

Abstract
AIMS
We evaluated whether ‘boosting’ doses of ritonavir can serve as a positive control inhibitor for pharmacokinetic drug–drug interaction studies involving cytochrome P450 3A (CYP3A). The study also determined whether 4,4-dimethyl-benziso-(2H)-selenazine (ALT-2074), an investigational organoselenium compound that acts as a catalytic mimic of glutathione oxidase, inhibits CYP3A metabolism in vivo.
METHODS
Thirteen healthy volunteers received single 3-mg oral doses of midazolam on three occasions: in the control condition, during co-treatment with low-dose ritonavir (three oral doses of 100 mg over 24 h), and during co-treatment with ALT-2074 (three oral doses of 80 mg over 24 h).
RESULTS
Ritonavir increased mean (±SE) total area under the curve (AUC) for midazolam by a factor of 28.4 ± 4.2 (P < 0.001), and reduced oral clearance to 4.2 ± 0.5% of control (P < 0.001). In contrast, ALT-2074 increased midazolam AUC by 1.25 ± 0.11 (P < 0.05), and reduced oral clearance to 88 ± 8% of control.
CONCLUSIONS
Low-dose ritonavir produces extensive CYP3A inhibition exceeding that of ketoconazole (typically 10- to 15-fold midazolam AUC enhancement), and is a suitable positive control index inhibitor for drug–drug interaction studies. ALT-2074 inhibits CYP3A metabolism to a small degree that is of uncertain clinical importance.
Keywords: cytochrome P450-3A, drug interaction, midazolam
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The viral protease inhibitor ritonavir is known to inhibit clearance of intravenous midazolam.
ALT-2074, a catalytic mimic of glutathione oxidase, inhibits human cytochrome P450 3A (CYP3A) isoforms in vitro.
WHAT THIS STUDY ADDS
Short-term administration of low-dose ritonavir increases area under the plasma concentration curve following oral midazolam by a factor of 28.
Therefore ritonavir is an appropriate positive control inhibitor for clinical drug interaction studies involving CYP3A substrates.
Midazolam clearance is weakly inhibited by ALT-2074, consistent with its in vitro profile.
Introduction
The benzodiazepine derivative midazolam, a substrate for biotransformation by cytochrome P450 3A (CYP3A) enzymes [1–3], is extensively used in drug development and clinical pharmacology as an index compound to profile the activity of hepatic and enteric CYP3A [4–12]. Under baseline conditions, midazolam undergoes extensive presystemic extraction after oral dosage, with net systemic bioavailability in the range of 30% [13–18]. It is established that incomplete oral bioavailability of midazolam results from a combination of hepatic and enteric CYP3A activity. An enteric-specific CYP3A inhibitor, such as grapefruit juice, has no effect on total area under the plasma concentration curve (AUC) of intravenous midazolam [19, 20], but increases AUC for oral midazolam by a factor of up to twofold [17, 20–23]. In contrast, an inhibitor such as ketoconazole, acting on both hepatic and enteric CYP3A, increases AUC of both intravenous and oral midazolam, but the effect on oral midazolam AUC is substantially greater [14, 18, 24–27].
In the course of drug development, new chemical entities suspected of being CYP3A inhibitors may be evaluated in clinical drug–drug interaction (DDI) studies using midazolam as the in vivo CYP3A probe compound [4–12]. The scientific value of such studies is strengthened by inclusion of a ‘positive control’ arm, intended to depict the ‘worst case scenario’ DDI. Ketoconazole is a possible choice as a positive control CYP3A inhibitor. However, recent studies suggest that CYP3A inhibition by ritonavir, even at relatively low ‘boosting’ doses, may produce CYP3A inhibition exceeding that of ketoconazole [28–35].
The present study evaluated low-dose ritonavir as an inhibitor of oral midazolam clearance, in the course of a DDI study of a medication under development. The candidate drug was 4,4-dimethyl-benziso-(2H)-selenazine (ALT-2074; formerly BXT-51072), a low-molecular-weight, orally active, organoselenium catalytic mimic of the enzyme glutathione peroxidase that is being developed for the treatment of inflammatory disorders characterized by the involvement of reactive oxygen species [36–41]. One possible indication is the treatment of acute coronary syndromes.
Previous in vitro studies have shown ALT-2074 is an inhibitor of human CYP3A, with an IC50 value in the range of 2.0–2.6 µM. This concentration might be achieved within the gastrointestinal tract or in the systemic circulation after oral administration of ALT-2074, raising the possibility that ALT-2074 might produce drug interactions with other CYP3A substrate drugs in vivo.
Methods and procedures
Study participants and design
The study protocol and consent form were reviewed and approved by the Western Institutional Review Board (Olympia, WA, USA). All subjects provided written informed consent prior to the study.
Participants were healthy male volunteers aged 18–55 years with no current or prior history of significant medical or psychiatric disease, and receiving no prescription medications. All subjects were within 25% of ideal body weight based on actuarial data incorporating height and frame size. Screening procedures included medical history, physical examination, electrocardiogram, haematology and chemistry screening, and urinalysis.
The DDI study was conducted using a three-way crossover design, with at least a 1-week interval elapsing between trials. After completing a screening period, subjects received each of the following three trial regimens in random sequence:
| Trial | Co-treatment | Midazolam |
|---|---|---|
| 1 | Placebo (three doses) | 3 mg |
| 2 | ALT-2074, 80 mg (three doses) | 3 mg |
| 3 | Ritonavir, 100 mg (three doses) | 3 mg |
The dosing schedule for ALT-2074 is consistent with typical therapeutic exposure. The dosage schedule for ritonavir represents exposure consistent with a ‘boosting’ regimen.
ALT-2074 and placebo (Synvista Therapeutics, Inc., Montvale, NJ, USA) were packaged identically and administered under double-blind conditions. Because of the shape and construction of the oral ritonavir dosage form (Abbott Laboratories, N. Chicago, IL, USA), a placebo to match ritonavir was not available, and ritonavir was given under nonblind conditions.
Subjects were admitted to the Study Unit for the duration of each trial period. Subjects received the first dose of the co-treatment between 16.00 and 18.00 h on the first study day. The second and third co-treatments were administered at 07.30 and 18.00 h on the second study day. Midazolam was prepared as 3 ml of the commercially available parenteral dosage form (1 mg ml−1) mixed with 240 ml of tap water. The midazolam was administered at 08.00 h on the second study day, followed by multiple pharmacokinetic blood samples. Subjects’ final blood samples were obtained at 08.00 h on the third study day. Subjects repeated the procedure, being randomized to a different co-treatment at approximately weekly intervals.
Experimental procedures
Subjects were admitted to the study unit on the afternoon prior to each midazolam trial. Between 16.00 and 18.00 h, the first dose of co-treatment was administered (placebo, ALT-2074, or ritonavir). On the following morning, a light breakfast was provided at 07.00 h. At 07.30 h, an indwelling cannula was inserted, and a predose blood sample was taken. The second dose of co-treatment (placebo, ALT-2074, or placebo) was then given. At 08.00 h, a 3-mg oral dose was administered, followed by blood sampling at 0.25, 0.5, 1.0, 2, 4, 6, 8, 10 and 12 h after dosage. The third dose of co-treatment was given at 18.00 h. The final blood sample was taken at 08.00 h on the following morning, 24 h after midazolam dosage. Subjects were then discharged from the study unit.
Venous blood samples were collected into heparinized tubes and stored on ice until centrifuged. The plasma was separated and frozen at −18°C until assay.
Analysis of samples
Plasma concentrations of midazolam in all samples were determined by liquid chromatography-mass spectroscopy, having a sensitivity limit of 0.5 ng ml−1[15]. All samples from a given subject's set of three trials were extracted and analysed together on the same day using the same calibration standards.
Plasma concentrations of ritonavir during the ritonavir co-treatment trial were determined by high-performance liquid chromatography [42]. Methods were not available for determination of ALT-2074 concentrations.
Pharmacokinetic analysis
The following pharmacokinetic parameters for midazolam were determined using standard model-independent (‘noncompartmental’) methods: maximum plasma concentration (Cmax), elimination rate constant (β), elimination half-life (T½), total AUC, and apparent oral clearance (CL).
Statistical analysis
A 50% difference in mean values of midazolam clearance between placebo or ALT-2074 co-treatments, or between placebo and ritonavir co-treatments, was assumed to be of potential clinical importance. Based on prior studies [13, 15, 23], the standard deviation of the difference between mean values was assumed to be 35% of the difference itself. Under these conditions, a sample size of n= 12 allows this difference to be detected with α= 0.05 and power of at least 0.8.
Arithmetic mean and SD/SE of untransformed pharmacokinetic variables were calculated and presented [43]. Differences among the three co-treatments (placebo, ALT-2074, or ritonavir) were evaluated using analysis of variance (anova) for repeated measures, followed by Dunnett's test to compare ALT-2074 vs. placebo and ritonavir vs. placebo individually. These analyses were done both without and with rank transformation (nonparametric analysis).
Ratios of pharmacokinetic variables with ALT-2074 or ritonavir co-treatment divided by the placebo value were also calculated. These were aggregated as arithmetic means and standard deviations, or geometric means and 90% confidence intervals (CIs).
Kinetic and statistical analyses were performed using Microsoft Excel or Statistical Analysis Systems (SAS Institute Inc., Cary, NC, USA).
Results
Subjects
Fifteen subjects initiated participation in the study, and completed the first of three trials. Two of these individuals did not complete subsequent trials for administrative reasons. Pharmacokinetic analysis was based on the 13 subjects that completed all three trials. Age ranges were 21–50 years, and weight ranged from 52 to 97 kg. The racial/ethnic distribution was eight White, five African-American.
Studies were completed with no adverse events reported.
Pharmacokinetics of midazolam
Figure 1 shows the mean plasma midazolam concentrations at corresponding times, comparing placebo with ALT-2074 and placebo with ritonavir.
Figure 1.
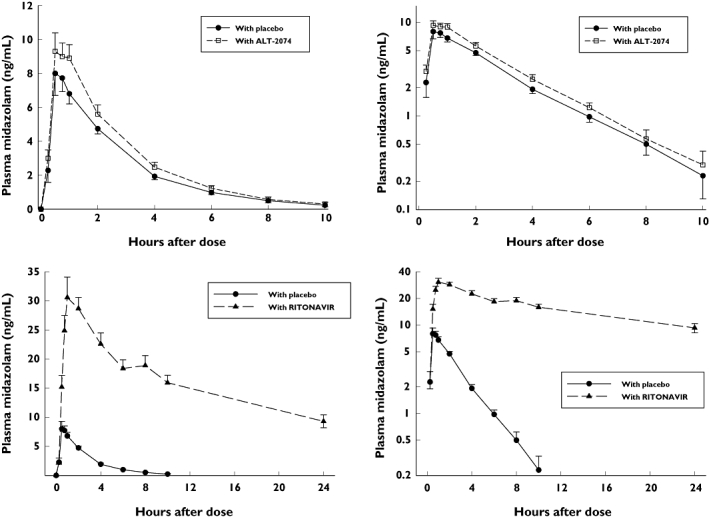
Mean (±SE) plasma midazolam concentrations at corresponding times. Top: placebo and ALT-2074 co-treatments (left: linear scale; right: logarithmic scale). Bottom: placebo and ritonavir co-treatments (left: linear scale; right: logarithmic scale)
Analysis of variance for repeated measures showed highly significant differences among the three treatments in all pharmacokinetic variables for midazolam (Table 1). Dunnett's test showed significant differences between ritonavir and placebo co-treatments. Ritonavir increased midazolam AUC by a factor of approximately 25, and correspondingly reduced clearance to about 4% of control values. Co-treatment with ALT-2074 increased midazolam AUC by a factor of about 1.25, and reduced clearance to 88% of control. Differences in mean values between ALT-2074 and placebo co-treatments were not significant.
Table 1.
Statistical analysis of midazolam pharmacokinetic variables*
| Mean (±SE, n= 13) for treatment conditions: | Dunnett's test | |||||
|---|---|---|---|---|---|---|
| Placebo | ALT-2074 | Ritonavir | Repeated measures anova | ALT-2074 vs. placebo | Ritonavir vs. placebo | |
| Cmax (ng ml−1) | 9.0 (±1.0) | 10.6 (±0.9) | 35.6 (±2.8) | F = 81.8, P < 0.001 | NS | P < 0.05 |
| t1/2 (h) | 2.06 (±0.15) | 2.08 (±0.17) | 18.07 (±2.25) | F = 49.4, P < 0.001 | NS | P < 0.05 |
| Total AUC (ng ml−1 h−1) | 24.65 (±1.93) | 29.87 (±2.62) | 651 (±77) | F = 64.9, P < 0.001 | NS | P < 0.05 |
| Clearance (ml min−1) | 2157 (±142) | 1844 (±167) | 85.9 (±6.9) | F = 92.2, P < 0.001 | NS | P < 0.05 |
| Clearance (ml min−1 kg−1) | 28.84 (±2.84) | 24.15 (±2.38) | 1.13 (±0.11) | F = 64.1, P < 0.001 | NS | P < 0.05 |
Analysis of actual values without transformation.
When the anova was done on rank-transformed variables, all conclusions were identical.
Table 2 summarizes the analysis of ratios for the pharmacokinetic variables. Based on untransformed ratios of untransformed values, all ritonavir/placebo ratios were different from 1.0 with a high level of statistical significance. The ALT-2074/placebo ratio for Cmax and clearance did not differ significantly from 1.0; the AUC ratio was significantly greater than 1.0 (P < 0.05, two-tailed test).
Table 2.
Analysis of ratios for midazolam pharmacokinetic variables
| Ritonavir/placebo ratio | ALT-2074/placebo ratio | |||||
|---|---|---|---|---|---|---|
| Cmax | AUC (total) | Clearance | Cmax | AUC (total) | Clearance | |
| Arithmetic | ||||||
| Mean | 4.47* | 28.4* | 0.042* | 1.34 | 1.25** | 0.88 |
| SD | 2.02 | 15.3 | 0.017 | 0.89 | 0.39 | 0.29 |
| SE | 0.56 | 4.2 | 0.005 | 0.19 | 0.11 | 0.08 |
| Geometric | ||||||
| Mean | 4.10 | 25.6 | 0.039 | 1.21 | 1.19 | 0.84 |
| 90% CI | 3.32, 5.07 | 20.5, 32.0 | 0.032, 0.049 | 0.96, 1.52 | 1.024, 1.40 | 0.72, 0.98 |
Student's t-test vs. 1.0:
P < 0.001;
P < 0.05.
Geometric mean ratios underestimated the arithmetic means. For the ritonavir/placebo ratios, the 90% CIs fell entirely outside the arbitrary 80–125% boundary. For ALT-2074/placebo, one extreme of the 90% CI fell outside the 80–125% boundaries.
Plasma ritonavir concentrations
Figure 2 shows mean plasma ritonavir concentrations at specific time points during the ritonavir co-administration trial. The results demonstrate systemic exposure to ritonavir consistent with the ritonavir dosage. However, there was no apparent relationship between the net exposure to ritonavir over the 0–10-h dosage interval (expressed as mean concentration over that interval) and the extent of midazolam clearance impairment relative to the control trial (expressed as midazolam total AUC ratio for Trial 3 divided by Trial 1) (Figure 3).
Figure 2.
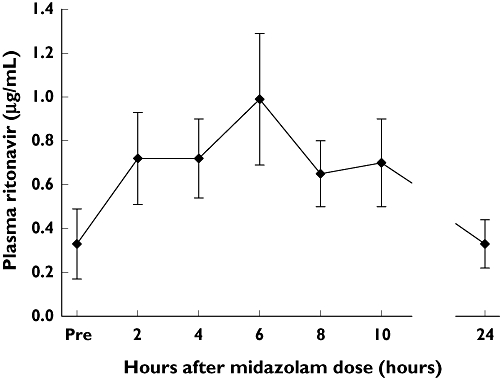
Mean (±SE) plasma ritonavir concentrations at individual time points
Figure 3.
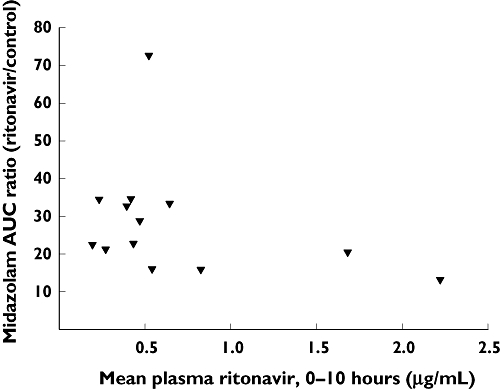
x-axis: Mean plasma ritonavir concentration across the time interval 0–10 h. y-axis: Ratio of midazolam total AUC during the ritonavir co-administration condition (Trial 3) divided by the control AUC value (Trial 1). Each point represents an individual subject
Discussion
In this study we evaluated the capacity of ALT-2074 and boosting doses of ritonavir to reduce the apparent oral clearance and thereby increase the systemic exposure of midazolam in human volunteers. Midazolam is cleared essentially exclusively via biotransformation by CYP3A isoforms (CYP3A4 and CYP3A5) [1–3]. After oral dosage, midazolam clearance is determined by a combination of enteric and hepatic CYP3A activity [13–18] and is considered to be a ‘sensitive’ CYP3A probe to indicate the capacity of drugs under clinical development to inhibit or induce CYP3A phenotype in vivo[4–12]. Although our study subjects were male volunteers, a review of available literature indicates that gender has only a small influence on the kinetics of midazolam and other CYP3A substrate drugs [44]. There is no evidence to indicate that susceptibility to metabolic inhibition is meaningfully influenced by gender [14, 17, 26].
A relatively low dose (100 mg given orally three times over 24 h) of the antiretroviral agent ritonavir was used as a positive control inhibitor. Ritonavir is a highly potent CYP3A inhibitor in vitro, with IC50 or Ki values in the low nanomolar range [45–48]. Since systemic exposure to ritonavir [I] with usual clinical dosage generally exceeds 1–2 µM, the ratio of [I]/Ki or [I]/IC50 will exceed 10.0, thereby predicting a high likelihood of clinical drug interactions involving ritonavir and CYP3A substrates [4–12]. This prediction has been verified in a number of previous clinical studies [28–35, 49–55]. It is also reported that CYP3A inhibition by ritonavir is reversible within a few days after discontinuation of ritonavir [55]. In the present study, a very large ritonavir–midazolam interaction was observed, in which relatively low ‘boosting’ doses of ritonavir increased midazolam systemic exposure by a factor of about 25. This exceeds the extent of midazolam clearance inhibition generally produced by ketoconazole, which typically increases midazolam exposure by a factor of 10–15 [14, 15, 24–27]. Thus the inclusion of ritonavir as a positive control inhibitor verified the validity of the clinical model, including the identification of midazolam as a ‘sensitive’ substrate. The fact that impairment of midazolam clearance was independent of systemic exposure to ritonavir indicates that all levels of ritonavir exposure were sufficient to produce extensive reduction of midazolam clearance [31].
The rationale for the clinical study was based on in vitro studies with ALT-2074 indicating an IC50 value for CYP3A inhibition (using triazolam hydroxylation as an index reaction) in the range of 2.0–2.6 µM. Assuming maximum systemic exposure to ALT-2074 to be in the range of 500 ng ml−1 (2.2 µM), the ratio of [I]/IC50 is approximately 1.0, indicating that a clinical drug interaction involving ALT-2074 is ‘possible’. However, the results of the clinical study indicated only a modest increase in midazolam AUC in the range of 20–25% with co-administration of ALT-2074. The effect of ALT-2074 was not statistically significant based on anova, with or without rank transformation of the values. Analysis of ratios indicated that the arithmetic mean midazolam AUC ratio (ALT-2074 divided by placebo) of 1.25 was significantly different from 1.0, and the 90% CI (1.02–1.40) for the geometric mean ratio (1.19) fell partially outside the ‘default’ upper boundary of 1.25 as specified in the Food and Drug Administration guidance. [56] Thus ALT-2074 could be considered a ‘weak’ CYP3A inhibitor at most. The clinical importance of this inhibition of CYP3A by ALT-2074 remains to be determined.
Competing interests
None to declare.
This work was supported in part by Grants AG-017880, GM-061384, DA-023861 and RR-025752 from the United States Department of Health and Human Services, and by Synvista Therapeutics, Inc.
REFERENCES
- 1.Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- 2.Perloff MD, von Moltke LL, Court MH, Kotegawa T, Shader RI, Greenblatt DJ. Midazolam and triazolam biotransformation in mouse and human liver microsomes: relative contribution of CYP3A and CYP2C isoforms. J Pharmacol Exp Ther. 2000;292:618–28. [PubMed] [Google Scholar]
- 3.von Moltke LL, Greenblatt DJ, Schmider J, Duan SX, Wright CE, Harmatz JS, Shader RI. Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J Clin Pharmacol. 1996;36:783–91. doi: 10.1002/j.1552-4604.1996.tb04251.x. [DOI] [PubMed] [Google Scholar]
- 4.Huang SM, Temple R, Throckmorton DC, Lesko LJ. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther. 2007;81:298–304. doi: 10.1038/sj.clpt.6100054. [DOI] [PubMed] [Google Scholar]
- 5.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–32. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 6.Tucker GT, Houston JB, Huang SM. Optimizing drug development: strategies to assess drug metabolism/transporter interaction potential—towards a consensus. Br J Clin Pharmacol. 2001;52:107–17. doi: 10.1046/j.0306-5251.2001.temp.1441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Al Habet S, Baweja RK, Burckart GJ, Chung S, Colangelo P, Frucht D, Green MD, Hepp P, Karnaukhova E, Ko HS, Lee JI, Marroum PJ, Norden JM, Qiu W, Rahman A, Sobel S, Stifano T, Thummel K, Wei XX, Yasuda S, Zheng JH, Zhao H, Lesko LJ. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–70. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 8.Galetin A, Ito K, Hallifax D, Houston JB. CYP3A4 substrate selection and substitution in the prediction of potential drug–drug interactions. J Pharmacol Exp Ther. 2005;314:180–90. doi: 10.1124/jpet.104.082826. [DOI] [PubMed] [Google Scholar]
- 9.Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug–drug interactions. J Pharmacol Exp Ther. 2006;316:336–48. doi: 10.1124/jpet.105.093229. [DOI] [PubMed] [Google Scholar]
- 10.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach SR, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug–drug interaction studies: a PhRMA perspective. J Clin Pharmacol. 2003;43:443–69. [PubMed] [Google Scholar]
- 11.Streetman DS, Bertino JS, Jr, Nafziger AN. Phenotyping of drug-metabolizing enzymes in adults: a review of in-vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000;10:187–216. doi: 10.1097/00008571-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Rogers JF, Rocci ML, Jr, Haughey DB, Bertino JS., Jr An evaluation of the suitability of intravenous midazolam as an in vivo marker for hepatic cytochrome P4503A activity. Clin Pharmacol Ther. 2003;73:153–8. doi: 10.1067/mcp.2003.23. [DOI] [PubMed] [Google Scholar]
- 13.Greenblatt DJ, Abernethy DR, Locniskar A, Harmatz JS, Limjuco RA, Shader RI. Effect of age, gender, and obesity on midazolam kinetics. Anesthesiology. 1984;61:27–35. [PubMed] [Google Scholar]
- 14.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66:461–71. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- 15.Farkas D, Oleson LE, Zhou Y, Harmatz JS, Zinny MA, Court MH, Greenblatt DJ. Pomegranate juice does not impair clearance of oral or intravenous midazolam, a probe for cytochrome P450-3A activity: comparison with grapefruit juice. J Clin Pharmacol. 2007;47:286–94. doi: 10.1177/0091270006298359. [DOI] [PubMed] [Google Scholar]
- 16.Gorski JC, Vannaprasaht S, Hamman MA, Ambrosius WT, Bruce MA, Haehner-Daniels B, Hall SD. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74:275–87. doi: 10.1016/S0009-9236(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 17.Gorski JC, Jones DR, Haehner-Daniels BD, Hamman MA, O’Mara EM, Hall SD. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64:133–43. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
- 18.Lee JI, Chaves-Gnecco D, Amico JA, Kroboth PD, Wilson JW, Frye RF. Application of semisimultaneous midazolam administration for hepatic and intestinal cytochrome P450 3A phenotyping. Clin Pharmacol Ther. 2002;72:718–28. doi: 10.1067/mcp.2002.129068. [DOI] [PubMed] [Google Scholar]
- 19.Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76:250–69. doi: 10.1016/j.clpt.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Kupferschmidt HHT, Ha HR, Ziegler WH, Meier PJ, Krähenbühl S. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58:20–8. doi: 10.1016/0009-9236(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 21.Andersen V, Pedersen N, Larsen NE, Sonne J, Larsen S. Intestinal first pass metabolism of midazolam in liver cirrhosis—effect of grapefruit juice. Br J Clin Pharmacol. 2002;54:120–4. doi: 10.1046/j.1365-2125.2002.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veronese ML, Gillen LP, Burke JP, Dorval EP, Hauck WW, Pequignot E, Waldman SA, Greenberg HE. Exposure-dependent inhibition of intestinal and hepatic CYP3A4 in vivo by grapefruit juice. J Clin Pharmacol. 2003;43:831–9. doi: 10.1177/0091270003256059. [DOI] [PubMed] [Google Scholar]
- 23.Greenblatt DJ, von Moltke LL, Harmatz JS, Chen G, Weemhoff JL, Jen C, Kelley CJ, LeDuc BW, Zinny MA. Time-course of recovery of cytochrome P450 3A function after single doses of grapefruit juice. Clin Pharmacol Ther. 2003;74:121–9. doi: 10.1016/S0009-9236(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 24.Chien JY, Lucksiri A, Ernest CS, II, Gorski JC, Wrighton SA, Hall SD. Stochastic prediction of CYP3A-mediated inhibition of midazolam clearance by ketoconazole. Drug Metab Dispos. 2006;34:1208–19. doi: 10.1124/dmd.105.008730. [DOI] [PubMed] [Google Scholar]
- 25.Eap CB, Buclin T, Cucchia G, Zullino D, Hustert E, Bleiber G, Golay KP, Aubert AC, Baumann P, Telenti A, Kerb R. Oral administration of a low dose of midazolam (75 microg) as an in vivo probe for CYP3A activity. Eur J Clin Pharmacol. 2004;60:237–46. doi: 10.1007/s00228-004-0762-z. [DOI] [PubMed] [Google Scholar]
- 26.Chung E, Nafziger AN, Kazierad DJ, Bertino JS., Jr Comparison of midazolam and simvastatin as cytochrome P450 3A probes. Clin Pharmacol Ther. 2006;79:350–61. doi: 10.1016/j.clpt.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 27.McCrea J, Prueksaritanont T, Gertz BJ, Carides A, Gillen L, Antonello S, Brucker MJ, Miller-Stein C, Osborne B, Waldman S. Concurrent administration of the erythromycin breath test (EBT) and oral midazolam as in vivo probes for CYP3A activity. J Clin Pharmacol. 1999;39:1212–20. doi: 10.1177/00912709922012015. [DOI] [PubMed] [Google Scholar]
- 28.Greenblatt DJ, von Moltke LL, Harmatz JS, Durol ALB, Daily JP, Graf JA, Mertzanis P, Hoffman JL, Shader RI. Differential impairment of triazolam and zolpidem clearance by ritonavir. J Acquir Immune Defic Syndr. 2000;24:129–36. doi: 10.1097/00126334-200006010-00007. [DOI] [PubMed] [Google Scholar]
- 29.Wyen C, Fuhr U, Frank D, Aarnoutse R, Klaassen T, Lazar A, Seeringer A, Doroshyenko O, Kirchheiner J, Abdulrazik F, Schmeisser N, Lehmann C, Hein W, Schomig E, Burger D, Fatkenheuer G, Jetter A. Effect of an antiretroviral regimen containing ritonavir boosted lopinavir on intestinal and hepatic CYP3A, CYP2D6 and P-glycoprotein in HIV-infected patients. Clin Pharmacol Ther. 2008;84:75–82. doi: 10.1038/sj.clpt.6100452. [DOI] [PubMed] [Google Scholar]
- 30.Knox TA, Oleson L, von Moltke LL, Kaufman RA, Wanke CA, Greenblatt DJ. Ritonavir greatly impairs CYP3A activity in HIV infection with chronic viral hepatitis. J Acquir Immune Defic Syndr. 2008;49:358–68. doi: 10.1097/qai.0b013e31818c7efe. [DOI] [PubMed] [Google Scholar]
- 31.Mathias AA, West S, Hui J, Kearney BP. Dose–response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther. 2009;85:64–70. doi: 10.1038/clpt.2008.168. [DOI] [PubMed] [Google Scholar]
- 32.Kharasch E, Bedynek P, Walker A, Whittington D, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: II. Ritonavir effects on CYP3A and P-glycoprotein activities. Clin Pharmacol Ther. 2008;84:506–12. doi: 10.1038/clpt.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh RF, Gaver VE, Patterson KB, Rezk NL, Baxter-Meheux F, Blake MJ, Eron JJ, Jr, Klein CE, Rublein JC, Kashuba AD. Lopinavir/ritonavir induces the hepatic activity of cytochrome P450 enzymes CYP2C9, CYP2C19, and CYP1A2 but inhibits the hepatic and intestinal activity of CYP3A as measured by a phenotyping drug cocktail in healthy volunteers. J Acquir Immune Defic Syndr. 2006;42:52–60. doi: 10.1097/01.qai.0000219774.20174.64. [DOI] [PubMed] [Google Scholar]
- 34.Mouly S, Rizzo-Padoin N, Simoneau G, Verstuyft C, Aymard G, Salvat C, Mahe I, Bergmann JF. Effect of widely used combinations of antiretroviral therapy on liver CYP3A4 activity in HIV-infected patients. Br J Clin Pharmacol. 2006;62:200–9. doi: 10.1111/j.1365-2125.2006.02637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fellay J, Marzolini C, Decosterd L, Golay KP, Baumann P, Buclin T, Telenti A, Eap CB. Variations of CYP3A activity induced by antiretroviral treatment in HIV-1 infected patients. Eur J Clin Pharmacol. 2005;60:865–73. doi: 10.1007/s00228-004-0855-8. [DOI] [PubMed] [Google Scholar]
- 36.Nogueira CW, Zeni G, Rocha JB. Organoselenium and organotellurium compounds: toxicology and pharmacology. Chem Rev. 2004;104:6255–85. doi: 10.1021/cr0406559. [DOI] [PubMed] [Google Scholar]
- 37.Moutet M, d’Alessio P, Malette P, Devaux V, Chaudiere J. Glutathione peroxidase mimics prevent TNFalpha- and neutrophil-induced endothelial alterations. Free Radic Biol Med. 1998;25:270–81. doi: 10.1016/s0891-5849(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 38.d’Alessio P, Moutet M, Coudrier E, Darquenne S, Chaudiere J. ICAM-1 and VCAM-1 expression induced by TNF-alpha are inhibited by a glutathione peroxidase mimic. Free Radic Biol Med. 1998;24:979–87. doi: 10.1016/s0891-5849(97)00396-1. [DOI] [PubMed] [Google Scholar]
- 39.Gosgnach W, Messika-Zeitoun D, Gonzalez W, Philipe M, Michel JB. Shear stress induces iNOS expression in cultured smooth muscle cells: role of oxidative stress. Am J Physiol Cell Physiol. 2000;279:C1880–8. doi: 10.1152/ajpcell.2000.279.6.C1880. [DOI] [PubMed] [Google Scholar]
- 40.Castagne V, Clarke PG. Neuroprotective effects of a new glutathione peroxidase mimetic on neurons of the chick embryo's retina. J Neurosci Res. 2000;59:497–503. doi: 10.1002/(SICI)1097-4547(20000215)59:4<497::AID-JNR4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 41.Blum S, Asaf R, Guetta J, Miller-Lotan R, Asleh R, Kremer R, Levy NS, Berger FG, Aronson D, Fu X, Zhang R, Hazen SL, Levy AP. Haptoglobin genotype determines myocardial infarct size in diabetic mice. J Am Coll Cardiol. 2007;49:82–7. doi: 10.1016/j.jacc.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 42.Granda BW, Giancarlo GM, von Moltke LL, Greenblatt DJ. Analysis of ritonavir in plasma/serum and tissues by high-performance liquid chromatography. J Pharmacol Toxicol Methods. 1999;40:235–9. doi: 10.1016/s1056-8719(99)00013-1. [DOI] [PubMed] [Google Scholar]
- 43.Greenblatt DJ. Preparation of scientific reports on pharmacokinetic drug interaction studies. J Clin Psychopharmacol. 2008;28:369–73. doi: 10.1097/JCP.0b013e31817e63cd. [DOI] [PubMed] [Google Scholar]
- 44.Greenblatt DJ, von Moltke LL. Gender has a small but statistically significant effect on clearance of CYP3A substrate drugs. J Clin Pharmacol. 2008;48:1350–5. doi: 10.1177/0091270008323754. [DOI] [PubMed] [Google Scholar]
- 45.von Moltke LL, Greenblatt DJ, Grassi JM, Granda BW, Duan SX, Fogelman SM, Daily JP, Harmatz JS, Shader RI. Protease inhibitors as inhibitors of human cytochromes P450: high risk associated with ritonavir. J Clin Pharmacol. 1998;38:106–11. doi: 10.1002/j.1552-4604.1998.tb04398.x. [DOI] [PubMed] [Google Scholar]
- 46.Granfors MT, Wang JS, Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT. Differential inhibition of cytochrome P450 3A4, 3A5 and 3A7 by five human immunodeficiency virus (HIV) protease inhibitors in vitro. Basic Clin Pharmacol Toxicol. 2006;98:79–85. doi: 10.1111/j.1742-7843.2006.pto_249.x. [DOI] [PubMed] [Google Scholar]
- 47.Eagling VA, Back DJ, Barry MG. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J Clin Pharmacol. 1997;44:190–4. doi: 10.1046/j.1365-2125.1997.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Moltke LL, Durol ALB, Duan SX, Greenblatt DJ. Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: comparison with ketoconazole. Eur J Clin Pharmacol. 2000;56:259–61. doi: 10.1007/s002280000125. [DOI] [PubMed] [Google Scholar]
- 49.Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, Kati W, Stewart K, Lal R, Hsu A, Betebenner D, Korneyeva M, Vasavanonda S, McDonald E, Saldivar A, Wideburg N, Chen X, Niu P, Park C, Jayanti V, Grabowski B, Granneman GR, Sun E, Japour AJ, Leonard JH, Plattner JJ, Norbeck DW. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother. 1998;42:3218–24. doi: 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muirhead GJ, Wulff MB, Fielding A, Kleinermans D, Buss N. Pharmacokinetic interactions between sildenafil and saquinavir/ritonavir. Br J Clin Pharmacol. 2000;50:99–107. doi: 10.1046/j.1365-2125.2000.00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merry C, Barry MG, Mulcahy F, Ryan M, Heavey J, Tjia JF, Gibbons SE, Breckenridge AM, Back DJ. Saquinavir pharmacokinetics alone and in combination with ritonavir in HIV-infected patients. AIDS. 1997;11:F29–33. doi: 10.1097/00002030-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen DF, McDonald E, Kukulka MJ, Hsu A, Granneman GR, Baroldi PA, Sun E, Pizzuti D, Plattner JJ, Norbeck DW, Leonard LM. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob Agents Chemother. 1997;41:654–60. doi: 10.1128/aac.41.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenblatt DJ, von Moltke LL, Harmatz JS, Fogelman SM, Chen G, Graf JA, Mertzanis P, Byron S, Culm KA, Granda BW, Daily JP, Shader RI. Short term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol. 2003;43:414–22. doi: 10.1177/0091270003251864. [DOI] [PubMed] [Google Scholar]
- 54.Greenblatt DJ, von Moltke LL, Harmatz JS, Durol ALB, Daily JP, Graf JA, Mertzanis P, Hoffman JL, Shader RI. Alprazolam–ritonavir interaction: implications for product labeling. Clin Pharmacol Ther. 2000;67:335–41. doi: 10.1067/mcp.2000.105757. [DOI] [PubMed] [Google Scholar]
- 55.Culm-Merdek KE, von Moltke LL, Gan L, Horan KA, Reynolds R, Harmatz JS, Court MH, Greenblatt DJ. Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: comparison with ritonavir. Clin Pharm Ther. 2006;79:243–54. doi: 10.1016/j.clpt.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 56.Guidance for Industry. Drug Interaction Studies. G:/6695dft.doc, 9/8/06. Available at http://www.FDA.gov/CEDR/guidance/index.htm (last accessed 9 October 2009.