

Abstract
Mitochondrial disease confirmation and establishment of a specific molecular diagnosis requires extensive clinical and laboratory evaluation. Dual genome origins of mitochondrial disease, multi-organ system manifestations, and an ever increasing spectrum of recognized phenotypes represent the main diagnostic challenges. To overcome these obstacles, compiling information from a variety of diagnostic laboratory modalities can often provide sufficient evidence to establish an etiology. These include blood and tissue histochemical and analyte measurements, neuroimaging, provocative testing, enzymatic assays of tissue samples and cultured cells, as well as DNA analysis. As interpretation of results from these multifaceted investigations can become quite complex, the Diagnostic Committee of the Mitochondrial Medicine Society developed this review to provide an overview of currently available and emerging methodologies for the diagnosis of primary mitochondrial disease, primarily focusing on disorders characterized by impairment of oxidative phosphorylation. The aim of this work is to facilitate the diagnosis of mitochondrial disease by geneticists, neurologists, and other metabolic specialists who face the challenge of evaluating patients of all ages with suspected mitochondrial disease.
Keywords: Mitochondrial Disease, Laboratory Diagnosis, Review
Introduction
Primary mitochondrial respiratory chain disease is a heterogeneous group of disorders characterized by impaired energy metabolism due to presumed genetically-based oxidative phosphorylation (OXPHOS) dysfunction. While the diagnosis is usually suspected on clinical grounds [1], biochemical and/or molecular evaluations are often necessary to confirm a specific diagnosis. Two diagnostic schemes have been proposed for the confirmation of mitochondrial disease, both of which require extensive laboratory testing to determine whether a given patient is ‘definitively’ affected with a mitochondrial disease [2, 3]. Molecular genetic confirmation is important, when possible, to solidify the diagnosis, provide guidance on management and prognosis, and permit accurate recurrence risk counseling. Primary mitochondrial disorders may follow patterns of maternal inheritance (i.e., mitochondrial DNA (mtDNA) mutations) or classical Mendelian inheritance (i.e., nuclear DNA (nDNA) mutations). However, the majority of mitochondrial disease in pediatric patients is caused by nDNA mutations [4], most commonly following an autosomal recessive pattern of inheritance. The challenge lies in determining which one of dozens, if not hundreds, of genes spanning two genomes are causative for mitochondrial dysfunction in a given patient.
No uniform or standardized set of guidelines for the biochemical and molecular evaluation of the suspected mitochondrial disease patient currently exist. Different methods are used by different laboratories worldwide, with basic enzymatic assays critical for the objective evaluation of mitochondrial function not standardized among diagnostic centers [5]. In addition, while fresh tissue is preferable for study, this is often not an option due to geographical constraints [6]. Molecular diagnostic techniques have greater inter-laboratory reproducibility [7]. However, mtDNA analysis presents a particular diagnostic challenge in that false negative results may result from low level heteroplasmy in peripheral blood samples [8]. Various molecular methodologies are under evaluation by different laboratories to overcome these diagnostic challenges.
The aim of this review is to enable the metabolism specialist to successfully navigate through the complicated and rapidly evolving evaluation of the suspected mitochondrial disease patient. First-tier diagnostic testing are beyond the scope of this paper but have been addressed elsewhere [1]. Rather, here is provided an overview of in-depth biochemical and molecular testing options for mitochondrial disease that are either currently available or emerging on the horizon. Algorithms providing a systematic overview of these evaluations are provided in Figure 1. Continually updated information on diagnostic modalities for mitochondrial disease is available at www.mitosoc.org.
Figure 1.


The in-depth laboratory evaluation of suspected primary mitochondrial disease involves both minimally-invasive and invasively obtained specimens.
Figure 1a. Analyte analyses of plasma, urine, and CSF samples may identify abnormalities suggestive of mitochondrial disease.
Figure 1b. Overview of tissue-based evaluations in suspected primary mitochondrial respiratory chain disease.
Minimally-Invasive Biochemical Analyte Interpretation
Lactate and Pyruvate Analysis
Lactic acid elevation in blood (typically considered > 2.1 mM) or CSF can be an important, albeit non-specific, marker of mitochondrial disease. Mitochondrial disease patients, may however, have normal lactate and pyruvate levels except when undergoing a metabolic crisis or following exercise [9]. Furthermore, many patients with mitochondrial disease consistently have normal or only minimally elevated lactic acid levels, as occurs in mitochondrial polymerase gamma (POLG1)-associated diseases, Leber Hereditary Optic Neuropathy (LHON), Leigh disease, Kearns-Sayre syndrome, and complex I deficiency [10]. Conversely, elevations of plasma lactate and/or pyruvate levels may be seen in a range of conditions other than primary mitochondrial disease, including spurious elevation due to poor collection or handling techniques, physiological elevation as a result of secondary mitochondrial dysfunction which may occur in a wide range of systemic diseases and metabolic disorders, as well as nutritional deficiency of thiamine [11]. It is well known that CSF lactate can be increased in CNS infection, stroke, malignancy, inflammation, and seizures, which limits the value of this laboratory finding in the absence of other objective confirmatory findings [12].
Spurious elevation of plasma lactate is indeed the most common cause, resulting from either a patient (usually a child) struggling or the prolonged use of a tourniquet during sample collection. A useful strategy to resolve this problem is to collect the sample 30 minutes after placement of an intravenous catheter. Obtaining accurate pyruvate measurements can also be challenging but is necessary to quantify blood lactate/pyruvate ratios, which indirectly reflect the NAD+/NADH cytoplasmic redox state [9]. Pyruvate is quite unstable, necessitating blood specimens be immediately transferred (i.e., within 30 seconds after drawing) into 8% perchlorate on ice and analyzed. Pyruvate levels may increase or decrease depending on sample handling and are elevated in the immediate postprandial period. Blood and/or CSF pyruvate may be elevated in pyruvate metabolism defects such as pyruvate dehydrogenase deficiency, pyruvate carboxylase deficiency, or biotinidase deficiency. Elevated plasma alanine may be a useful indicator of long-standing pyruvate accumulation. Similarly, CSF lactate or pyruvate levels may be elevated without blood elevation in mitochondrial disease patients with predominant brain manifestations [13]. While defects of pyruvate metabolism are genetically-based mitochondrial diseases that clearly fall high in the differential of primary oxidative phosphorylation dysfunction, excellent discussions of these disorders are available elsewhere [14, 15].
Amino Acid Analysis
Amino acid analysis is performed using several methodologies, each with its own strengths and weaknesses. The most accurate method uses ion exchange chromatography with post-column derivitization using an automated amino acid analyzer. Although this method allows for the most comprehensive amino acid quantifications, it has the longest analysis time. Tandem mass spectrometry (MS/MS) is an increasingly popular method for amino acid analysis due to its relative low cost and high throughput, though it poorly quantifies glycine [16], cystine, homocystine, lysine, tryptophan, and GABA; most of these are neurotransmitters and/or involved in methylation reactions. Reverse-phase HPLC is another relatively sensitive and high throughput method for amino acid analysis, but also poorly quantifies tryptophan and cystine.
Quantitative amino acid analysis is not utilized as often as it might be to specifically diagnose disorders of mitochondrial metabolism. However, this testing can be a useful adjunct when alanine elevations are present in both plasma and CSF, especially when hyperalaninemia occurs in a fasting specimen (since mild elevations can occur from recent ingestion of a carbohydrate-rich meal). It is possible to discern relative alanine elevation by comparing it with the essential amino acids lysine (a normal alanine: lysine ratio < 3:1, with values above this indicating true hyperalaninemia) and alanine: phenylalanine + tyrosine (normal ratio < 4:1) [17]. An absolute elevation in alanine above 450 uM is a factor utilized to determine the likelihood of mitochondrial disease in the Nijmegen diagnostic protocol [3]. Urine amino acid analysis is typically performed when low serum bicarbonate is present. Generalized aminoaciduria along with renal tubular acidosis and glycosuria comprises renal Fanconi syndrome, which can be seen in mitochondrial disease, particularly those involving mtDNA deletions [18, 19].
Similar to other biochemical markers of mitochondrial dysfunction, the sensitivity of an elevated alanine for mitochondrial disease is low, as it may only be elevated at certain times as during a physiologic stress or regression. Thus, normal alanine levels do not exclude the diagnosis of mitochondrial disease. The strength of incorporating alanine measurement in the evaluation of OXPHOS disease is the relative resistance of amino acid quantitation to artifact from improper specimen collection. Other amino acids whose elevations have been associated with mitochondrial dysfunction include proline, glycine, and sarcosine [17]. It is becoming increasingly recognized that some mitochondrial disease patients with severe infantile-onset liver disease due to one of the mtDNA depletion syndromes have elevated tyrosine on newborn screening studies, but without a rise in succinylacetone as would be diagnostic of type 1 Tyrosinemia.
Amino acid quantification can be performed on blood (plasma or serum), urine, and CSF, although testing of urine is typically only helpful in diagnosing a mitochondrial disease-associated tubulopathy. Blood specimens are collected in a heparin or EDTA tube with plasma separated promptly. All specimens need to be refrigerated. Quantitative methods are of higher diagnostic yield and more clinically meaningful than is qualitative analysis. Test results may indeed be affected by a variety of conditions, including improper specimen handling, hemolysis, and dietary intake. Improperly stored specimens cause artifactual elevations in glutamate (especially in relation to glutamine), aspartate, and ornithine, with decreases of glutamine, cystine, asparagine, and homocystine. Hemolysis causes artificially low arginine and elevated aspartate, glutamate, ornithine, phosphoserine, and taurine. Post-prandial specimens commonly result in generalized aminoacidemia including elevations in the branched chain amino acids (valine, isoleucine, and leucine) and alanine; a fasting specimen can avoid this pitfall.
Organic Acid Analysis
Organic acids are byproducts of protein, carbohydrate, and fat catabolism. Organic acid analysis can be performed in one of two ways. Qualitative analysis will identify inborn errors of metabolism with large excretions of diagnostic organic acids, while quantitative analysis permits identification of more subtle abnormalities and comparison between time points. Analysis is often performed by column gas chromatography followed by mass spectrometry, with quantification provided by ion ratio analysis or stable isotope dilution analysis. While the latter method is most accurate, it is not practical for routine screening and is only utilized to quantify specific organic acids (e.g. mevalonate quantitation for the diagnosis of mild mevalonic kinase deficiency or hyper IgD syndrome). Methods of organic acid quantification more suited for screening purposes include the mass spectrometry extracted ion ratio method and semi-quantitative analysis utilizing gas chromatography flame ionization detection with mass spectrometry for peak identification [20].
Urine is the preferred specimen for organic acid screening owing to the ready excretion and greater extraction efficiency in urine compared to plasma. Limitations to urine organic acid analysis are the artificially lower accuracy in very dilute urine specimens, and rarely, interfering peaks caused by drugs. Certain analytes with greater volatility such as propionic acid may be missed due to loss during sample preparation or elution with the solvent peak. Organic acid analysis can also be performed on CSF and plasma in select clinical scenarios. Plasma organic acid analysis rarely provides diagnostic information that would be missed by urine analysis, but may be of use for simultaneous quantification of lactate and ketone bodies in plasma and urine. Similarly, CSF organic acid analysis has utility in select clinical scenarios, such as in disorders of GABA metabolism.
Urine organic acid analysis is routinely obtained in infants with sudden-onset encephalopathy in an effort to identify amino and organic acidopathies, as well as certain fatty acid oxidation disorders. Urine organic analysis during a period of clinical stability may have a low sensitivity for detecting mitochondrial disease, as abnormalities are often only present when a patient is acutely symptomatic. Surprisingly, lactic acid elevation on urine organic acid analysis is not a good discriminator for mitochondrial cytopathies [6]. Increased excretion of tricarboxylic acid (TCA) cycle intermediates, ethylmalonic acid, and 3-methyl glutaconic acid commonly occur in mitochondrial disease, but are rarely diagnostic of a specific mitochondrial disorder [21-23]. Of note, renal immaturity is a common cause of TCA cycle intermediates elevation in the urine. Thus, abnormal urine organic acid results in infants less than 1 year of age should be interpreted with caution to assure reported normal values are age-specific. Another common finding on urine organic acid analysis in individuals with mitochondrial disease is dicarboxylic aciduria, arising as a result of microsomal fatty acid metabolism. This is caused by impairment of mitochondrial fatty acid β-oxidation due to primary deficiency or secondary inhibition. However, dicarboxylic aciduria may also result from dietary artifact (i.e., medium chain triglycerides), drugs, and prolonged fasting. Low muscle mass (caused by starvation or a variety of primary and secondary muscle disorders) and creatine synthesis defects can result in apparent false elevations of organic acids as the results are normalized to the concentration of creatinine in the urine.
Carnitine Analysis
Carnitine serves as a mitochondrial shuttle for free fatty acids and a key acceptor of potentially toxic coenzyme A (CoA) esters. It permits restoration of intramitochondrial CoA and removal from the mitochondrion of esterified intermediates by enabling their urinary excretion. Quantification of blood total and free carnitine levels, along with acyl-carnitine profiling permits identification of fatty acid oxidation defects, as well as some primary aminoacidemias and organic acidemias. In addition, this analysis enables identification of secondary fatty acid oxidation defects and carnitine deficiency which may occur in primary OXPHOS disorders.
Quantitative acyl-carnitine analysis is performed by either tandem mass spectrometry (MS/MS) or HPLC followed by electrospray ionization (ESI)-MS/MS. Traditional MS/MS uses a butyryl group for derivitization, which leads to loss of one-third of the acetyl-carnitine species. It may also result in either over- or under-quantification of certain acyl-species (e.g., valproyl and octanoyl carnitine) due to their sharing the same mass [24]. Plasma is obtained for analysis from specimens collected in an EDTA or heparin tube and frozen until time of analysis. A filter paper blood spot can be utilized, but different normal ranges must be used since long-chain acyl-carnitine species are present in higher proportions in cellular elements included on the spot. Urine acyl-carnitine profiles are often difficult to interpret because various acyl-forms are produced by the kidney and very long chain acyl-carnitines are not routinely excreted.
Once quantified, both the absolute values of the acyl-carnitine species and the ratios of certain acyl-carnitine esters may be useful in supporting a specific diagnosis. The test is limited by false negatives in individuals with total carnitine deficiency and elevations in carnitine treated patients, as well as in cases where the biochemical abnormality is of mild or fluctuating severity. See Figure 1a for an overview of findings present on minimally-invasive analyte screening in mitochondrial disease.
MRS-based Central Nervous System Biochemical Analysis
Nuclear magnetic resonance imaging (MRI) is an important modality in the evaluation of anatomical structures in neurometabolic disorders. Unfortunately, mitochondrial disease represents a class of disease in which MRI findings, if present, are non-specific or change over time, greatly lowering its diagnostic sensitivity [25-27]. However, a new neuroimaging technique, proton magnetic resonance spectroscopy (MRS) has evolved from which important metabolic information can be derived utilizing the same acquisition parameters needed for MRI. MRS detects the ability of small molecules to emit radio waves based on the nuclear spin of protons, neutrons, and atomic nuclei. Proton (1H) MRS is the most commonly used spectroscopy technique for neurometabolic evaluations.
Several compounds involved in mitochondrial physiology are detectable by MRS based upon variations in their chemical properties within electrical fields, thus making MRS a useful adjunct in the evaluation of suspected mitochondrial disease [26, 28-30]. Detectable metabolic compounds each emit a unique resonance frequency called chemical shift, which is expressed in parts per million (ppm) and is derived from Larmor frequency and coupling. The chemical shift peaks most commonly studied are lactate (1.33 ppm), N-acetyl-L-aspartate (2.02 ppm), succinate (2.39/2.40 ppm), total creatine (3.03 ppm), choline (3.22 ppm), and myo-inositol (3.55 pm). Peak area is proportional to the number of spins that produce the signal and is a very rough estimate of metabolite concentration.
MRS interpretation based simply upon visual peak inspection is not a reliable method, as molecules may share similar chemical shifts or have altered magnetic behavior at various time to echo (TE) measurements. The use of varying TE times may allow certain peaks to become more evident, depending on the magnet strength (1.5 Tesla versus 3.0 Tesla MRI machines). When 1.5 Tesla MRI machines are used, the lipid resonance at 1.0 – 1.7 ppm may mask the lactate peak resonance at 1.33 ppm at short TE times (i.e., 35 milliseconds). However, when combined with intermediate TE times (i.e., 135 milliseconds), the lactate peak inverts to become distinct. Due to a narrower bandwidth, the inversion of lactate at 135 TE is not as distinct when using 3.0 Tesla machines; therefore, a longer TE time (i.e., 288 milliseconds) may be required to reduce the contaminating lipid material and define lactate. Currently, most clinical spectroscopy centers report peak area ratios because the MRS signals they acquire are not calibrated (Figures 2 and 3). Ideally, each machine should be calibrated prior to clinical use with pure compounds of known concentrations to standardize each metabolite measurement both within a particular machine and between different machines. In addition, precise computer models to determine area under the peak should be utilized to properly interpret pure metabolite concentrations and remove possible artifactual contaminants. Until such precision becomes routine in signal interpretation and reporting, comparisons will not be fully reliable between different spectroscopy centers and the full utility of MRS will not be achieved. Lactate is not detected above the lipid background in normal infants, children, and adults. When metabolism shifts to anaerobic glycolysis in mitochondrial respiratory chain deficiencies, however, lactate levels become increased in brain tissue (Figure 2 and 3). Barkovich et al detected lactate (Lac) peaks in 5 of 5 patients diagnosed with either Leigh syndrome or mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) [26]. In additional studies by 2 differerent groups, Lac peaks were seen in all 13 patients with MELAS who were studied [31, 32], as well as in 18 of 21 patients with a variety of respiratory chain deficiencies [33]. However, just as blood lactate levels have only a moderate sensitivity for mitochondrial disease, brain Lac peaks on MRS are also not highly sensitive. Two studies showed the sensitivity of Lac peaks in mitochondrial disease to be 18 % to 27% [34, 35]. Lac peaks vary depending on whether a patient is undergoing an exacerbation of their disease [30], as well as whether their lesions are acute, subacute, or chronic [28]. Detection of Lac signal may also depend on its concentration, as the threshold of detection by MRS is 0.5 mM to 1 mM [34, 35]. Absence of a Lac peak similarly does not rule-out mitochondrial disease, as the specific tissues involved in mitochondrial disease vary, as does the location of brain involvement. MRS specificity for mitochondrial disease is further limited by the possibility of other conditions giving rise to Lac peaks [35]. However, in the scenario of clinical and biochemical suspicion for mitochondrial disease, identification of a Lac peak would add weight to its possibility and potentially provide evidence to substantiate a more invasive diagnostic evaluation.
Figure 2.
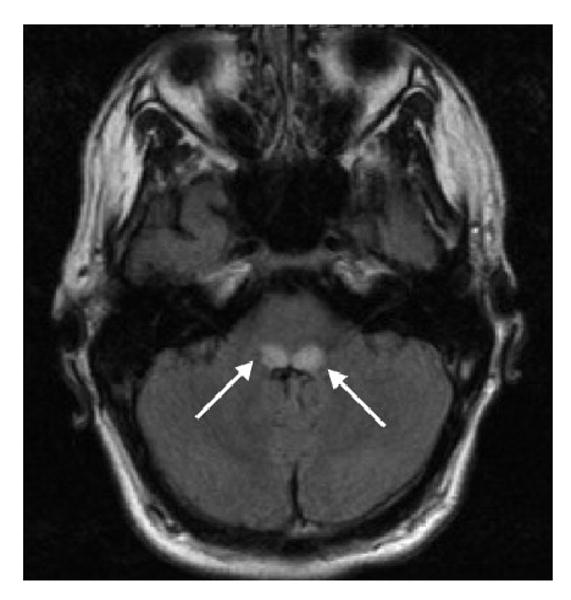
Brain MRI demonstrating an axial FLAIR sequence of a 17-year-old boy with new-onset respiratory difficulty, episodic vomiting, and extreme fatigue. This figure represents the symmetric regions of elevated T2/FLAIR signal located within the tegmentum (arrows). The diagnosis was Leigh syndrome.
Figure 3.
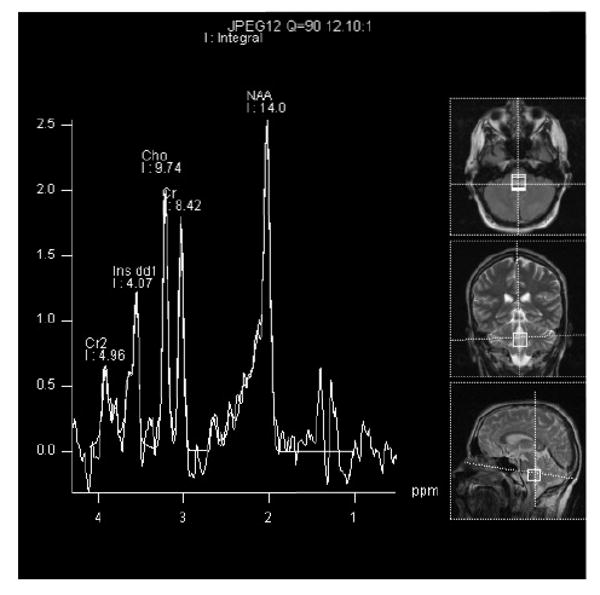
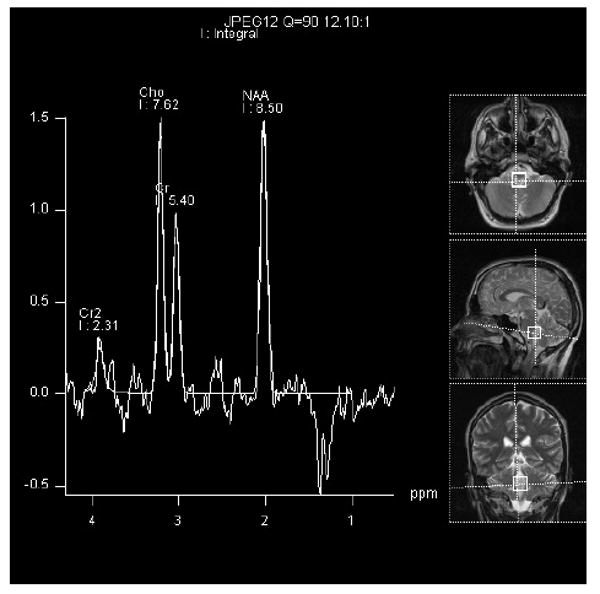
Figure 3A. This brain MRS scan represents a 1.6 × 1.6 cm voxel within the hyperintense FLAIR signal region. The exact location can be seen on the MRI images represented on the right of the picture. The time to echo (TE) was 35 msec. There is a upward lactate peak (Lac) located at 1.33 ppm, N-acetyl-L-aspartate (NAA) peak at 2.02 ppm, total creatine peak (Cr) at 3.03 ppm, choline peak (Cho) at 3.2 ppm, and inositol peak (Ins) at 3.8 ppm. It is likely that the voxel may include some cerebral spinal fluid. There was no detectable lactate in the cerebral spinal fluid several days before the image was acquired.
Figure 3b. This MRS scan represents the same 1.6 × 1.6 cm voxel as figure 3A. The time to echo (TE) was 135 msec. Notice the downward Lac peak at 1.3 ppm, while the NAA peak at 2.02 ppm, Cr peak at 3.03 ppm, and Cho peak at 3.2 ppm remain upward. At this TE, the Ins peak is not well visualized.
N-acetyl-L-aspartate (NAA) predominately localizes to neurons, axons, and dendrites [36]. MRS measurements of NAA appear to be one of the best surrogate biomarkers for neuronal integrity. NAA is synthesized within the mitochondria in an energy dependent fashion and may represent a functional mitochondrial marker within neuronal populations. A decrease in NAA levels when normalized to creatine may reflect mitochondrial disease [37]. The ratio of each metabolite to creatine is used as a standard reporting value due to fairly uniform CNS creatine levels in most individuals. In 15 patients with mitochondrial disease syndromes, NAA/Cr ratio reductions were seen in cerebellum in 93% and in cortical gray matter regions in 87%, even at times when these areas appeared normal on MRI [29]. Furthermore, a strict regional expression of NAA was seen, with no decrease in the NAA/Cr signal in white matter. Yet, in another series of 16 patients with definitive mitochondrial disease consisting of either known syndromes or respiratory chain deficiencies, a decreased NAA/Cr ratio was seen in 11 patients (69%) in both gray and white matter regions [30]. Interestingly, the NAA/Cr ratio signal changes were only found in those patients with detectable Lac peaks. In some studies, the apparent decrease in the NAA/Cr signal was reversible [38, 39]. As with elevated Lac, decreased NAA is not specific for mitochondrial, or even metabolic disease because other disorders may also present with NAA signal alterations [35, 40]. However, additional clinical and biochemical evaluation may often segregate these disorders from primary mitochondrial disease.
The choline (Cho) signal on MRS is comprised of a group of components including free choline, phosphorylcholine, and phosphatidylcholine which constitute brain myelin and allow fluidity of cell membranes [41, 42]. However, the complete metabolite profile contributing to the Cho signal is not certain [43]. Cho elevation reflects membrane turnover and demyelination. Cho signal changes have not been well reported in patients with mitochondrial disease. Whether this represents no change or a heterogeneous change that has not been appreciated due to small sample sizes is unclear. One study did demonstrate reductions in the Cho/Cr ratio in 40% of patients with reduced NAA/Cr and in 53% of patients with reduced NAA/Cr ratio and elevated Lac [30].
Succinate is a tricarboxylic acid cycle component present in brain at low concentrations, on the order of 0.5 mmol/kg wet weight [44]. In proton MRS, the resonance of succinate overlaps with those of glutamate and glutamine [43]. Under normal conditions, the succinate peak is not visible on MRS [45]. Deficient respiratory chain activity produces significant increases in succinate concentration to permit its detection by MRS. In 3 patients with complex II deficiency, a very large succinate signal along with reduced NAA/Cr and increased Lac signals were observed within white matter, while only a decrease in NAA/Cr ratio was seen within cortical gray matter [45]. This highlights the importance of utilizing multivoxel acquisition, or at least using single voxels over both gray and white matter in the MRS evaluation of respiratory chain disease. This study also raises the possibility that an enlarged 2.4 ppm succinate peak may be specific for complex II disease.
Proton MRS represents a useful tool for the non-invasive investigation of neurometabolic disorders, in particular mitochondrial disease. Additional information is obtained over conventional MRI, as metabolic changes can be visualized even in areas of brain that appear structurally normal. Conventional MRI acquisition is used and can be added to routine MRI studies when MRS is requested in advance [46]. Similar to other modalities of investigation for mitochondrial disease, the disease acuity, specific disease type, involvement of specific brain regions, and timing of testing influence the utility of recovered data. In the correct context, MRS can greatly increase the sensitivity of the diagnostic evaluation for mitochondrial disease.
Provocative Clinical Testing
Carbohydrate loading with glucose or fructose followed by serial measurements of plasma lactate, pyruvate, and alanine can unmask mitochondrial disease. Fasting studies carry greater risk but can reveal a tendency toward hypoglycemia or secondary fatty acid oxidation defects. Both of these diagnostic approaches can be used to assist in the selection of a diet that will minimize lactic or pyruvic acidosis. In addition, they can help to confirm the safety in a given patient of standard interventions, such as high fat or ketogenic diets [47].
Exercise testing is increasingly employed as a clinical diagnostic test for mitochondrial myopathy [48]. Bicycle and treadmill ergometry may demonstrate decreased exercise tolerance, excessive lactate production, and slow clearance of accumulated plasma lactate. In addition, performing forearm exercise testing coupled with venous oxygen saturation measurements in a patient with mitochondrial disease will reveal “arterialized” venous blood. This occurs because mitochondrial dysfunction in skeletal muscle results in its inability to extract oxygen from blood [49, 50].
Invasive Tissue Investigations
The first decision to be made when invasive procedures are deemed necessary is the selection of which tissue to investigate for evidence of mitochondrial dysfunction. The best choice is the tissue most profoundly affected by the disease process in a given patient [51]. Although skin biopsy seems preferable to muscle biopsy due to its relatively lower invasiveness, it is common for many children who have an OXPHOS defect detectable in skeletal muscle to have normal respiratory chain enzyme activities in cultured skin fibroblasts [52]. Skeletal muscle is commonly affected in primary mitochondrial disease. While this has made skeletal muscle the most widely used tissue for OXPHOS enzyme studies, it also has limitations. Many patients have been reported with detectable enzyme defects in liver [53] or cardiac muscle [54] but normal skeletal muscle activities. In general, whatever the affected organ, it is important to perform a skin biopsy at the time of tissue biopsy to obtain cultured fibroblasts for possible later study. The following discussion largely focuses on the multifaceted investigation of skeletal muscle, with subsequent brief overview of the utility of cardiac and liver analyses.
Skeletal Muscle Analysis
Muscle biopsy allows for pathological, molecular and biochemical study. Portions of the biopsy are utilized for histochemical, immunohistochemical, and even ultrastructural study to evaluate for morphologic evidence of primary OXPHOS disease, evidence of other conditions in the differential diagnosis, or pathological changes in the muscle that could produce secondary abnormalities in muscle biochemistry. Muscle tissue is also used for respiratory chain enzyme activity analysis and high resolution respirometry studies of integrated OXPHOS capacity, for mitochondrial isolation for biochemical testing including analysis of enzyme activities and integrated OXPHOS capacity, and for the extraction of DNA for genetic testing.
Morphologic Analysis of Skeletal Muscle
The presence of mitochondrial proliferation within myofibers is highly suggestive of a mitochondrial OXPHOS disorder. Traditionally, this has been demonstrated as ragged-red fibers (RRF) seen on modified Gomori trichrome staining as red granular deposits of mitochondria in the subsarcolemmal space in the background of a varying degree of muscle fiber atrophy. Despite the importance of this finding in mitochondrial disease, ragged red fibers may occur in many other primary muscle disorders. Other useful histochemical stains for analysis of mitochondrial enzyme activity are NADH dehydrogenase, succinate dehydrogenase (SDH), and cytochrome c oxidase (COX) [55, 56]. SDH staining evaluates complex II, which is a respiratory chain component encoded entirely by nuclear genes, and may also identify subsarcolemmal mitochondrial accumulation. In comparison, the COX reaction evaluates complex IV, which is a respiratory chain component encoded by both mitochondrial and nuclear genomes. With sequential application of these two reactions to a single muscle section, abnormal COX-deficient fibers will appear blue among normal COX activity fibers which appear brown. This approach facilitates the detection of abnormal fibers which might otherwise go undetected in a background of normal COX activity [57].
RRF are rarely seen in childhood, as it seems to take time for mitochondrial accumulation and muscle fiber deterioration to reach this stage. Subsarcolemmal accumulations of mitochondria, representing a milder manifestation of mitochondrial proliferation, are more common than RRF in pediatric patients. Although a valuable finding when present, mitochondrial proliferation was absent in 35% of 113 pediatric patients with proven mitochondrial dysfunction [58]. Especially in children, COX-deficient fibers sometimes outnumber RRF and may be the only abnormal finding in the muscle biopsy [59]. Neither the presence of RRF or focal loss of COX activity is disease-specific. Rather, they may appear in skeletal muscle as an age-related phenomenon as well as a secondary phenomenon infrequently seen in other disorders such as muscular dystrophies, myotonic dystrophy, inflammatory myopathies, glycogenoses, and congenital myopathies [59]. Other pathological features which may be seen in skeletal muscle in OXPHOS disorders are more non-specific, including neurogenic atrophy, internal nuclei, abnormal variation in fiber size, and accumulations of glycogen or lipid [60, 61]. Staining for glycogen and lipid remain important to evaluate for primary glycogen or lipid storage disorders. Rhabdomyolysis and dystrophic changes are rare in mitochondrial OXPHOS disorders.
The presence of RRF having strong subsarcolemmal SDH activity and low COX activity is typical of disorders due to mtDNA deletions (i.e., Kearns-Sayre Syndrome or progressive external ophthalmoplegia (PEO)) or tRNA mutations (i.e., myoclonic epilepsy and ragged red fibers (MERRF)) which impair mitochondrial protein synthesis [58]. COX deficiency occurs when wildtype mtDNA levels fall below the threshold necessary for COX protein subunit expression. None or only a few COX-deficient fibers may be present despite high percentages of an mtDNA mutation if there is an even distribution of mutant and wild-type mtDNA throughout the fiber. An example of this is classic MELAS due to an A3243G tRNALeu gene mutation in which RRF are often COX-positive. An increase in vascular smooth muscle SDH activity frequently is also seen in MELAS [62].
A mosaic and segmental pattern of COX activity is highly indicative of a heteroplasmic mtDNA disorder. In contrast, a global decrease in the activity of COX throughout the length of the muscle fibers is usually suggestive of a mutation in a nuclear gene encoding one of the proteins required for COX assembly and function, such as SURF1. However, a similar pattern could be observed in some patients presenting with homoplasmic tRNA mutations. If COX activity is diffusely decreased but spares muscle spindles and vascular smooth muscle, diagnostic considerations should include both the fatal and benign forms of infantile COX-deficient myopathy [63]. Mutations in the mtDNA protein-encoding genes are occasionally associated with RRF but have COX-positive fibers (unless the COX genes themselves are the site of mutation). This is the case for disorders associated with mutations in cytochrome b, COX I-III, and some mutations in complex I (ND1-6, ND4L) genes. In contrast, mutations associated with NARP/MILS (T8993G/C mutation in the ATPase 6 mtDNA gene) and ND mutations associated with LHON do not usually reveal any specific changes on muscle biopsy.
Mitochondrial myopathy is uncommon in OXPHOS disorders due to nDNA mutations and is usually not seen in association with mutations in nuclear-encoded complex I and II subunits, which are often but not exclusively associated with Leigh syndrome. Exceptions are some patients with adPEO, mtDNA depletion disorders, and myopathic forms of CoQ10 deficiency, as well as early biopsies from children with the benign form of infantile COX-deficient myopathy [64].
Electron microscopy study of ultrastructural changes in muscle has been questioned by some authors regarding its value in the diagnostic evaluation of mitochondrial myopathies [58]. Although characteristic ultrastructural abnormalities such as increased mitochondrial number and size, distorted or absent cristae, and osmophilic or paracrystalline inclusions are well-documented in patients with mitochondrial disorders [65], these changes are non-specific and may be seen in other myopathic and neuropathic diseases [60]. However, electron microscopy can identify structurally abnormal mitochondria when histochemistry is unhelpful. Ultrastructural changes of mitochondria have been described in muscle biopsies of patients who have no RRF or COX deficient fibers [60].
Biochemical Analysis of Skeletal Muscle
Only a small proportion of children investigated for a suspected mitochondrial disorder have classical syndromic presentations (i.e., Kearns-Sayre syndrome, Pearson syndrome, Leigh syndrome, MELAS, MERRF, or LHON). Furthermore, a pediatric patient's clinical picture is rarely specific for a single genetic defect and muscle histology is most frequently normal or shows only non-specific changes [51]. It is estimated that over 1,000 genes are involved in the proper functioning of the mitochondrion, many of which have not yet been characterized in humans [66]. All of this taken together generally makes biochemical study of tissue a prerequisite to direct further molecular genetic investigations in pediatric mitochondrial disease.
Biochemical investigations commonly include spectrophotometric assays of enzyme activity as well as functional studies of intact mitochondria, each providing useful and complementary clues to the diagnosis of an OXPHOS disorder [67]. The measurement of enzyme activities by spectrophotometric methods can be performed in isolated mitochondria from tissues or cultured cells, in tissue homogenate, or in whole cells. Spectrophotometric data gives valuable information about maximal enzyme activities of the catalytic component of the various respiratory complexes following either detergent- or freeze-thaw disruption of the inner mitochondrial membrane and are generally quite easy to reproduce and interpret within a given laboratory [68, 69]. Unfortunately, no universally agreed standardization exists for these assays or assay conditions, and inter-laboratory variability in test results is common. Indeed, a recent sample exchange program among European laboratories found wide variation in assay results on the same samples [5]. Another important limitation is that activities are determined under in vitro conditions, which do not correspond to the physiological, cytosolic environment of mitochondria.
Spectrophotometric-based activities of the different electron transport chain (ETC) enzyme complexes can be studied in isolation as complex I (NADH-ubiquinone oxidoreductase), complex II (succinate-ubiquinone oxidoreductase), complex III (decylubiquinone-cytochrome c reductase) or complex IV (cytochrome c oxidase), or they can be studied together as complex I+III (NADH-cytochrome c reductase) or complex II+III (succinate-cytochrome c reductase). While not often pursued routinely, direct assays of ATP synthase (complex V) hydrolytic activity are available by spectrophotometric analysis [22]. To help distinguish primary electron transport chain defects from secondary deficiencies, activity measurements are reported relative to a marker enzyme, such as citrate synthase, or as internal ratios rather than relative to protein concentration [52, 69, 70]. These analyses may yield a wide range of results in patients with mitochondrial OXPHOS disorders, with some patient samples showing normal ETC enzyme activities. This apparent paradox may result from the fact that assays use artificial substrates in disrupted mitochondrial preparations, as compared to intact cellular mitochondrial respiration that takes place in a stereochemical environment of multi-enzyme complexes and supercomplexes. In addition, high biological variability of these assays can make clear identification of a single deficiency difficult. However, isolated defects involving one complex may suggest a mutation of either an mtDNA-encoded or nDNA encoded subunit or assembly factor of that particular complex (i.e., SURF1 mutation in COX deficiency). Partial enzyme deficiencies involving complex I, III and IV are typical of patients with a generalized translation defect due to either mtDNA deletions or tRNA mutations [71, 72]. Multiple enzyme defects are also seen in mitochondrial polymerase defects. Exceptions exist, as complex I activity may be preferentially decreased in muscle from patients with MELAS and COX activity may be preferentially decreased in patients with MERRF, MELAS and in mitochondrial polymerase deficiency. The combined measurements of I+III and II+III provide useful information in the diagnosis of the potentially treatable coenzyme Q10 deficiency disorders or disorders that affect CoQ10 binding/electron exchange, which can subsequently be confirmed with coenzyme Q10 quantification in muscle [73, 74].
Polarographic studies measure oxygen consumption by isolated muscle mitochondria using a Clark electrode in the presence of various substrates (e.g., malate+pyruvate or malate+glutamate to donate NADH, succinate to donate FADH2, or TMPD+ascorbate to donate electrons directly to cytochrome C) [61, 70]. In patients with complex I deficiency, impaired respiration with NADH–producing substrates is seen while oxidation rates in the presence of the other substrates are normal. Patients with complex II deficiency show reduced oxygen consumption only with FADH2 producing substrates. Low respiratory rates with both NADH- and FADH2-producing substrates but normal oxidation of durohydroquinone and TMPD+ascorbate suggest a deficiency of coenzyme Q10, whereas low respiratory rates with both NADH- and FADH2-producing substrates with low oxidation of durohydroquinone suggests a deficiency of complex III. Diminished oxidation rates in the presence of all substrates tested is suggestive of a complex IV deficiency. In addition, polarographic studies may indicate a possible pyruvate dehydrogenase complex (PDHC) deficiency by finding a reduced oxidation of pyruvate in the presence of normal oxidation of glutamate [75].
Another means of measuring respiratory chain function is to use radioactively labeled substrates (e.g., [1-14C]pyruvate, [U-14C]malate and [1,4-14C]succinate) in the absence or presence of various inhibitors to measure production of 14CO2 and ATP in relation to citrate synthase activity as a marker of mitochondrial content. Various ratios can be determined in order to pinpoint the deficiency to a single respiratory chain complex. In experienced laboratories, this is an excellent and very exact approach [76].
In order to measure muscle mitochondria in their physiologic environment and to minimize sample sizes, a method using permeabilized muscle fibers has been developed. Single muscle fibres are permeabilised with saponin and placed in a high-resolution polarographic chamber where various substrates (as above) can be added to quantify oxygen consumption rates. Fifty mg of muscle is sufficient for multiple assays with up to 7 different substrates. Originally, the results were expressed normalized to wet weight of the muscle fibers, although some investigators have found this approach is not as sensitive as oxidation of radioactively labeled substrates in the diagnosis of respiratory chain deficiency (Wolf NI, unpublished results). Whether these results can be improved in comparison to one of the established techniques by using citrate synthase as a reference enzyme, has yet to be demonstrated [77].
While fresh muscle tissue is widely regarded as preferable for biochemical analyses, due to many factors including geographical constraint, frozen muscle must frequently suffice. A fresh muscle biopsy has the advantage that integrated functional studies of isolated mitochondria can be performed by various methods, such as by measuring oxygen consumption with polarography, substrate oxidation, or ATP production rates. In addition, high resolution respirometry of permeabilized muscle fibers offers the opportunity to use polarography to study fresh, intact muscle biopsy specimens obtained by needle biopsy of muscle or liver that weigh as little as 2 milligrams [78, 79]. All of these functional studies permit the ability to assess integrated activities of all complexes in the OXPHOS system in either intact permeabilized cells [78] or in isolated mitochondria under conditions which are much closer to the in vivo situation than are isolated ETC enzyme assays [80, 81]. This type of measurement may also identify mitochondrial defects that are not detectable with enzymatic analysis of the ETC on frozen samples [82, 83]. Polarographic assay permits identification of coenzyme Q10 synthetic defects when low oxygen utilization of both NADH and FADH-producing substrates is seen [84, 85]. Functional studies may also detect abnormalities in PDHC deficiency, tricarboxylic acid cycle enzymes, beta-oxidation, coenzymes, complex V, and transmembrane carriers [51]. Indeed, deficiency of two different mitochondrial membrane transporters involving the carriers for pyruvate and phosphate were demonstrated using functional measurements in fresh muscle [86, 87]. In a recent study, 30% of patients with abnormal functional studies on fresh mitochondria had normal single enzyme activities; these would have been missed had only frozen muscle had been studied [76].
Coenzyme Q10 Quantification
Coenzyme Q10 serves multiple intracellular redox functions, with an important role in mitochondrial electron transport as a mobile electron carrier to shuttle electrons to complex III from I, II, electron transfer factor (ETF), or other electron donors. It also has proxidant functions [88], as well as the ability to recycle other antioxidants including tocopherol and ascorbate. Primary coenzyme Q10 deficiency, which results from deficiencies in enzymes needed for its synthesis, has four currently recognized phenotypes: i) an encephalomyopathy with exercise intolerance, myopathy, myoglobinuria, seizures, and ataxia, ii) severe infantile encephalopathy with renal tubulopathy, iii) myopathic form with exercise intolerance, myopathy, and rhabdomyolysis, and iv) encephalopathy with ataxia, seizures, and basal ganglia disease [89, 90]. While primary deficiencies of this critical cofactor results in these serious disorders, primary coenzyme Q10 deficiency is rare. In addition to clinical findings, establishing this diagnosis requires detection of impaired coenzyme Q10-dependent respiratory chain activity and a tissue-specific reduction of coenzyme Q10 levels. Plasma levels of coenzyme Q10 are usually normal in primary muscle coenzyme Q10 deficiency. Secondary coenzyme Q10 deficiency in muscle was first reported along with a treatment response in 1986 in Kearns-Sayre syndrome [91]. Since then evidence has accumulated in several studies that a subset of mitochondrial disease patients have muscle coenzyme Q10 deficiency [92].
The concentration of coenzyme Q10 varies greatly in tissues, with heart muscle having the highest content at 114 ug/gram [93]. Analysis of coenzyme Q10 in human plasma has been available in commercial laboratories for over a decade. Plasma control ranges vary between laboratories [94]. Because coenzyme Q10 is primarily lipoprotein bound, the free coenzyme Q10 is related to the concentration of cholesterol, with normative values established as a function of age [95]. Skeletal muscle coenzyme Q10 concentrations in healthy children have been reported over a two-fold range [96]. Leukocytes (and/or lymphocytes) and platelets are other tissues in which coenzyme Q10 can be measured, and may more accurately reflect, compared with plasma levels, the true tissue concentration of coenzyme Q10. A good correlation between mononuclear white blood cells and muscle coenzyme Q10 levels was demonstrated in one study performed on 12 control samples and one deficient patient [94].
Cardiac Muscle Analysis
A variety of defects in mitochondrial oxidative phosphorylation have been described in both hypertrophic and dilated cardiomyopathy [97, 98]. In some cases, dilated cardiomyopathy merely reflects a final common pathway of pump failure arising from many different causes, including hypertrophic cardiomyopathies. Chamber dilation, with its resultant increase in wall tension, represents a final common physiological response to balance cardiac output and blood pressure through equilibrium of Laplace (pressure-volume) and Frank-Starling (wall tension-force) principles. Most patients have multisystemic dysfunction with isolated cardiomyopathy rarely described [70, 99]. What has been difficult to prove is in which patients, was mitochondrial dysfunction primary or secondary to a failing heart [100, 101]. Several studies have demonstrated mitochondrial dysfunction in both cardiac aging and cardiac disease [102, 103]. Therefore, cardiac muscle biopsy may reveal respiratory chain defects that are secondary to primary mechanical or coronary vascular insufficiency. Only by including the evaluation of skeletal muscle or other affected tissues for respiratory chain defects can primary and secondary OXPHOS defects be resolved [98]. In a severely ill neonate, infant or young child, direct gene testing may be helpful when conditions preclude biopsy as mutations in SCO2, COX-10, and COX-15 can give rise to cardiac hypertrophy. Animal studies suggest that both structural and respiratory chain differences may be related to intermyofibillar and subsarcolemmal location of heart mitochondria [102, 104]. Increased yield for respiratory chain defects would therefore, include isolation of intermyofibillar and subsarcolemmal mitochondria for assay. Given the high rate of mitochondrial dysfunction in cardiac disease, isolated abnormalities derived solely from heart tissue should be viewed with caution.
Liver Analysis
When liver dysfunction is clinically evident and a diagnosis of hepatic or hepatocerebral mitochondrial disease is considered, the least invasive initial assessment is blood DNA testing for POLG, dGUOK, or MPV17 mutations. Although less common, SCO1 and SUCLA2 may also be considered if ketoacidosis (SCO1) or myopathy (SCO1 and SUCLA2) is also present (Table 1). A liver biopsy is indicated either when the diagnosis cannot be established with these initial DNA studies or when the severity of clinical liver disease demands simultaneous biopsy and DNA studies. DNA studies may take up to two months for results, whereas biopsy can produce histological results in a few days. Needle biopsy typically yields sufficient tissue for histology, electron microscopy, and miniaturized respiratory chain polarography and biochemistry.
Table 1.
Detailed compilation of biochemical and clinical findings associated with pathogenic mutations in nuclear DNA genes currently implicated in primary mitochondrial diseases.
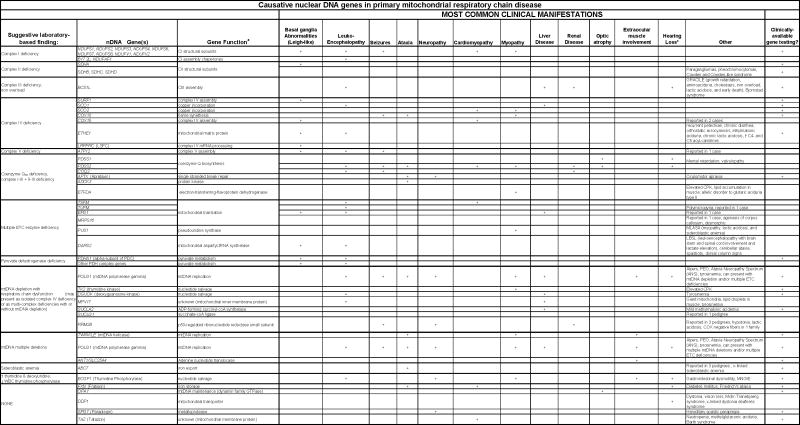 |
Sensorineural hearing loss (most commonly localized to the central brainstem or peripheral cochlear in pediatric and adult patients, respectively) can be seen in virtually any mitochondrial disease, although it is rarely the presenting complaint
Footnote: Citations for Table 1. Complex I structural subunits [151], B17.2L & NDUFAF1 [151], SDHA [152], SDHC [153], SDHD [153], BCS1L [154, 155], SURF1 [156], SCO1 [157], SCO2 [158], COX10 [159], COX15 [160, 161], ETHE1 [162], LRPPRC (LSFC) [138], ATP12 [163], PDSS1 [164], PDSS2 [165], COQ2 [164], APTX [166], ADCK3 [167], ETFDH [168], TSFM [169], TUFM [170], EGF1 [170], MRPS16 [171], PUS1 [172], DARS2 [173], PDHA1 [14], POLG1 [107], TK2 [174, 175], DGUOK [176], MPV17 [137], SUCLA2 [177],SUCLG1 [177],RRM2B [178], TWINKLE [179], ANT1 [180], ABC7 [181], FXN [182], OPA1 [183], DDP1 [184], SPG7 [185, 186], TAZ [187]
A wide range of abnormal liver histologies can be produced by mitochondrial disease [105, 106]. Isolated COX deficiency in respiratory chain biochemical testing can be seen in disease caused by any of the 5 nuclear genes listed above. Multiple ETC deficiencies can be seen with mtDNA depletion associated with POLG, dGUOK, MPV17, or SUCLA2 mutations. Quantitative Southern analysis or real-time quantitative PCR to detect mtDNA depletion should be a routine part of liver biopsy testing. However, a word of caution is warranted. Similar to the case for interpretation of blood lactate, it is helpful to know when mtDNA is depleted, but a normal result provides no information either for or against mitochondrial disease. For example, it is common to have normal mtDNA content on biopsies obtained early in the course of POLG disease, as in Alpers syndrome presentations, while repeat testing on biopsies obtained later in the natural history of the disease will show depletion [107].
Fibroblast Analysis
A skin biopsy is frequently useful in the diagnostic evaluation of metabolic disease. This simple office procedure can be done with local anesthesia (either topical local anesthetic cream or intradermal injection of a local anesthetic) using a disposable 3 or 4 mm punch biopsy, without the need for stitches. This contrasts sharply with a muscle biopsy, which is an invasive procedure that requires sedation, if not general anesthesia. Fibroblasts have a long tract record of utility in the study of mitochondrial disease biochemistry [108]. Added benefits of cultured skin fibroblasts are that they can be stored indefinitely, used as a renewable source of DNA, and recultured for testing as new tests become available [109, 110]. The major disadvantage is that not all mitochondrial phenotypes are expressed in skin fibroblasts [6, 109]. Unfortunately, fibroblasts are not as useful as muscle as a tissue in which to identify mitochondrial disease since respiratory chain defects expressed in muscle tissue may not be expressed in fibroblasts. This is due in part to altered heteroplasmy and a high tissue regeneration rate of fibroblasts when compared with muscle cells [109]. It is important to culture fibroblasts in the presence of uridine and pyruvate to avoid the potential loss of mtDNA mutant-harboring cells [111]. Residual ETC enzyme activity in fibroblasts is often substantial – even in genetically proven cases of mitochondrial disease [6]. With the additional concern of inter- and intra-laboratory variability, especially for the complex I enzyme activity assay, fibroblast ETC enzyme analysis has a high false-negative rate. Thus, an absence of defects in fibroblast biochemical analysis does not exclude a disorder of mitochondrial metabolism. An excellent review of the utility of and indications for fibroblast studies in mitochondrial diagnosis is available [109].
Biochemical testing which can be informative in cultured skin fibroblasts includes electron transport chain enzyme analysis via spectrophotometry and radiolabeled isotope fatty acid oxidation studies. Additional evaluation can include measurements of lactate and pyruvate [110]. ATP production and consumption can be studied in fibroblasts [112], although ATP analysis is more reliable in lymphoblasts [113]. Fibroblasts can also provide excellent material for protein studies, including western blot and blue native polyacrylamide gel electrophoresis (BN-PAGE) analysis [114]. BN-PAGE permits analysis of multisubunit, integral membrane protein complexes like those of the mitochondrial electron transport complexes. It is used to identify complex assembly abnormalities such as SURF1 defects [115] or tissue-specific mitochondrial translation defects [116]. Additional abnormalities can be detected when 2-dimensional BN-PAGE/SDS-PAGE is performed in combination with Western analysis using antibodies directed at proteins of special interest [117,118].
Genetic Diagnostic Testing
Mitochondrial DNA Analysis
Performing thorough mitochondrial DNA analysis necessitates understanding the breadth of mitochondrial DNA mutation types, the relative merits and drawbacks of the various analytic methods available, and the appropriate tissue for testing in a given patient. The types of mitochondrial DNA abnormalities that can be causative of human mitochondrial disease include single basepair point mutations, several basepair insertions or deletions, large scale deletions on the order of 100s to 1000s of basepairs, or relative depletion of total mitochondrial DNA content. Laboratory methods that can be used to screen for known point mutations include PCR with restriction fragment length polymorphism (RFLP) analysis to screen for specific mutations on a single basis or multiplex PCR with allele specific oligonucleotide (ASO) analyis to screen for multiple known mutations simultaneously. Methods that can be used to screen for unknown mtDNA point mutations include single-strand conformation polymorphism (SSCP) [119], heteroduplex screening assays (such as temporal temperature gradient gel electrophoresis (TTGE) [120-122], temperature gradient gel electrophoresis (TGGE) [123], denaturant gradient gel electrophoresis (DGGE) [124, 125], denaturing high performance liquid chromatography (dHPLC) [126]), and sequencing. As direct DNA sequencing is generally considered to be the gold standard for mutation detection in nuclear genes, the advent of rapid sequencing tools has resulted in some laboratories including mtDNA sequencing in their clinical diagnostic approach by one of several different sequencing methodologies available. Interference by nuclear pseudogenes should be excluded by use of rhoo cells during the development of new PCR-based assays. Point mutations or deletions may not be present in all mtDNA genomes within a cell or tissue, but rather be heteroplasmic with wildtype to varying degrees. The sensitivity of different analytic methods for detecting homoplasmic or heteroplasmic mutations is a driving factor in determining their relative utility. Quantification of mutant heteroplasmy may be performed by densitometer scanning utilizing RFLP or real-time ARMS qPCR [127]. Southern blot analysis is the method most often used for the detection of large-scale mtDNA deletions and duplications, although these can also be detected by long-range polymerase chain reaction (PCR) analysis. Finally, depletion testing utilizes either Southern densitometry scanning or real time qPCR to quantify mtDNA copy number in reference to a nuclear gene [127]. A full discussion of the relative merits and drawbacks of each of these assays is available on www.mitosoc.org.
Nuclear DNA Analysis
An estimated 75%-90% of pediatric primary mitochondrial disease results from nDNA mutations. The ability to identify a causative genetic mutation in a given patient has substantially increased the ability to definitively diagnose primary mitochondrial disease. When classical symptoms of mitochondrial disease create a high index of suspicion, or patient acuity limits a step-wise approach, one can sometimes proceed directly to DNA-testing without having to pursue extensive biochemical-based testing for preliminary diagnostic guidance. Figure 4 provides a sample algorithm of how one clinical molecular laboratory approaches DNA-based analyses in mitochondrial disease (courtesy of L-J Wong). However, it is often prudent to use the biochemical evaluation to provide sufficient information to guide the choice of genetic diagnostic testing to pursue.Table 1 provides a detailed overview of how results from laboratory-based investigation can suggest specific nuclear genes among those implicated in mitochondrial disease to date as possible pathogenic candidates in a given patient.
Figure 4.

Clinical algorithm for genetic diagnostic testing of mtDNA and nDNA genes in patients suspected of mitochondrial disorders currently followed by Baylor College of Medicine, Mitochondrial Diagnostics Laboratory.
*This algorithm reflects currently available clinical testing and experience of Baylor College of Medicine, Mitochondrial Diagnostics Laboratory.
Regular reassessment of the evolving clinical signs and symptoms is required. Mitochondrial disorders are developmental and degenerative. New organ system involvement occurs step-wise over time. Consequently, it is rare for even the most clear-cut and classic mitochondrial disorders like Alpers or Kearns-Sayre syndrome, to present the full clinical spectrum of disease at the time of first medical contact. The extent of recognized clinical manifestations for any single gene disorder is quite likely to change over time as less “classical” cases become identified. Establishing a genetic diagnosis permits accurate etiologic, prognostic, recurrence risk, and management counseling. In the future, it is hoped that this will also permit for accurate subgrouping among the heterogeneous category of mitochondrial disease patients in an effort to develop disease-focused interventions.
Emerging Technologies
DNA-based Technologies
mtDNA Sequencing Microarray
The latest version of the Affymetrix oligonucleotide resequencing microarray (MitoChip 2.0) provides a method that yields sequence data on the complete coding region and 15,452 of the 15,569 bp. mtDNA [128]. It reduces the possibility of amplifying pseudogenes by first amplifying mtDNA in 3 long overlapping fragments that are not present in the nDNA. In addition, it now appears to be able to detect heteroplasmy down to about 2-5% [129].
ESI-TOF Mass Spectrometry
A variety of mass spectrometry platforms are suitable for rapid screening of PCR-amplified fragments of mtDNA. These include electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS), Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) [130] and ion-pair reverse phase HPLC time-of-flight mass spectrometry (ICEMS) [131]. While the instrumentational costs of FTICR-MS put this particular platform beyond the reach of many labs, both ESI-TOF-MS and ICEMS are more economical and well-suited for rapid sequence screening and the quantitation of heteroplasmy in the range of 1-2%.
Protein-based Technologies
Immunocytochemistry
With the commercial availability of a monoclonal antibodies directed to a number of mitochondrial proteins, it is now possible to use immunocytological methods for diagnosis of selected OXPHOS defects [117]. In most cases, the success of these methods has depended on the expression of the mitochondrial protein defect in cultured skin fibroblasts. The diagnostic images are compelling when this occurs, as in the case of pyruvate dehydrogenase E1α defects [118]. Background autofluorescence complicates the use of many immunofluorescent probes in muscle biopsies, but the use of cocktails of carefully selected OXPHOS antibodies in Western immunoblot analysis [132] and immunocapture of assembled complexes [133] have proven to be powerful analytical technologies.
Proteomics and Systems Biology
Careful purification of mitochondria from different tissues, combined with gel and chromatography-enhanced mass spectrometry methods, has allowed a number of tissue-specific mitochondrial proteomes to be characterized [134] and the physiologic significance of the tissue-specific heterogeneities to be examined [135]. This work is nicely complemented by the development of powerful bioinformatics tools by Mootha and colleagues [136]. Using a systems biology approach, they developed the trainable software program, “Maestro”. This uses eight different parameters to estimate the probability of mitochondrial localization to identify 1,451 mitochondrial proteins out of 33,860 proteins in the Ensembl human protein data set with an estimated false discovery rate of just 10% [136]. For example, application of these methods has led to the identification of MPV17 as a cause of mtDNA depletion syndromes [137] and LRPPRC as the cause of one form of Leigh syndrome [138].
Functional Spectroscopy
31P and 13C Spectroscopy
More than any static measure, the dynamic capacity of cells and mitochondria to respond to and recover from changing physiologic conditions in real-time determines how well they perform and survive. One method for measuring this dynamic capacity in vivo is 31phosphorus magnetic resonance spectroscopy (31P-MRS). This method allows for baseline, exercise, and recovery measurements of ATP and phosphocreatine that can reveal very subtle derangements in mitochondrial function [139]. 13C-MRS is also an emerging technology. However, the low natural abundance of 13C (about 1.1% of 12C) creates some challenges for its routine use in clinical metabolic testing [140]. Nevertheless, its versatility in enrichment and tracer studies continues to drive new developments in our understanding of cell-specific differences in mitochondrial function [141].
Optics, Biophotonics, and Nanotechnologies
Biocavity Laser Spectroscopy
The biocavity laser is a semiconductor nanolaser about the size of a dime. Cells or mitochondria that flow through the biocavity laser trigger the emission of photons of laser light. Measurement of the red-shift of this emitted laser light provides information of the biophysical composition of healthy and diseased mitochondria. Unlike the large numbers of chemical, enzymologic, or polarographic measurements currently required for diagnosis, the biocavity laser represents a single test that can measure the overall biophysical state of the cell and mitochondria. In the first studies of this new technology in human disease, the biocavity laser was able to detect mitochondrial dysfunction in disorders as varied as cancer [142] and primary OXPHOS disease [143].
Nanomedicine
Mitochondria have the unique electrophysical property of concentrating or attracting amphipathic, charge-delocalized, cationic molecules and nanoparticles. This allows for the rational design of a variety of cargo-delivery systems. Indeed, mitochondriotropic tags incorporated into paclitaxel-bearing nanoliposomes inhibited tumor cell growth in a mouse model, whereas free paclitaxel had no effect [144]. The selective delivery of DNA to mitochondria, at least in cell culture, may not be far behind [145].
Mitochondria Light Interactions
The alpha proteobacterial protoendosymbiont from which mitochondria were derived is a descendant of an ancestral purple, non-sulfur, photosynthetic group of bacteria [146]. Traces of important light interactions of mitochondria linger today. For example, Otto Warburg was the first to show that flashes of light of specific wavelengths could be used to restore normal oxygen consumption in preparations of mitochondria that were inhibited with carbon monoxide [147]. Today, specific wavelengths in the red and near infrared ranges (ca 600-1200 nm) are known to stimulate a wide array of biological processes ranging from the expansion of cardiac stem cells in culture [148] to accelerated healing of chemotherapy-associated mucositis [149]. Low level laser treatment using an 808 nm light source has even shown promise in a blinded, prospective clinical trial in acute stroke patients [150].
Summary
An in-depth evaluation of mitochondrial disease is possible and indeed warranted to obtain a clear diagnosis for individual patients. This is necessary to minimize cost of duplicate investigations, correct misinformation to families and health care providers, and provide accurate prognostic and recurrence risk counseling. Recognized mitochondrial diagnostic centers working in close collaboration with referring clinicians can successfully navigate the complexities of the diagnostic evaluation and work towards the development of quality assessment programs in the laboratory-based investigations of mitochondrial disease.
Acknowledgments
The authors thank Drs. Salvatore DiMauro, Wolfgang Sperl, and Jan Smeitink for their helpful comments on this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haas RH, P S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH. Mitochondrial Disease: A practical approach for primary care physicians. Pediatrics. 2007 doi: 10.1542/peds.2007-0391. in press. [DOI] [PubMed] [Google Scholar]
- 2.Bernier FP, et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59(9):1406–11. doi: 10.1212/01.wnl.0000033795.17156.00. [DOI] [PubMed] [Google Scholar]
- 3.Wolf NI, Smeitink JA. Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59(9):1402–5. doi: 10.1212/01.wnl.0000031795.91814.d8. [DOI] [PubMed] [Google Scholar]
- 4.DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul Disord. 2005;15(4):276–86. doi: 10.1016/j.nmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Gellerich FN, et al. The problem of interlab variation in methods for mitochondrial disease diagnosis: enzymatic measurement of respiratory chain complexes. Mitochondrion. 2004;4(56):427–39. doi: 10.1016/j.mito.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Thorburn DR, Smeitink J. Diagnosis of mitochondrial disorders: clinical and biochemical approach. J Inherit Metab Dis. 2001;24(2):312–6. doi: 10.1023/a:1010347808082. [DOI] [PubMed] [Google Scholar]
- 7.Hammond EL, et al. Assessment of precision and concordance of quantitative mitochondrial DNA assays: a collaborative international quality assurance study. J Clin Virol. 2003;27(1):97–110. doi: 10.1016/s1386-6532(02)00134-8. [DOI] [PubMed] [Google Scholar]
- 8.Moraes CT, et al. Techniques and pitfalls in the detection of pathogenic mitochondrial DNA mutations. J Mol Diagn. 2003;5(4):197–208. doi: 10.1016/S1525-1578(10)60474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debray FG, et al. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin Chem. 2007;53(5):916–21. doi: 10.1373/clinchem.2006.081166. [DOI] [PubMed] [Google Scholar]
- 10.Triepels RH, et al. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45(6):787–90. doi: 10.1002/1531-8249(199906)45:6<787::aid-ana13>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Haas RH. Thiamin and the brain. Annu Rev Nutr. 1988;8:483–515. doi: 10.1146/annurev.nu.08.070188.002411. [DOI] [PubMed] [Google Scholar]
- 12.Chow SL, et al. The significance of elevated CSF lactate. Arch Dis Child. 2005;90(11):1188–9. doi: 10.1136/adc.2005.075317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finsterer J. Cerebrospinal-fluid lactate in adult mitochondriopathy with and without encephalopathy. Acta Med Austriaca. 2001;28(5):152–5. doi: 10.1046/j.1563-2571.2001.01036.x. [DOI] [PubMed] [Google Scholar]
- 14.Cameron JM, et al. Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1alpha subunit. Am J Med Genet A. 2004;131(1):59–66. doi: 10.1002/ajmg.a.30287. [DOI] [PubMed] [Google Scholar]
- 15.Maj MC, Cameron JM, Robinson BH. Pyruvate dehydrogenase phosphatase deficiency: orphan disease or an under-diagnosed condition? Mol Cell Endocrinol. 2006;249(12):1–9. doi: 10.1016/j.mce.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Tan ES, et al. Non-ketotic hyperglycinemia is usually not detectable by tandem mass spectrometry newborn screening. Mol Genet Metab. 2007;90(4):446–8. doi: 10.1016/j.ymgme.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Zschocke J, Hoffmann GF. Vademecum Metabolicum. 2nd. Friedrichsdorf, Germany: Milupa; 2004. p. 32. [Google Scholar]
- 18.Campos Y, et al. Mitochondrial DNA deletion in a patient with mitochondrial myopathy, lactic acidosis, and stroke-like episodes (MELAS) and Fanconi's syndrome. Pediatr Neurol. 1995;13(1):69–72. doi: 10.1016/0887-8994(95)00082-q. [DOI] [PubMed] [Google Scholar]
- 19.Niaudet P, et al. Deletion of the mitochondrial DNA in a case of de Toni-Debre-Fanconi syndrome and Pearson syndrome. Pediatr Nephrol. 1994;8(2):164–8. doi: 10.1007/BF00865468. [DOI] [PubMed] [Google Scholar]
- 20.Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38(4):296–309. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Barshop BA. Metabolomic approaches to mitochondrial disease: correlation of urine organic acids. Mitochondrion. 2004;4(56):521–7. doi: 10.1016/j.mito.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Sperl W, et al. Deficiency of mitochondrial ATP synthase of nuclear genetic origin. Neuromuscul Disord. 2006;16(12):821–9. doi: 10.1016/j.nmd.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Gibson KM, et al. Phenotypic heterogeneity in the syndromes of 3-methylglutaconic aciduria. J Pediatr. 1991;118(6):885–90. doi: 10.1016/s0022-3476(05)82199-7. [DOI] [PubMed] [Google Scholar]
- 24.Minkler PE, Hoppel CL. Quantification of free carnitine, individual short- and medium-chain acylcarnitines, and total carnitine in plasma by high-performance liquid chromatography. Anal Biochem. 1993;212(2):510–8. doi: 10.1006/abio.1993.1361. [DOI] [PubMed] [Google Scholar]
- 25.van der Knaap MS, Jakobs C, Valk J. Magnetic resonance imaging in lactic acidosis. J Inherit Metab Dis. 1996;19(4):535–47. doi: 10.1007/BF01799114. [DOI] [PubMed] [Google Scholar]
- 26.Barkovich AJ, et al. Mitochondrial disorders: analysis of their clinical and imaging characteristics. AJNR Am J Neuroradiol. 1993;14(5):1119–37. [PMC free article] [PubMed] [Google Scholar]
- 27.Valanne L, et al. Neuroradiologic findings in children with mitochondrial disorders. AJNR Am J Neuroradiol. 1998;19(2):369–77. [PMC free article] [PubMed] [Google Scholar]
- 28.Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in the diagnostic evaluation of suspected mitochondrial disease. AJNR Am J Neuroradiol. 2003;24(1):33–41. [PMC free article] [PubMed] [Google Scholar]
- 29.Bianchi MC, et al. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic relevance. AJNR Am J Neuroradiol. 2003;24(10):1958–66. [PMC free article] [PubMed] [Google Scholar]
- 30.Dinopoulos A, et al. Brain MRI and proton MRS findings in infants and children with respiratory chain defects. Neuropediatrics. 2005;36(5):290–301. doi: 10.1055/s-2005-872807. [DOI] [PubMed] [Google Scholar]
- 31.Castillo M, Kwock L, Green C. MELAS syndrome: imaging and proton MR spectroscopic findings. AJNR Am J Neuroradiol. 1995;16(2):233–9. [PMC free article] [PubMed] [Google Scholar]
- 32.Wilichowski E, et al. Quantitative proton magnetic resonance spectroscopy of cerebral metabolic disturbances in patients with MELAS. Neuropediatrics. 1999;30(5):256–63. doi: 10.1055/s-2007-973500. [DOI] [PubMed] [Google Scholar]
- 33.Saneto RP, et al. MRS detection of CNS lactate peaks in primary mitochondrial cytopathies. Neurology. 2001;56(suppl 3):A42. [Google Scholar]
- 34.Ross BD. A biochemistry primer for the neuroradiologist, in Advanced imaging symposium: Preparing the neuroradiologist for the new millenium. American Society of Neuroradiology; Oak Brook IL: 2000. [Google Scholar]
- 35.Kingsley PB, Shah TC, Woldenberg R. Identification of diffuse and focal brain lesions by clinical magnetic resonance spectroscopy. NMR Biomed. 2006;19(4):435–62. doi: 10.1002/nbm.1039. [DOI] [PubMed] [Google Scholar]
- 36.Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetylaspartate with monoclonal antibodies. Neuroscience. 1991;45(1):37–45. doi: 10.1016/0306-4522(91)90101-s. [DOI] [PubMed] [Google Scholar]
- 37.Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20(45):271–6. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- 38.Pavlakis SG, et al. Magnetic resonance spectroscopy: use in monitoring MELAS treatment. Arch Neurol. 1998;55(6):849–52. doi: 10.1001/archneur.55.6.849. [DOI] [PubMed] [Google Scholar]
- 39.De Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995;34(5):721–7. doi: 10.1002/mrm.1910340511. [DOI] [PubMed] [Google Scholar]
- 40.Martin E, et al. Absence of N-acetylaspartate in the human brain: impact on neurospectroscopy? Ann Neurol. 2001;49(4):518–21. [PubMed] [Google Scholar]
- 41.Tan J, et al. Lack of effect of oral choline supplement on the concentrations of choline metabolites in human brain. Magn Reson Med. 1998;39(6):1005–10. doi: 10.1002/mrm.1910390619. [DOI] [PubMed] [Google Scholar]
- 42.Miller BL, et al. In vivo 1H MRS choline: correlation with in vitro chemistry/histology. Life Sci. 1996;58(22):1929–35. doi: 10.1016/0024-3205(96)00182-8. [DOI] [PubMed] [Google Scholar]
- 43.Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13(3):129–53. doi: 10.1002/1099-1492(200005)13:3<129::aid-nbm619>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 44.Klunk WE, et al. Quantitative 1H and 31P MRS of PCA extracts of postmortem Alzheimer's disease brain. Neurobiol Aging. 1996;17(3):349–57. doi: 10.1016/0197-4580(96)00035-8. [DOI] [PubMed] [Google Scholar]
- 45.Brockmann K, et al. Succinate in dystrophic white matter: a proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Ann Neurol. 2002;52(1):38–46. doi: 10.1002/ana.10232. [DOI] [PubMed] [Google Scholar]
- 46.Lin A, et al. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx. 2005;2(2):197–214. doi: 10.1602/neurorx.2.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haas RH, Barshop BA. Diet change in the management of metabolic encephalomyopathies. Biofactors. 1998;7(3):259–62. doi: 10.1002/biof.5520070323. [DOI] [PubMed] [Google Scholar]
- 48.Tarnopolsky M. Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion. 2004;4(56):529–42. doi: 10.1016/j.mito.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 49.Taivassalo T, et al. Venous oxygen levels during aerobic forearm exercise: An index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol. 2002;51(1):38–44. doi: 10.1002/ana.10027. [DOI] [PubMed] [Google Scholar]
- 50.Jensen TD, et al. A forearm exercise screening test for mitochondrial myopathy. Neurology. 2002;58(10):1533–8. doi: 10.1212/wnl.58.10.1533. [DOI] [PubMed] [Google Scholar]
- 51.Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106(1):4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- 52.Thorburn DR, Chow CW, Kirby DM. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion. 2004;4(56):363–75. doi: 10.1016/j.mito.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Cazorla A, et al. Mitochondrial respiratory chain deficiencies expressing the enzymatic deficiency in the hepatic tissue: a study of 31 patients. J Pediatr. 2006;149(3):401–405. doi: 10.1016/j.jpeds.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 54.Taylor RW, et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003;41(10):1786–96. doi: 10.1016/s0735-1097(03)00300-0. [DOI] [PubMed] [Google Scholar]
- 55.Bourgeois JM, Tarnopolsky MA. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion. 2004;4(56):441–52. doi: 10.1016/j.mito.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 56.Tanji K, Bonilla E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods Cell Biol. 2007;80(26):135–54. doi: 10.1016/S0091-679X(06)80006-3. [DOI] [PubMed] [Google Scholar]
- 57.Sciacco M, et al. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3(1):13–9. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 58.Scaglia F, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–31. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto M, et al. Focal cytochrome c oxidase deficiency in various neuromuscular diseases. J Neurol Sci. 1989;91(12):207–13. doi: 10.1016/0022-510x(89)90088-9. [DOI] [PubMed] [Google Scholar]
- 60.Rollins S, et al. Diagnostic yield muscle biopsy in patients with clinical evidence of mitochondrial cytopathy. Am J Clin Pathol. 2001;116(3):326–30. doi: 10.1309/WATB-W4QV-NA53-B9MY. [DOI] [PubMed] [Google Scholar]
- 61.Tulinius MH, et al. Mitochondrial encephalomyopathies in childhood. I. Biochemical and morphologic investigations. J Pediatr. 1991;119(2):242–50. doi: 10.1016/s0022-3476(05)80734-6. [DOI] [PubMed] [Google Scholar]
- 62.Hasegawa H, et al. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601–5. doi: 10.1002/ana.410290606. [DOI] [PubMed] [Google Scholar]
- 63.Tanji K, Bonilla E. Neuropathologic aspects of cytochrome C oxidase deficiency. Brain Pathol. 2000;10(3):422–30. doi: 10.1111/j.1750-3639.2000.tb00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oldfors A, Tulinius M. Mitochondrial encephalomyopathies. J Neuropathol Exp Neurol. 2003;62(3):217–27. doi: 10.1093/jnen/62.3.217. [DOI] [PubMed] [Google Scholar]
- 65.Tassin S, et al. Histochemical and ultrastructural analysis of the mitochondrial changes in a familial mitochondrial myopathy. Neuropathol Appl Neurobiol. 1980;6(5):337–47. doi: 10.1111/j.1365-2990.1980.tb00670.x. [DOI] [PubMed] [Google Scholar]
- 66.Cotter D, et al. MitoProteome: mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004;32(Database issue):D463–7. doi: 10.1093/nar/gkh048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thorburn DR, et al. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta. 2004;1659(23):121–8. doi: 10.1016/j.bbabio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Birch-Machin MA, et al. An evaluation of the measurement of the activities of complexes I-IV in the respiratory chain of human skeletal muscle mitochondria. Biochem Med Metab Biol. 1994;51(1):35–42. doi: 10.1006/bmmb.1994.1004. [DOI] [PubMed] [Google Scholar]
- 69.Chretien D, et al. Reference charts for respiratory chain activities in human tissues. Clin Chim Acta. 1994;228(1):53–70. doi: 10.1016/0009-8981(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 70.Rustin P, et al. Endomyocardial biopsies for early detection of mitochondrial disorders in hypertrophic cardiomyopathies. J Pediatr. 1994;124(2):224–8. doi: 10.1016/s0022-3476(94)70308-6. [DOI] [PubMed] [Google Scholar]
- 71.Bindoff LA, et al. Multiple defects of the mitochondrial respiratory chain in a mitochondrial encephalopathy (MERRF): a clinical, biochemical and molecular study. J Neurol Sci. 1991;102(1):17–24. doi: 10.1016/0022-510x(91)90088-o. [DOI] [PubMed] [Google Scholar]
- 72.Moraes CT, et al. The mitochondrial tRNA(Leu(UUR)) mutation in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am J Hum Genet. 1992;50(5):934–49. [PMC free article] [PubMed] [Google Scholar]
- 73.Montero R, et al. Muscle coenzyme Q10 concentrations in patients with probable and definite diagnosis of respiratory chain disorders. Biofactors. 2005;25(14):109–15. doi: 10.1002/biof.5520250112. [DOI] [PubMed] [Google Scholar]
- 74.Quinzii CM, D S, Hirano M. Human Coenzyme Q(10) Deficiency. Neurochem Res. 2006 doi: 10.1007/s11064-006-9190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tulinius M, et al. A family with pyruvate dehydrogenase complex deficiency due to a novel C>T substitution at nucleotide position 407 in exon 4 of the X-linked Epsilon1alpha gene. Eur J Pediatr. 2005;164(2):99–103. doi: 10.1007/s00431-004-1570-2. [DOI] [PubMed] [Google Scholar]
- 76.Janssen AJ, et al. Measurement of the energy-generating capacity of human muscle mitochondria: diagnostic procedure and application to human pathology. Clin Chem. 2006;52(5):860–71. doi: 10.1373/clinchem.2005.062414. [DOI] [PubMed] [Google Scholar]
- 77.Wenchich L, et al. Polarographic evaluation of mitochondrial enzymes activity in isolated mitochondria and in permeabilized human muscle cells with inherited mitochondrial defects. Physiol Res. 2003;52(6):781–8. [PubMed] [Google Scholar]
- 78.Sperl W, et al. High resolution respirometry of permeabilized skeletal muscle fibers in the diagnosis of neuromuscular disorders. Mol Cell Biochem. 1997;174(12):71–8. [PubMed] [Google Scholar]
- 79.Kuznetsov AV, et al. Evaluation of mitochondrial respiratory function in small biopsies of liver. Anal Biochem. 2002;305(2):186–94. doi: 10.1006/abio.2002.5658. [DOI] [PubMed] [Google Scholar]
- 80.Chowdhury SK, et al. Activities of mitochondrial oxidative phosphorylation enzymes in cultured amniocytes. Clin Chim Acta. 2000;298(12):157–73. doi: 10.1016/s0009-8981(00)00300-4. [DOI] [PubMed] [Google Scholar]
- 81.Villani G, Attardi G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic Biol Med. 2000;29(34):202–10. doi: 10.1016/s0891-5849(00)00303-8. [DOI] [PubMed] [Google Scholar]
- 82.Janssen AJ, Smeitink JA, van den Heuvel LP. Some practical aspects of providing a diagnostic service for respiratory chain defects. Ann Clin Biochem. 2003;40(Pt 1):3–8. doi: 10.1258/000456303321016114. [DOI] [PubMed] [Google Scholar]
- 83.Puchowicz MA, et al. Oxidative phosphorylation analysis: assessing the integrated functional activity of human skeletal muscle mitochondria--case studies. Mitochondrion. 2004;4(56):377–85. doi: 10.1016/j.mito.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 84.Boitier E, et al. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J Neurol Sci. 1998;156(1):41–6. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 85.Rotig A, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356(9227):391–5. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 86.Brivet M, et al. Impaired mitochondrial pyruvate importation in a patient and a fetus at risk. Mol Genet Metab. 2003;78(3):186–92. doi: 10.1016/s1096-7192(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 87.Mayr JA, et al. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet. 2007;80(3):478–84. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Linnane AW, Kios M, Vitetta L. Coenzyme Q(10) - Its role as a prooxidant in the formation of superoxide anion/hydrogen peroxide and the regulation of the metabolome. Mitochondrion. 2007;7 1:S51–61. doi: 10.1016/j.mito.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 89.Rotig A, et al. Infantile and pediatric quinone deficiency diseases. Mitochondrion. 2007;7 1:S112–21. doi: 10.1016/j.mito.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 90.Quinzii C, et al. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am J Hum Genet. 2006;78(2):345–9. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ogasahara S, et al. Treatment of Kearns-Sayre syndrome with coenzyme Q10. Neurology. 1986;36(1):45–53. doi: 10.1212/wnl.36.1.45. [DOI] [PubMed] [Google Scholar]
- 92.Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7 1:S136–45. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 93.Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660(12):171–99. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 94.Duncan AJ, et al. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin Chem. 2005;51(12):2380–2. doi: 10.1373/clinchem.2005.054643. [DOI] [PubMed] [Google Scholar]
- 95.Miles MV, et al. Age-related changes in plasma coenzyme Q10 concentrations and redox state in apparently healthy children and adults. Clin Chim Acta. 2004;347(12):139–44. doi: 10.1016/j.cccn.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 96.Pastore A, et al. Simultaneous determination of ubiquinol and ubiquinone in skeletal muscle of pediatric patients. Anal Biochem. 2005;342(2):352–5. doi: 10.1016/j.ab.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 97.Lev D, et al. Clinical presentations of mitochondrial cardiomyopathies. Pediatr Cardiol. 2004;25(5):443–50. doi: 10.1007/s00246-003-0490-7. [DOI] [PubMed] [Google Scholar]
- 98.Marin-Garcia J, et al. Mitochondrial dysfunction in skeletal muscle of children with cardiomyopathy. Pediatrics. 1999;103(2):456–9. doi: 10.1542/peds.103.2.456. [DOI] [PubMed] [Google Scholar]
- 99.Marin-Garcia J, Goldenthal MJ, Filiano JJ. Cardiomyopathy associated with neurologic disorders and mitochondrial phenotype. J Child Neurol. 2002;17(10):759–65. doi: 10.1177/08830738020170101701. [DOI] [PubMed] [Google Scholar]
- 100.Jarreta D, et al. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res. 2000;45(4):860–5. doi: 10.1016/s0008-6363(99)00388-0. [DOI] [PubMed] [Google Scholar]
- 101.Stanley WC, Hoppel CL. Mitochondrial dysfunction in heart failure: potential for therapeutic interventions? Cardiovasc Res. 2000;45(4):805–6. doi: 10.1016/s0008-6363(99)00419-8. [DOI] [PubMed] [Google Scholar]
- 102.Lesnefsky EJ, et al. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33(6):1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 103.Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J. 2000;139(2 Pt 3):S70–85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 104.Riva A, et al. Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol Heart Circ Physiol. 2005;289(2):H868–72. doi: 10.1152/ajpheart.00866.2004. [DOI] [PubMed] [Google Scholar]
- 105.Nguyen KV, et al. Molecular diagnosis of Alpers syndrome. J Hepatol. 2006;45(1):108–16. doi: 10.1016/j.jhep.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 106.Labarthe F, et al. Clinical, biochemical and morphological features of hepatocerebral syndrome with mitochondrial DNA depletion due to deoxyguanosine kinase deficiency. J Hepatol. 2005;43(2):333–41. doi: 10.1016/j.jhep.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 107.Nguyen KV, et al. POLG mutations in Alpers syndrome. Neurology. 2005;65(9):1493–5. doi: 10.1212/01.wnl.0000182814.55361.70. [DOI] [PubMed] [Google Scholar]
- 108.Robinson BH, et al. The use of skin fibroblast cultures in the detection of respiratory chain defects in patients with lacticacidemia. Pediatr Res. 1990;28(5):549–55. doi: 10.1203/00006450-199011000-00027. [DOI] [PubMed] [Google Scholar]
- 109.van den Heuvel LP, Smeitink JA, Rodenburg RJ. Biochemical examination of fibroblasts in the diagnosis and research of oxidative phosphorylation (OXPHOS) defects. Mitochondrion. 2004;4(56):395–401. doi: 10.1016/j.mito.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 110.Cameron JM, et al. Respiratory chain analysis of skin fibroblasts in mitochondrial disease. Mitochondrion. 2004;4(56):387–94. doi: 10.1016/j.mito.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 111.Bourgeron T, et al. Fate and expression of the deleted mitochondrial DNA differ between human heteroplasmic skin fibroblast and Epstein-Barr virus-transformed lymphocyte cultures. J Biol Chem. 1993;268(26):19369–76. [PubMed] [Google Scholar]
- 112.Carrozzo R, et al. The T9176G mtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology. 2001;56(5):687–90. doi: 10.1212/wnl.56.5.687. [DOI] [PubMed] [Google Scholar]
- 113.Tatuch Y, Robinson BH. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem Biophys Res Commun. 1993;192(1):124–8. doi: 10.1006/bbrc.1993.1390. [DOI] [PubMed] [Google Scholar]
- 114.McKenzie M, et al. Analysis of mitochondrial subunit assembly into respiratory chain complexes using Blue Native polyacrylamide gel electrophoresis. Anal Biochem. 2007;364(2):128–37. doi: 10.1016/j.ab.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 115.Tiranti V, et al. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum Mol Genet. 1999;8(13):2533–40. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- 116.Antonicka H, et al. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15(11):1835–46. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 117.Capaldi RA, et al. Immunological approaches to the characterization and diagnosis of mitochondrial disease. Mitochondrion. 2004;4(56):417–26. doi: 10.1016/j.mito.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 118.Margineantu DH, et al. Heterogeneous distribution of pyruvate dehydrogenase in the matrix of mitochondria. Mitochondrion. 2002;1(4):327–38. doi: 10.1016/s1567-7249(01)00033-2. [DOI] [PubMed] [Google Scholar]
- 119.Suomalainen A, et al. Use of single strand conformation polymorphism analysis to detect point mutations in human mitochondrial DNA. J Neurol Sci. 1992;111(2):222–6. doi: 10.1016/0022-510x(92)90074-u. [DOI] [PubMed] [Google Scholar]
- 120.Wong LJ, et al. Comprehensive scanning of the entire mitochondrial genome for mutations. Clin Chem. 2002;48(11):1901–12. [PubMed] [Google Scholar]
- 121.Wong LJ, Chen TJ, Tan DJ. Detection of mitochondrial DNA mutations using temporal temperature gradient gel electrophoresis. Electrophoresis. 2004;25(15):2602–10. doi: 10.1002/elps.200406016. [DOI] [PubMed] [Google Scholar]
- 122.Chen TJ, Boles RG, Wong LJ. Detection of mitochondrial DNA mutations by temporal temperature gradient gel electrophoresis. Clin Chem. 1999;45(8 Pt 1):1162–7. [PubMed] [Google Scholar]
- 123.Wartell RM, Hosseini SH, Moran CP., Jr Detecting base pair substitutions in DNA fragments by temperature-gradient gel electrophoresis. Nucleic Acids Res. 1990;18(9):2699–705. doi: 10.1093/nar/18.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sternberg D, et al. Exhaustive scanning approach to screen all the mitochondrial tRNA genes for mutations and its application to the investigation of 35 independent patients with mitochondrial disorders. Hum Mol Genet. 1998;7(1):33–42. doi: 10.1093/hmg/7.1.33. [DOI] [PubMed] [Google Scholar]
- 125.Michikawa Y, et al. Comprehensive, rapid and sensitive detection of sequence variants of human mitochondrial tRNA genes. Nucleic Acids Res. 1997;25(12):2455–63. doi: 10.1093/nar/25.12.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Den Bosch BJ, et al. Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Res. 2000;28(20):E89. doi: 10.1093/nar/28.20.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bai RK, Wong LJ. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem. 2004;50(6):996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- 128.Zhou S, et al. An oligonucleotide microarray for high-throughput sequencing of the mitochondrial genome. J Mol Diagn. 2006;8(4):476–82. doi: 10.2353/jmoldx.2006.060008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coon KD, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6(4):194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 130.Jiang Y, et al. Mitochondrial DNA mutation detection by electrospray mass spectrometry. Clin Chem. 2007;53(2):195–203. doi: 10.1373/clinchem.2006.074823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oberacher H, et al. Profiling 627 mitochondrial nucleotides via the analysis of a 23-plex polymerase chain reaction by liquid chromatography-electrospray ionization time-of-flight mass spectrometry. Anal Chem. 2006;78(22):7816–27. doi: 10.1021/ac061210i. [DOI] [PubMed] [Google Scholar]
- 132.Kramer KA, et al. Automated spectrophotometric analysis of mitochondrial respiratory chain complex enzyme activities in cultured skin fibroblasts. Clin Chem. 2005;51(11):2110–6. doi: 10.1373/clinchem.2005.050146. [DOI] [PubMed] [Google Scholar]
- 133.Keeney PM, et al. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26(19):5256–64. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Forner F, et al. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics. 2006;5(4):608–19. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 135.Johnson DT, et al. Functional consequences of mitochondrial proteome heterogeneity. Am J Physiol Cell Physiol. 2007;292(2):C698–707. doi: 10.1152/ajpcell.00109.2006. [DOI] [PubMed] [Google Scholar]
- 136.Calvo S, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38(5):576–82. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 137.Spinazzola A, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38(5):570–5. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 138.Mootha VK, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100(2):605–10. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Saft C, et al. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington's disease. Mov Disord. 2005;20(6):674–9. doi: 10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- 140.Ross B, et al. Clinical experience with 13C MRS in vivo. NMR Biomed. 2003;16(67):358–69. doi: 10.1002/nbm.852. [DOI] [PubMed] [Google Scholar]
- 141.Sonnewald U, et al. Intracellular metabolic compartmentation assessed by 13C magnetic resonance spectroscopy. Neurochem Int. 2004;45(23):305–10. doi: 10.1016/j.neuint.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 142.Gourley PL, et al. Mitochondrial correlation microscopy and nanolaser spectroscopy - new tools for biophotonic detection of cancer in single cells. Technol Cancer Res Treat. 2005;4(6):585–92. doi: 10.1177/153303460500400602. [DOI] [PubMed] [Google Scholar]
- 143.Gourley CR, Naviaux RK. Optical phenotyping of human mitochondria in a biocavity laser. IEEE J Selected Topics Quantum Electronics. 2005;11:818–826. [Google Scholar]
- 144.Weissig V, et al. Liposomes and liposome-like vesicles for drug and DNA delivery to mitochondria. J Liposome Res. 2006;16(3):249–64. doi: 10.1080/08982100600851169. [DOI] [PubMed] [Google Scholar]
- 145.D'Souza GG, Boddapati SV, Weissig V. Gene therapy of the other genome: the challenges of treating mitochondrial DNA defects. Pharm Res. 2007;24(2):228–38. doi: 10.1007/s11095-006-9150-y. [DOI] [PubMed] [Google Scholar]
- 146.Cavalier-Smith T. Origin of mitochondria by intracellular enslavement of a photosynthetic purple bacterium. Proc Biol Sci. 2006;273(1596):1943–52. doi: 10.1098/rspb.2006.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Warburg O. Nobel Lecture, December 10, 1931. Amsterdam, Netherlands: Elsevier Publishing Company; 1965. [Google Scholar]
- 148.Tuby H, Maltz L, Oron U. Low-level laser irradiation (LLLI) promotes proliferation of mesenchymal and cardiac stem cells in culture. Lasers Surg Med. 2007;39(4):373–8. doi: 10.1002/lsm.20492. [DOI] [PubMed] [Google Scholar]
- 149.Desmet KD, et al. Clinical and experimental applications of NIR-LED photobiomodulation. Photomed Laser Surg. 2006;24(2):121–8. doi: 10.1089/pho.2006.24.121. [DOI] [PubMed] [Google Scholar]
- 150.Lampl Y, et al. Infrared laser therapy for ischemic stroke: a new treatment strategy: results of the NeuroThera Effectiveness and Safety Trial-1 (NEST-1) Stroke. 2007;38(6):1843–9. doi: 10.1161/STROKEAHA.106.478230. [DOI] [PubMed] [Google Scholar]
- 151.Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest. 2005;115(10):2784–92. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Horvath R, et al. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA) J Neurol Neurosurg Psychiatry. 2006;77(1):74–6. doi: 10.1136/jnnp.2005.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bayley JP, Devilee P, Taschner PE. The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med Genet. 2005;6:39. doi: 10.1186/1471-2350-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fernandez-Vizarra E, et al. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet. 2007;16(10):1241–52. doi: 10.1093/hmg/ddm072. [DOI] [PubMed] [Google Scholar]
- 155.Hinson JT, et al. Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med. 2007;356(8):809–19. doi: 10.1056/NEJMoa055262. [DOI] [PubMed] [Google Scholar]
- 156.Zhu Z, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20(4):337–43. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 157.Valnot I, et al. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet. 2000;67(5):1104–9. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Papadopoulou LC, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23(3):333–7. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 159.Valnot I, et al. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9(8):1245–9. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- 160.Antonicka H, et al. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72(1):101–14. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bugiani M, et al. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J Med Genet. 2005;42(5):e28. doi: 10.1136/jmg.2004.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tiranti V, et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet. 2004;74(2):239–52. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.De Meirleir L, et al. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J Med Genet. 2004;41(2):120–4. doi: 10.1136/jmg.2003.012047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mollet J, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117(3):765–72. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lopez LC, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79(6):1125–9. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mosesso P, et al. The novel human gene aprataxin is directly involved in DNA single-strand break repair. Cell Mol Life Sci. 2005;62(4):485–91. doi: 10.1007/s00018-004-4441-0. [DOI] [PubMed] [Google Scholar]
- 167.Lagier-Tourenne C, et al. Mitochondrial ADCK3, an ancestral prokaryotic kinase involved in Coenzyme Q biosynthesis, is mutant in a new form of recessive ataxia. Presented at the Am Soc Human Genetics; San Diego, CA. 2007. [Google Scholar]
- 168.Gempel K, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130(Pt 8):2037–44. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Smeitink JA, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79(5):869–77. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Valente L, et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80(1):44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Miller C, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56(5):734–8. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- 172.Bykhovskaya Y, et al. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) Am J Hum Genet. 2004;74(6):1303–8. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Scheper GC, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39(4):534–9. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 174.Saada A, et al. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29(3):342–4. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 175.Galbiati S, et al. New mutations in TK2 gene associated with mitochondrial DNA depletion. Pediatr Neurol. 2006;34(3):177–85. doi: 10.1016/j.pediatrneurol.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 176.Mandel H, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29(3):337–41. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 177.Ostergaard E, et al. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81(2):383–7. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bourdon A, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39(6):776–80. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 179.Spelbrink JN, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28(3):223–31. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 180.Holt IJ. Oxford monographs on medical genetics; no. 47. Oxford; New York: Oxford University Press; 2003. Genetics of mitochondrial diseases; p. xxiii.p. 350. [Google Scholar]
- 181.Allikmets R, et al. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A) Hum Mol Genet. 1999;8(5):743–9. doi: 10.1093/hmg/8.5.743. [DOI] [PubMed] [Google Scholar]
- 182.Palau F. Friedreich's ataxia and frataxin: molecular genetics, evolution and pathogenesis (Review) Int J Mol Med. 2001;7(6):581–9. doi: 10.3892/ijmm.7.6.581. [DOI] [PubMed] [Google Scholar]
- 183.Delettre C, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26(2):207–10. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 184.Roesch K, et al. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum Mol Genet. 2002;11(5):477–86. doi: 10.1093/hmg/11.5.477. [DOI] [PubMed] [Google Scholar]
- 185.Wilkinson PA, et al. A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain. 2004;127(Pt 5):973–80. doi: 10.1093/brain/awh125. [DOI] [PubMed] [Google Scholar]
- 186.Casari G, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93(6):973–83. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 187.Johnston J, et al. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am J Hum Genet. 1997;61(5):1053–8. doi: 10.1086/301604. [DOI] [PMC free article] [PubMed] [Google Scholar]