

Abstract
Alzheimer’s disease is characterized by two types of fibrous aggregates in the affected brains, the amyloid fibers (consisting of the Aβ-peptide, generating the amyloid plaques), and paired helical filaments (PHFs; made up of tau protein, forming the neurofibrillary tangles). Hence, tau protein, a highly soluble protein that normally stabilizes microtubules, becomes aggregated into insoluble fibers that obstruct the cytoplasm of neurons and cause a loss of microtubule stability. We have developed recently a rapid assay for monitoring PHF assembly and show here that PHFs arise from a nucleated assembly mechanism. The PHF nucleus comprises about 8–14 tau monomers. A prerequisite for nucleation is the dimerization of tau because tau dimers act as effective building blocks. PHF assembly can be seeded by preformed filaments (made either in vitro or isolated from Alzheimer brain tissue). These results suggest that dimerization and nucleation are the rate-limiting steps for PHF formation in vivo.
Alzheimer’s disease, the most common age-relate dementia, is characterized by two pathological protein deposits in the brain, the amyloid plaques, consisting largely of amyloid fibers assembled from the Aβ-peptide [a derivative of the membrane protein APP (amyloid precursor protein); reviewed in ref. 1] and the neurofibrillary tangles (NFT), which are bundles of paired helical filaments (PHFs) whose main constituent is the microtubule-associated protein tau (for reviews, see refs. 2 and 3). The uncontrolled precipitation of these aggregates is believed to be largely responsible for the neuronal degeneration, and the disease has been classified into several stages on the basis of the spreading of neurofibrillary deposits (4). It is therefore important to understand the factors underlying the abnormal aggregation of Aβ and tau. Both form filaments of ≈10 nm in width; in the case of Aβ they are smooth, while most tau filaments from Alzheimer brains show a characteristic “paired helical” structure, resembling two strands wound around one another, with a crossover periodicity of ≈80 nm and width varying between 10 and 20 nm (for review, see ref. 5).
The membrane-derived Aβ-peptide is partly hydrophobic so that its tendency to aggregate is intuitively understandable (6, 7). By contrast, the cytosolic tau has a very hydrophilic character and is highly soluble (8, 9). Thus, it shows hardly any tendency to aggregate in physiological buffer conditions, and the formation of aggregates is very slow (days or weeks; ref. 10). Soluble tau protein contains very little secondary structure (α-helix or β-sheet content < 5%), and the same holds for Alzheimer PHFs, in spite of their long-range periodicity (11). It therefore is not obvious why tau should aggregate in a specific manner and which structural principle could be responsible for this. Progress in understanding the mechanism has been correspondingly slow. Several factors supporting assembly have emerged in recent years. (i) The repeat domain in the C-terminal half of tau forms PHFs more readily than full-length tau isoforms (10), consistent with its position at the core of Alzheimer PHFs (12). (ii) Assembly proceeds more rapidly if the protein subunits are crosslinked into dimers, e.g., by an oxidized disulfide bridge at Cys322 (13), suggesting that dimers are important building blocks for PHFs. (iii) Polyanions further enhance the efficiency of assembly, presumably by compensating the positive charges of tau in a coacervate-like fashion. This can be achieved by extracellular components such as heparin (14–16) or intracellular factors such as RNA or acidic peptides (17, 18).
However, the kinetic mechanism and the intermediate steps of PHF assembly have remained undefined. We have developed recently a new assay for rapidly inducing and monitoring PHF assembly in solution and report here that tau protein obeys a nucleation-dependent polymerization process, with a nucleus containing about 4–7 tau dimers. In this regard, PHF assembly resembles that of amyloid fibers or cytoskeletal fibers (e.g., F-actin or microtubules) but differs from the mechanism of template-directed prion protein assembly (for reviews, see refs. 19 and 20).
MATERIALS AND METHODS
Chemicals and Proteins.
Heparin [average molecular weight (MW) of 6000], polyglutamate (average MW of 600 or 1,000), and thioflavine S (ThS) were obtained from Sigma. Full-length tau isoform htau23 and constructs of tau protein (see Fig. 1) were expressed in Escherichia coli and purified by making use of the heat stability and fast protein liquid Mono S (Pharmacia) chromatography, as described (21). The purity of the proteins were analyzed by SDS/PAGE, and protein concentrations were determined by the Bradford assay.
Figure 1.

Bar diagram of tau isoforms and constructs. From top to bottom: htau40 (the longest isoform in human central nervous system, 441 residues; ref. 39), htau23 (the shortest isoform, lacking the two near-N-terminal inserts and the second repeat), construct K12, K12A322, and construct K19, containing only the repeat region of tau. Numbering follows that of the longest human isoform, htau40. The position of Cys322, crucial for dimerization, is indicated.
Preparation of Dimeric Tau.
Dimeric tau protein (which is a building block of PHFs; ref. 13) was obtained by air oxidation. Tau dimers were separated from monomers by gel filtration on a Superdex 75 column (Pharmacia) equilibrated with 100 mM NH4Ac, pH 7.0. Fractions were collected and assayed for dimeric tau protein by SDS/PAGE under nonreducing conditions.
ThS Assay.
The assay was performed essentially as described recently (18). Briefly, fluorescence was measured with a Spex Fluoromax (Instruments S.A., Edison, NJ) with excitation at 440 nm and emission at 500 nm. Measurements were carried out at room temperature in 20 mM Mops⋅NaOH, pH 6.5, with 5 μM ThS. Aliquots of assembly reaction mixtures were added at 1 μM total tau protein. Background fluorescence and light scattering of the sample without thioflavine was subtracted.
Electron Microscopy.
Protein solutions diluted to 0.1–10 μM were placed on 600-mesh carbon-coated copper grids for 1 min and negatively stained with 2% uranyl acetate for 45 sec. The specimens were examined on a Philips CM12 electron microscope at 100 kV.
PHF Formation in Vitro.
Assembly of synthetic PHFs from tau protein (1–40 μM) was performed at 37°C in the presence of polyanions (heparin or polyglutamate, 0.1–400 μM) in 20 mM Mops⋅NaOH, pH 7.0. Assembly was followed either qualitatively by electron microscopy or quantitatively by fluorescence assay using ThS. Although RNA is also capable of inducing PHF formation in vitro (17), it was not included in this study as RNA interferes with the ThS assay (18).
Preparation of Seeds.
Filaments assembled from tau (see above) were diluted to 10 μM monomeric protein in 20 mM Mops⋅NaOH in a final volume of 200 μl. Samples were placed on ice and sonicated with a Branson Sonifier for 2 min (output level 5; duty cycle, 50%). PHF concentrations and length distributions were checked by electron microscopy and thioflavine fluorescence.
Polymerization Kinetics.
Samples of tau or tau constructs in the absence and presence of polyanions and/or seeds at the indicated concentrations were incubated im 20 mM MOPS⋅NaOH, pH 7.0, at 37°C (see Results for details). After suitable time intervals, aliquots were withdrawn and assayed with the ThS assay or with electron microscopy. Alternatively, at low protein concentration (<5 μM tau monomer) polymerization kinetics were followed directly in the presence of 5 μM ThS. Photobleaching of ThS was minimized by setting excitation slit width to 0.4 nm and the emission slit width to 21 nm.
Isolation of PHF from Alzheimer’s Disease (AD) Brain.
PHFs were isolated from AD brain as described, including an immunopurification step using PHF-specific antibodies (22, 23).
RESULTS
PHF Assembly Is a Nucleation-Dependent Process.
The tau construct K19 (containing the repeat region of tau, Fig. 1) can be dimerized by crosslinking at Cys322 and polymerized into PHFs in the presence of different polyanions; it is therefore a suitable model peptide for PHF formation (Fig. 2 A and B). The reaction M + M →k0 S generates the effective subunits from two tau monomers (S = M2), and the rate k0 is governed by the concentration of tau and the conditions of oxidation. The efficiency of PHF assembly is enhanced by complexing the subunits with polyanions. Other tau isoforms or constructs also can be crosslinked into dimers and polymerized (at slower rates), and the type of polyanion affects the rate of assembly (heparin > RNA > poly-Glu; ref. 18). The assembly behavior is qualitatively similar in these cases and points to a common underlying mechanism. In particular, at higher protein concentrations one observes a roughly exponential approach to equilibrium, but at lower protein concentrations the rapid phase is preceded by a lag phase (Fig. 3A). This delay is not due to a lack of sensitivity of the assay but rather to a retarded onset of assembly. Such a behavior is suggestive of a nucleation phase that must precede the elongation phase. In analogy with other self-assembly systems (24), the overall reaction could be approximated by the scheme
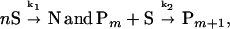 |
where k1 is the rate-limiting step of nucleation, resulting in a nucleus comprising n subunits S, and k2 is the rate of elongation of the polymer P by subunits S. Because the tau constructs initially were crosslinked by disulfide bridges, the effective subunit of the assembly in Fig. 3 consists of dimers, i.e., S = M2.
Figure 2.

Electron micrographs of PHFs formed by spontaneous or seeded assembly. (A) PHFs formed by spontaneous assembly of tau construct K19 (20 μM) after dimerization in the presence of poly-Glu. (B) PHFs formed by seeded assembly of K19 dimers (with poly-Glu). The concentration of seeds corresponds to 1 μM K19 monomers. The estimated seed number concentration is ≈2–20 nM (based on apparent PHF fragment lengths of 10–80 nm and a mass density of ≈60–70 kDa/nm, data not shown). (C and D) Seeds made by sonication for 2 or 5 min of filaments shown in A. Arrowheads indicate crossover points of the two strands of a PHF, spaced about 80 nm. The variations in stain accumulation were compensated by adjusting the contrast by image processing to make the filament structure more visible. (Bar = 100 nm.)
Figure 3.

Kinetics of spontaneous and seeded PHF assembly. (A) Dimeric tau protein (construct K19) was incubated in assembly buffer at a concentration of 10 μM in the presence of poly-Glu (MW 600, 80 μM) and varying concentrations of seeds obtained by sonication of preassembled PHFs. Assembly was followed by ThS fluorescence. Note that in the presence of added seeds the assembly curves can be fitted by a simple exponential approach to equilibrium, while in the absence of seeds (bottom curve) there is a lag phase because of the rate-limiting spontaneous nucleation. (B) The maximum assembly rates obtained from A increase linearly as a function of seed concentration. (C) Concentration dependence of maximum velocities for dimeric tau protein (K19) with either heparin at various concentration ratios [K19/heparin, 1:1, (○), 1:0.5 (▿), 1:0.25 (□)], or polyglutamate [MW 1,000; 1:4 (•)] without added seeds (spontaneous nucleation). The size of the nucleus (n = 2x) can be estimated from the slope of the curve (x) (24). The slope varies between 2 and 3.5, depending on the polyanion and the stoichiometry, so that the nucleus contains about 4–7 subunits (= tau dimers) or 8–14 tau monomers.
The Nucleation Barrier Can Be Overcome by Seeding.
The validity of the above reaction scheme can be ascertained by several experiments. First, it should be possible to circumvent the self-nucleation step by adding external seeds, and the increase in polymer mass after seeding, d[P]/dt, should be proportional to the concentration of seeds [N] and subunits [S], according to d[P]/dt = −d[S]/dt = k2[N][S], where the added seeds represent the effective nuclei.
Seeds can be obtained by sonication of PHFs made from K19 or other tau constructs. Depending on the duration and intensity of sonication, the PHFs were broken into short filaments, typically 50- to 200-nm long (Fig. 2 C and D). Longer exposures resulted in the disappearance of the filaments and the loss of seeding capacity. When aliquots of seeds were added to tau dimer solutions at the beginning of the assembly experiments a lag phase was no longer observed, even at low protein concentrations (Fig. 3A), and the initial rate of assembly increased linearly with seed concentration as predicted (Fig. 3B). Similarly, at a constant concentration of seeds the initial assembly rate increased linearly with the concentration of subunits (not shown). This behavior demonstrates that the assembly of PHFs follows a nucleated assembly mechanism. The seeded PHFs were structurally indistinguishable from those obtained by self-nucleation (compare Fig. 2 A and B).
Although the fragments of PHFs obtained by sonication were effective as seeds for assembly it is not possible to derive an absolute concentration of seeds since the fragments were of heterogeneous size and since it is possible that even smaller units not visible by electron microscopy could act as seeds. However, it is possible to derive an approximate value for the size of the nucleus from the concentration dependence of the maximum rate of self-assembly. This can be approximated by the equation (d[P]/dt)max = K[S]x, where x = n/2 equals half the number of subunits, n, which are incorporated into a nucleus (24). Thus, a double log plot of the rate of assembly vs. the concentration of subunits should yield a straight line with a slope of x = n/2. Such linear plots indeed are obtained (Fig. 3C). Their slopes are remarkably similar for different ratios of tau/polyanion and a range of between 2.2 and 3.5 so that the nucleus comprises between ≈4 and 7 subunits (= tau dimers), depending on the nature and the stoichiometry of the polyanion used. Thus, elongation can proceed from nuclei as small as 8–14 tau monomers, which is much smaller than the typical short filaments visible after sonication (Fig. 2).
Nucleation and Growth Are Both Dependent on Dimerization of Tau Protein.
In the experiments described thus far we have made use of the fact that dimerization of tau strongly promotes PHF assembly so that tau dimers can be regarded as effective subunits (13). However, the role of tau monomers vs. dimers in nucleated assembly can be addressed directly since dimers are formed by oxidation of Cys322 in the third repeat. We used a pair of tau constructs (K12 and K12A322, Fig. 1) that differ in their ability to dimerize covalently. K12 was used in its dimeric form (disulfide linkage via Cys322) while the mutant K12A322 exists mainly (>95%) as a monomer in solution (13). As seen in Fig. 4, in the absence of seeds dimeric K12 (20 μM monomer units) forms filaments within a few hours (t1/2 = 40 min), while monomeric K12A322 makes hardly any filaments even at elevated concentration after several days (t1/2 > 6 days at 100 μM protein). However, upon addition of small amounts of seeds (5% wt/wt) PHF formation is accelerated significantly with both constructs, displaying half-times of polymerization of <4 min for K12 and 2 hr for K12A322, respectively. Nevertheless, there was still a difference between monomeric and dimeric tau, showing that dimerization of tau is important not only for nucleation but also for elongation.
Figure 4.

Nucleation and growth are both dependent on dimeric tau protein. Monomeric tau (20 μM; K12A322) or 10 μM dimeric tau (K12 crosslinked via disulfide linkage of Cys322) was incubated in assembly buffer with 5 μM heparin in the absence or presence of seeds (corresponding to 1 μM tau protein). The time course of assembly shows that seeding accelerates assembly in both cases. Note that dimeric K12 without seeds is even faster than monomeric K12A322 in the presence of seeds, illustrating a high tendency of dimeric tau to self-assemble. The final extent of assembly is the same in all cases.
Seeding of PHFs from Full-Length Tau.
The above experiments showed results for tau constructs comprising the repeat region of tau (K19 or K12). Since the major component of PHFs is full-length tau (25), we asked whether the above finding also holds true for full-length tau. We repeated the nucleation experiments with tau23, the smallest human isoform (Fig. 1), either in its monomeric or dimeric form. PHF formation from both constructs indeed can be accelerated by seeding (Fig. 5) and results in the typical twisted filaments (not shown). These results show that the mechanism of PHF formation from either truncated or full-length tau are, in principle, similar.
Figure 5.

Seeded assembly of full-length tau (htau23) with seeds made from recombinant tau or from Alzheimer PHFs. (A) Dimeric full-length tau protein (htau23) was incubated at 2.5 μM with 20 μM low-molecular-weight polyglutamate (MW 600) in 10 mM Mops/5 μM ThS, pH 7.0 at 37°C in the absence or presence of 1/10 (wt/wt) sonicated PHF made either in vitro from tau construct K19 (dashed line) or isolated ex vivo from Alzheimer brains (dotted line). Filament formation was followed in real time by thioflavine fluorescence. The controls (bottom curves) show that seeds alone, made either by sonication of K19 PHFs (dashed) or from AD-PHFs (dotted), or htau23 without seeds (solid), do not show appreciable assembly during the experiments. (B) Electron micrograph of seeds obtained by sonication of PHFs prepared from Alzheimer brain tissue (same as in A). (C) PHFs formed in vitro by seeded assembly of htau23 dimers on seeds obtained by sonication of Alzheimer PHFs (same as in B). Note that PHFs formed by htau23 seeded by sonicated AD-PHFs exhibit the same characteristic twist as AD-PHFs and PHFs formed by construct K19 in vitro. (Bar = 100 nm.)
PHF Formation Can Be Seeded by PHFs Isolated from AD Brain.
Finally, a crucial question for the seeded assembly mechanism is whether it can be demonstrated with PHFs isolated from Alzheimer brains. PHFs were obtained from autopsy tissue as described (22, 23), sonicated, and used as seeds for both truncated and full-length tau. As shown in Fig. 5A, Alzheimer PHFs are fully capable of seeding the assembly of recombinant tau, resulting in bona fide PHFs as judged by electron microscopy (Fig. 5 B and C). This experiment also illustrates that it is the tau component of PHFs that is responsible for PHF assembly, rather than some other component(s) that might be present in PHFs isolated from Alzheimer tissue.
DISCUSSION
The neurofibrillary pathology, based on the aggregation of tau protein into paired helical filaments, is one of the major hallmarks of Alzheimer’s disease. The distribution and density of the tau deposits correlate strongly with the loss of neurons and impairment of cognitive functions and provide a basis for defining the stages of the disease (26–28). While the formation of amyloid deposits in AD brains from the Aβ-peptide has been determined in some detail (1, 19) and is understandable in terms of hydrophobic interactions typical of membrane proteins, the aggregation of tau is much less understood and even counterintuitive since tau is cytosolic and one of the most highly soluble proteins known. Several modifications of tau have been proposed to trigger the aggregation, such as phosphorylation, glycation, proteolysis, oxidation, or interaction with polyanions (for reviews, see refs. 2 and 3), but their role in vivo has remained ambiguous. Therefore, one has to rely on in vitro studies to map out the parameters that govern the assembly process of tau. The following principles have emerged thus far. (i) In physiological buffer conditions the polymerization of tau into PHFs is exceedingly slow, consistent with its hydrophilic nature, but it can be speeded up with forms of tau containing only the repeats (10, 29). This agrees with the observation that the core of PHFs contains the repeat domain of tau (12, 30). (ii) The dimerization of tau via a disulfide linkage of Cys322 in the third repeat enhances aggregation by providing ready-made building blocks (“subunits”; ref. 13). This is consistent with the impaired reducing capacity observed in aging brain tissue (31). (iii) Polyanionic cofactors such as heparin (14–16), RNA (17), or poly-Glu (18) accelerate filament formation dramatically. The latter two factors are particularly intriguing; mRNA is enriched in neurofibrillary tangles (32), and tau interacts with the C terminus of tubulin resembling poly-Glu, opening up the possibility that PHF assembly can be stimulated by its natural partner (and indeed tubulin is found as a component of neurofibrillary tangles; ref. 33). Optimal assembly rates occur in a fairly broad range of stoichiometries between tau and polyanions but decrease when polyanions are too high or too low (18). Analogous features have been observed with the coacervate-like assembly of polymorphic tubulin aggregates (polyanionic) in the presence of polycationic factors (34). The interaction can be thought of in terms of an activation of subunits generated by tau’s interaction with polyanions, which, however, need not have a precisely fixed stoichiometry. Nevertheless, even in oxidative conditions and in the presence of polyanions the assembly of PHFs is extremely slow at the typical concentrations of tau in neurons (μM or less; ref. 35). This implies that there is another rate-limiting step in PHF assembly that ensures that this process does not occur in normal, healthy cells.
We have addressed this problem with kinetic methods that recently have become available and show that PHF assembly follows a nucleation–elongation mechanism. This means that even if there are assembly-competent subunits, their polymerization is rate-limited by a nucleation step. The overall assembly reaction is illustrated in Fig. 6 and can be broken down into the following stages:
Figure 6.

Model of intermediate stages of nucleated PHF assembly. Tau monomers form homodimers by oxidation of SH groups; these dimers represent the effective assembling species of PHFs (“subunits”). About 4–7 dimers can form nuclei, which then elongate into PHFs, but this process normally is very slow (days to weeks). Nucleation and elongation of PHFs are greatly accelerated by anionic cofactors. Note that the stoichiometry between tau and polyanions can be variable, and that the arrangement of tau molecules in the nucleus of in PHFs is not known. The rate-limiting step of nucleation can be circumvented by adding external seeds obtained by fragmentation of preformed PHFs.
(1) Dimerization of tau to form subunits: 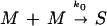
(2) Nucleation of PHFs: 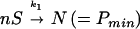
(3) Elongation of PHFs: 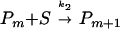 .
.
The rate constants depend strongly on the environment of the cell and can only be estimated since the overall process takes several years. From the in vitro experiments it appears that the major rate limitation lies in steps 1 and 3. Dimerization of tau by oxidation of sulfhydrils is unlikely in the reducing environment of a healthy cell but will become possible with age when oxidative damage accumulates. This generates the potential subunits of PHF assembly, which can be activated further by interaction with polyanions, thus facilitating the subsequent steps (i.e., increasing k1 and k2). The next kinetic barrier is nucleation, which requires 4–7 subunits to interact cooperatively and therefore depends on high tau concentrations. After that, elongation can proceed with relative ease as a bimolecular reaction.
These features make PHF assembly broadly similar to that of amyloid fiber aggregation in Alzheimer’s disease or other aggregation processes in different neuronal disorders that also follow a nucleation–elongation mechanism (even though the nature of the subunits, the nuclei, and the type of interaction are different; refs. 6, 36, and 37, and reviewed in ref. 19). As in the case of amyloid, the necessity of a cooperative nucleation reaction opens new possibilities of studying PHF assembly and for developing strategies of preventing it in Alzheimer’s and other neurodegenerative diseases. It is likely that neurons could tolerate relatively high concentrations of soluble tau if nucleation could be blocked. Aiming to prevent nucleation by specific inhibitors therefore would appear to be a preferred strategy against PHF assembly. In general, the reaction pathway suggests several possibilities for interfering with PHFs. The first is to boost the cellular reducing potential since this will prevent the dimerization of tau and thus the generation of assembly-competent subunits. Second, the ratio of tau to cytosolic polyanions should remain in a regime where complex formation and acceleration of assembly is minimized (theoretically, this could be achieved by an increase or decrease of the polyanions, away from the range of optimal interaction; ref. 18). Third, keeping the concentration of tau low would reduce all steps in the assembly pathway, particularly the nucleation step (note that in AD, tau appears to be locally up-regulated because of aberrant neurite sprouting; ref. 38). Fourth, since the nucleus of PHF assembly is likely to have a special structure and interaction, it should be possible to target it with specific inhibitory compounds. Fourth, we note that there are other modes of tau assembly that depend on different conditions (e.g., fatty acids) and result in different filaments (40). In these cases, the mechanism of nucleation and elongation is likely to be different as well.
Acknowledgments
We thank Dr. J. Biernat and A. Konopatzki for providing the tau constructs used in this study, and C. Weaver (Albert Einstein College) for preparing the Alzheimer PHFs. This project was supported by a grant from the Deutsche Forschungsgemeinschaft.
ABBREVIATIONS
- AD
Alzheimer’s disease
- PHF
paired helical filaments
- ThS
thioflavine S
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Selkoe D J. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 2.Johnson G V, Jenkins S M. Alz Dis Rev. 1996;1:38–54. [Google Scholar]
- 3.Friedhoff P, Mandelkow E. In: Guidebooks to the Cytoskeletal and Motor Proteins and to the Extracellular Matrix and Adhesion Proteins. Kreis T, Vale R, editors. Oxford: Oxford Univ. Press; 1998. [Google Scholar]
- 4.Braak H, Braak E. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 5.Crowther R A. Curr Opin Struct Biol. 1993;3:202–206. doi: 10.1016/j.sbi.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 6.Fraser P E, McLachlan D R, Surewicz W K, Mizzen C A, Snow A D, Nguyen J T, Kirschner D A. J Mol Biol. 1994;244:64–73. doi: 10.1006/jmbi.1994.1704. [DOI] [PubMed] [Google Scholar]
- 7.Jarrett J T, Lansbury P T. Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 8.Cleveland D W, Hwo S Y, Kirschner M W. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 9.Lee G, Cowan N, Kirschner M. Science. 1988;239:285–288. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- 10.Wille H, Drewes G, Biernat J, Mandelkow E M, Mandelkow E. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schweers O, Schönbrunn-Hanebeck E, Marx A, Mandelkow E. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 12.Wischik C, Novak M, Thogersen H, Edwards P, Runswick M, Jakes R, Walker J, Milstein C, Roth M, Klug A. Proc Natl Acad Sci USA. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schweers O, Mandelkow E M, Biernat J, Mandelkow E. Proc Natl Acad Sci USA. 1995;92:8463–8467. doi: 10.1073/pnas.92.18.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goedert M, Jakes R, Spillantini M G, Hasegawa M, Smith M, Crowther R. Nature (London) 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 15.Perez M, Valpuesta J M, Medina M, Degarcini E M, Avila J. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- 16.Hasegawa M, Crowther R A, Jakes R, Goedert M. J Biol Chem. 1997;272:33118–33124. doi: 10.1074/jbc.272.52.33118. [DOI] [PubMed] [Google Scholar]
- 17.Kampers T, Friedhoff P, Biernat J, Mandelkow E M. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- 18.Friedhoff P, Schneider A, Mandelkow E-M, Mandelkow E. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 19.Lansbury P J. Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner S B, Scott M R, DeArmond S J, Cohen F E. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 21.Biernat J, Mandelkow E M, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H E, Mercken M, Vandermeeren A, Goedert M, et al. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent I J, Davies P. Proc Natl Acad Sci USA. 1992;89:2878–2882. doi: 10.1073/pnas.89.7.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jicha, G. A., O’Donnell, A., Weaver, C., Angeletti, R. & Davies, P. (1998) J. Neurochem., in press. [DOI] [PubMed]
- 24.Wegner A, Engel J. Biophys Chem. 1975;3:215–225. doi: 10.1016/0301-4622(75)80013-5. [DOI] [PubMed] [Google Scholar]
- 25.Lee V M Y, Balin B J, Otvos L, Trojanowski J Q. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Braak E. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 27.Arriagada P V, Growdon J H, Hedley-Whyte E, Hyman B T. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 28.Terry R D. J Neuropathol Exp Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- 29.Crowther R A, Olesen O F, Smith M J, Jakes R, Goedert M. FEBS Lett. 1994;337:135–138. doi: 10.1016/0014-5793(94)80260-2. [DOI] [PubMed] [Google Scholar]
- 30.Novak M, Kabat J, Wischik C M. EMBO J. 1993;12:365–370. doi: 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shigenaga M K, Hagen T M, Ames B N. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ginsberg S D, Crino P B, Lee V M Y, Eberwine J, Trojanowski J Q. Ann Neurol. 1997;41:200–209. doi: 10.1002/ana.410410211. [DOI] [PubMed] [Google Scholar]
- 33.Schwab C, McGeer P L. Neurobiol Aging. 1998;19:41–45. doi: 10.1016/s0197-4580(97)00165-6. [DOI] [PubMed] [Google Scholar]
- 34.Erickson H P, Voter W A. Proc Natl Acad Sci USA. 1976;73:2813–2817. doi: 10.1073/pnas.73.8.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drubin D, Kirschner M. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilbich C, Kisters-Woike B, Reed J, Masters C, Beyreuther K. J Mol Biol. 1992;228:460–473. doi: 10.1016/0022-2836(92)90835-8. [DOI] [PubMed] [Google Scholar]
- 37.Jarrett J T, Berger E P, Lansbury P T. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 38.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Deteresa R, Terry R, Baudier J, Saitoh T. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 39.Goedert M, Spillantini M, Potier M, Ulrich J, Crowther R. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson D, Binder L I. Am J Pathol. 1997;150:2181–2219. [PMC free article] [PubMed] [Google Scholar]