer patients are currently unknown. The work presented in this study raises the possibility that doxorubicin-induced cardiac failure is a stem cell disease mediated by a severe loss of resident cardiac progenitor cells (CPCs). Doxorubicin results in CPC apoptosis which with time affects myocardial homeostasis and tissue repair. The dramatic reduction in the pool of CPCs conditions myocyte renewal promoting the accumulation of senescent poorly functional cardiomyocytes. Chronically, these abnormalities in cardiac structure and myocyte performance markedly impair ventricular hemodynamics and animal survival. Importantly, repopulation assays with syngeneic CPCs rescue the cardiac phenotype and restore largely the functional properties of the diseased heart. Although extreme caution has to be exercised in the translation of these animal studies to human beings, the possibility is raised that myocardial biopsies may be obtained before antineoplastic drugs are given to cancer patients. Autologous CPCs can be isolated and expanded from these myocardial samples for the treatment of heart failure in individuals who are particularly sensitive to the cardiotoxicity of these chemotherapeutic agents.
References
- 1.van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. 2008:CD003917. doi: 10.1002/14651858.CD003917.pub3. [DOI] [PubMed] [Google Scholar]
- 2.Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330–352. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Intern Med. 1996;125:47–58. doi: 10.7326/0003-4819-125-1-199607010-00008. [DOI] [PubMed] [Google Scholar]
- 4.Nithipongvanitch R, Ittarat W, Cole MP, Tangpong J, Clair DK, Oberley TD. Mitochondrial and nuclear p53 localization in cardiomyocytes: redox modulation by doxorubicin (Adriamycin)? Antioxid. Redox Signal. 2007;9:1001–1008. doi: 10.1089/ars.2007.1632. [DOI] [PubMed] [Google Scholar]
- 5.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 6.Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31- but Not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res. 2005;97:52–61. doi: 10.1161/01.RES.0000173297.53793.fa. [DOI] [PubMed] [Google Scholar]
- 7.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Regene rative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 8.Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, Yasuzawa-Amano S, Trofimova I, Siggins RW, Lecapitaine N, Cascapera S, Beltrami AP, D’Alessandro DA, Zias E, Quaini F, Urbanek K, Michler RE, Bolli R, Kajstura J, Leri A, Anversa P. Human cardiac stem cells. Proc Natl Acad Sci USA. 2007;104:14068–14073. doi: 10.1073/pnas.0706760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs JJ, de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4:1364–1368. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- 11.Siveski-Iliskovic N, Kaul N, Singal PK. Probucol promotes endogenous antioxidants and provides protection against adriamycin-induced cardiomyopathy in rats. Circulation. 1994;89:2829–2835. doi: 10.1161/01.cir.89.6.2829. [DOI] [PubMed] [Google Scholar]
- 12.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- 13.Sorrentino BP, Brandt SJ, Bodine D, Gottesman M, Pastan I, Cline A, Nienhuis AW. Selection of drug-resistant bone marrow cells in vivo after retroviral transfer of human MDR1. Science. 1992;257:99–103. doi: 10.1126/science.1352414. [DOI] [PubMed] [Google Scholar]
- 14.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 15.Gan Q, Yoshida T, McDonald OG, Owens GK. Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells. 2007;25:2–9. doi: 10.1634/stemcells.2006-0383. [DOI] [PubMed] [Google Scholar]
- 16.Tang H, Rompani SB, Atkins JB, Zhou Y, Osterwalder T, Zhong W. Numb proteins specify asymmetric cell fates via an endocytosis- and proteasome-independent pathway. Mol Cell Biol. 2005;25:2899–2909. doi: 10.1128/MCB.25.8.2899-2909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurdon JB, Melton DA. Nuclear reprogramming in cells. Science. 2008;322:1811–1815. doi: 10.1126/science.1160810. [DOI] [PubMed] [Google Scholar]
- 18.Levasseur DN, Wang J, Dorschner MO, Stamatoyannopoulos JA, Orkin SH. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes Dev. 2008;22:575–580. doi: 10.1101/gad.1606308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, Amano K, Gonzalez A, Vitale S, Ojaimi C, Rizzi R, Bolli R, Yutzey KE, Rota M, Kajstura J, Anversa P, Leri A. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Schatteman GC, Awad O. Hemangioblasts, angioblasts, and adult endothelial cell progenitors. Anat Rec A Discov Mol Cell Evol Biol. 2004;276:13–21. doi: 10.1002/ar.a.10131. [DOI] [PubMed] [Google Scholar]
- 21.Rota M, Kajstura J, Hosoda T, Bearzi C, Vitale S, Esposito G, Iaffaldano G, Padin-Iruegas ME, Gonzalez A, Rizzi R, Small N, Muraski J, Alvarez R, Chen X, Urbanek K, Bolli R, Houser SR, Leri A, Sussman MA, Anversa P. Bone marrow cells adopt the cardiomyogenic fate in vivo. Proc Natl Acad Sci USA. 2007;104:17783–17788. doi: 10.1073/pnas.0706406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rota M, Hosoda T, De Angelis A, Arcarese ML, Esposito G, Rizzi R, Tillmanns J, Tugal D, Musso E, Rimoldi O, Bearzi C, Urbanek K, Anversa P, Leri A, Kajstura J. The young mouse heart is composed of myocytes heterogeneous in age and function. Circ Res. 2007;101:387–399. doi: 10.1161/CIRCRESAHA.107.151449. [DOI] [PubMed] [Google Scholar]
- 23.Greco SJ, Liu K, Rameshwar P. Functional similarities among genes regulated by OCT4 in human mesenchymal and embryonic stem cells. Stem Cells. 2007;25:3143–3154. doi: 10.1634/stemcells.2007-0351. [DOI] [PubMed] [Google Scholar]
- 24.Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 26.Braunwald E. Biomarkers in heart failure. N Engl J Med. 2008;358:2148–2159. doi: 10.1056/NEJMra0800239. [DOI] [PubMed] [Google Scholar]
- 27.Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P. Senescence and death of primitive cells and myocytes leads to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.