

Abstract
Several recurrent, constitutional genomic disorders are present on chromosome 22q. These include the translocations and deletions associated with DiGeorge and velocardiofacial syndrome and the translocations that give rise to the recurrent t(11;22) supernumerary der(22) syndrome (Emanuel syndrome). The rearrangement breakpoints on 22q cluster around the chromosome-specific segmental duplications of proximal 22q11, which are involved in the etiology of these disorders. While the deletions are the result of nonallelic homologous recombination (NAHR) between low copy repeats or segmental duplications within 22q11, the t(11;22) is the result of rearrangement between palindromic AT-rich repeats on 11q and 22q. Here we describe the mechanisms responsible for these recurrent rearrangements, discuss the recurrent deletion endpoints that are the result of NAHR between chromosome 22q specific low copy repeats as well as present current diagnostic approaches to deletion detection.
Keywords: 22q11.2 rearrangement mechanisms, segmental duplications, 22q11.2 deletion diagnosis
INTRODUCTION
The frequency of the 22q11.2 deletion and complexity of the phenotype makes this disorder a significant health problem. The 22q11.2 deletion results in a syndromic phenotype that can include significant cardiac defects, thymic, parathyroid, craniofacial, developmental, neurological and behavioral manifestations. Using fluorescence in situ hybridization (FISH) and/or PCR of polymorphic markers, the position of the proximal and distal deletion endpoints have been localized in several large patient groups to determine the size of the deletion [Lindsay et al.,1995a; Carlson et al., 1997; Kerstjens-Frederikse et al., 1999; Shaikh et al., 2000; Saitta et al., 2004; Weksberg et al., 2007]. On the basis of the observation of recurrent proximal and distal deletion endpoints, it has been demonstrated that the deletion endpoints cluster and that there is a typically deleted region (TDR) of ~3 Mb in >85% of patients. In addition, there are recurrent, nested, variant deletions mostly with different distal deletion endpoints. Although there are rare, unique, smaller atypical deletions and deletions that do not overlap the TDR, many of these variant deletion endpoints tend to occur in similar regions and with clustered breakpoints [Amati et al., 1999; Saitta et al., 1999; Shaikh et al., 2007].
The chromosome 22q11.2 deletion syndrome (22q11.2DS) is considered a genomic disorder [Lupski, 1998]. The term genomic disorder is typically used to describe a gain (duplication) or loss (deletion) of a specific chromosomal region, associated with a clinical genetic syndrome that may present with congenital anomalies, or with impairment in neurological and cognitive function. In general, the clinical features are believed to reflect changes in normal copy number or dosage of the genes contained within a given genomic interval, one or more of which contribute to the resulting phenotype. Less frequently the aberrant phenotype may be the result of gene disruption, creation of a fusion gene, a position effect or the unmasking of a recessive gene mutation. Technological improvements, coupled with higher quality sequence and better annotation, have revealed a number of underlying mechanistic etiologies for genomic disorders. In this article, we highlight the impact that genomic sequence and new molecular approaches have had on progress toward understanding the mechanisms underlying the genomic rearrangements responsible for the 22q11.2DS and their diagnosis.
MECHANISMS OF RECURRENT REARRANGEMENTS
Geneticists and cytogeneticists have long been interested in understanding the mechanisms and consequences of genomic rearrangement. Chromosomal deletions and duplications, many visible at the cytological level, are the result of DNA alterations at the molecular level. Nonrandom chromosomal changes have been demonstrated in association with a number of human diseases. The well studied nonrandom, constitutional abnormalities of 22q include the duplications associated with the supernumerary bisatellited marker chromosome of Cat Eye syndrome (CES) [McDermid et al., 1986], the translocations which give rise to the recurrent t(11;22) mal-segregation-derived Supernumerary der(22)t(11;22) syndrome or Emanuel syndrome (MIM# 609029) [Fraccaro et al., l980; Iselius et al., 1983; Zackai and Emanuel, 1980], and the translocations and deletions associated with DiGeorge, Velocardiofacial, and conotruncal anomaly face syndromes (DGS/VCFS/CAFS) [De La Chapelle et al., l98l; Kelley et al., l982; Driscoll et al., 1992a,b, 1993; Carey et al., 1992; Burn et al., 1993; Summar et al., 1995].
Several chromosomal regions, including 22q11, are characterized by the presence of chromosome specific, low-copy repeats, or segmental duplications. Many of these segmental duplications flank the genomic regions associated with known human deletion and duplication syndromes and have been regarded as part of the underlying basis for genomic disorders. Segmental duplications share a high level of sequence identity predisposing the regions they occupy to nonallelic homologous recombination (NAHR). The result of NAHR can be deletions, duplications, and inversions [Shaffer and Lupski, 2000; Feuk et al., 2006]. Such segmental duplications might allow for mispairing and unequal crossing over between homologous chromosomes (interchromosomal) or allow intrachromosomal recombination events to take place within a single or between sister chromatids. There is significant evidence that such a mechanism is responsible for the 22q11.2 deletion [Baumer et al., 1998; Saitta et al., 2004]. In addition, there is evidence of nonrandom asynchronous replication at 22q11.2 with earlier replication of paternal alleles [Baumer et al., 2004]. This has been proposed as a risk factor for formation of the deletion by increasing the likelihood of mispairing at paternal alleles of the low copy repeats and fostering an aberrant exchange. Further, chromosome 22, contains unusual DNA configurations within its segmental duplications, palindromic AT-rich repeats that appear to predispose 22q11.2 to engage in significant numbers of both random and recurrent translocations [Li et al., 1995; Kurahashi et al., 2000a,b; Edelmann et al., 2001, 2003; Nimmakayalu et al., 2003; Spiteri et al., 2003; Gotter et al., 2004, 2007].
The 22q11.2DS has been characterized as one of the most frequent of the genomic disorders. On the molecular level, the 22q11.2 deletion results from variable-sized deletions of regions of 22q11, ranging in size from small deletions (the minimum size is not yet known) to deletions spanning approximately 3 megabases (Mb). Several groups, including our own, have found that only between 6% and 25% of deletions are inherited [Leana-Cox et al., 1996; Ryan et al., 1997; Matsuoka et al., 1998; McDonald-McGinn et al., 1997, 2001]. Instead, the overwhelming majority of deletions occur de novo. The prevalence of these de novo 22q11.2 deletions indicates an extremely high “mutation” rate within this region (~2.5 × 10−4). Investigators have evaluated possible factors that predispose 22q11.2 to frequent deletional events in order to determine the mechanisms involved. A factor that appears to be very much involved is the presence of the “recombination permissive” duplicated DNA sequences in 22q11.2 [Edelmann et al., 1999b; Shaikh et al., 2000, 2001].
The 22q11 deletion interval contains at least four large blocks of duplicated DNA sequence, which appear to coincide with the common recurrent deletion endpoints. The location and organization of these segmental duplications in the vicinity of the deletion endpoints strongly implicated them in the events leading to deletion. Once the entire 3 Mb TDR was sequenced, detailed investigation of the duplicated sequence blocks and their involvement in the 22q11.2 deletion became possible. Sequence analysis characterized the four distinct segments of duplicated sequence within the 3 Mb TDR and several additional segments of duplicated sequence distal to the TDR (Fig. 1). Although the blocks of duplicated sequence differ in content and organization of shared modules, those modules that are common between blocks share 97–98% sequence identity with one another (Fig. 2). By FISH analysis, it was found that the end-points of the recurrent 22q11.2 deletions leading to the 22q11.2DS appear to localize to the blocks of duplicated sequence [Edelmann et al., 1999b; Shaikh et al., 2000, 2001]. Pulsed-field gel electrophoresis (PFGE) and Southern hybridization were used to identify rearranged junction fragments from several recurrent variant deletions. Analysis of junction fragments by PCR and sequencing of the PCR products implicated the duplicated sequence blocks directly in the formation of 22q11.2 deletions [Shaikh et al., 2000, 2001].
Fig. 1.
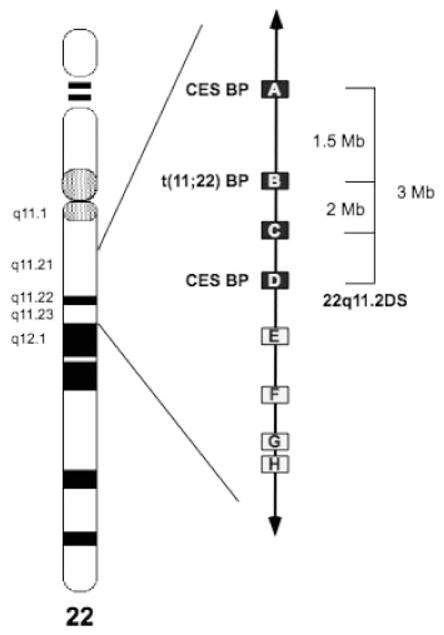
An ideogram of chromosome 22 indicating the position of the low copy repeats (LCRs) in 22q11.2. The chromosome bands involved in the rearrangements of 22q11 are as indicated. Boxes indicate the position of the LCRs. Filled boxes (LCRs through D) are those that have been documented to be involved in numerous recurrent rearrangements. In brackets are indicated the recurrent nested deletions of chromosome 22q11.2. Open boxes (LCRs E through H) indicate LCRs that have only rarely been implicated in rearrangements. The proximal and distal breakpoints for the marker chromosomes seen in association with the cat eye syndrome and the t(11;22) breakpoint region are also noted.
Fig. 2.

A schematic depiction of low copy repeats (LCRs) that mediate genomic instability on chromosome 22. The 22q11 region is enlarged, represented by the blue line with arrows at both ends, and is drawn from centromere (left) to telomere (right). The complex modular LCRs that characterize this region are represented by the multicolored vertical rectangular boxes, each color representing a stretch of DNA sequence repeated in multiple LCRs. The BCRL module is depicted by a royal blue box that is present in all of the LCRs except LCR-B. Zigzag lines represent the locations of multiple recurrent breakpoints including those for the deletions seen in the 22q11.2 deletion syndrome patients, the duplications seen in the Cat Eye syndrome, and the translocations involving 22q. Light blue horizontal rectangles on the lower portion of the diagram indicate the extent of the prevalent, recurrent LCR-mediated 22q deletions seen in the 22q11.2 deletion syndrome. It has been demonstrated that palindromic AT rich repeats in the white “gap” indicated in LCR-B of 22q11 can form hairpins and cruciforms that mediate translocations between chromosome 22 and several other partner chromosomes. Concept and design courtesy of Tamim H. Shaikh, Ph.D.
The complexity and large size of the low copy repeats responsible for the typical recurrent 22q11.2 deletions has prevented the precise, to the nucleotide, localization of the deletion breakpoints within the four proximal 22q11 low copy repeats. Recently, a study was performed to elucidate the deletion mechanism by utilizing two patients with less common distal deletions of 22q11 that were mediated by the distal, lower complexity low copy repeats [Shaikh et al., 2007]. This approach allowed for the successful identification of deletion breakpoints within these 22q11 low copy repeats. Both deletions share a common endpoint, LCR-E (Figs. 1 and 2). The LCR-E breakpoints of each of the two patients are located ~8 kb apart within a duplicated module containing the BCR-like (BCRL) marker, suggesting that the DNA sequence within this module may predispose the region to rearrangement. Interestingly, the sequences that correspond to the BCRL module within the low copy repeats in 22q11 had previously been suggested as a potential rearrangement hotspot within LCRs-A (LCR22-2) and -D (LCR22-4) [Pavlicek et al., 2005]. This region, designated ΨBCR, is enriched in shared polymorphic sites (SPSs) and poor in paralogous sequence variants, making it a hotspot for gene conversion and consequently for meiotic crossover and rearrangement [Hurles et al., 2004; Pavlicek et al., 2005]. The coincidence between these sequenced breakpoints and the predicted rearrangement hotspot is highly suggestive that this BCRL motif plays a role in the deletion mechanism.
Although the deletions whose breakpoints have been isolated and sequenced do not overlap, it is of interest that their breakpoints localize to the BCRL module, the only duplicated module common to almost all (7/8) of the 22q11 low copy repeats that have been entirely sequenced (Fig. 2). LCR-B still contains a gap in its sequence and it is still hypothetically possible that a BCRL module resides within the remaining gap. The orientation of the BCRL modules within the seven low copy repeats known to contain them may predict which low copy repeats are likely to mediate NAHR since it has been suggested that elements within the low copy repeats that have a direct orientation with respect to one another are likely to mediate deletions and duplications [Shaffer and Lupski, 2000]. The ability to generate and utilize oligonucleotide based array CGH data should permit the direct localization of breakpoint junctions in the near future.
It has been well-documented that LCR-B in 22q11 is one of the most rearrangement prone sites in the human genome, where the breakpoints of a number of constitutional translocations cluster. This breakage sensitive region is located within an unclonable gap left from the human genome project, suggestive of a specific sequence recalcitrant to cloning. Recently, a part of this gap has been cloned and found to contain a novel 595 bp palindromic AT-rich repeat [Kurahashi et al., 2007]. The breakpoints of numerous unrelated t(11;22) cases have been consistently shown to be located within the palindromic AT-rich repeats located in 22q11, as well as on 11q23 [Kurahashi et al., 2000a,b, 2007, Edelmann et al., 2001, Tapia-Paez et al., 2001]. The majority of the t(11;22) breakpoints have been localized at the center of the palindromic AT-rich repeats, suggesting that the center of the palindrome is susceptible to double strand breaks inducing illegitimate chromosomal rearrangement. Junction fragments often possess small size deletions at the breakpoints on both chromosomes as well as an occasional insertion of a single nucleotide at the junction. This supports repair via a nonhomologous end-joining pathway.
The breakpoints of a number of translocations involving 22q11 cluster within this palindromic AT-rich region, suggesting that this region is highly unstable in the human genome as well as in the host organisms used for cloning. Recent findings of palindromic sequences at the translocation breakpoints of other chromosome 22q11 partner chromosomes supports the conclusion that palindrome-mediated chromosomal translocation appears to be one of the universal pathways for human genomic rearrangements [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2003; Nimmakayalu et al., 2003; Gotter et al., 2004, 2007]. To date, several translocations involving chromosome 22q11 in addition to the t(11;22) have been analyzed in detail [Kurahashi et al., 2003; Nimmakayalu et al., 2003; Gotter et al., 2003, 2007]. Although the complete extended breakpoint regions from 22q11 have not been cloned, junction fragments from both translocation derivative chromosomes have been isolated. A putative structure for the 22q11.2 breakpoint region has been inferred from sequence information of the junction fragments of the numerous t(11;22) and the less frequent translocations of 22q11 with other autosomes. These data suggest the presence of palindromic sequences at all of the breakpoints that occur within LCR-B. Some, but not all are AT-rich in nature. Although LCR-B is the second most frequent 22q11.2 deletion endpoint, the role that the palindromic AT-rich repeats play in the generation of the 22q11.2DS has yet to be determined.
RECURRENT DELETION ENDPOINTS IN THE 22Q11.2 DELETION SYNDROME
We have had the opportunity to determine the size of the 22q11.2 deletion in a significant number of affected individuals. These experiments were performed using dual color FISH with the single copy probes that we generated as the tiling path for sequencing of 22q11 as part of the Genome Center for Chromosome 22 [Dunham et al., 1999]. Families of patients with clinical features of DGS/VCFS and a deletion of 22q11.2 as determined by FISH with the N25 (D22S75) diagnostic probe (Vysis) were the starting material for these studies. FISH was used to study peripheral blood lymphocyte chromosome preparations from the deleted patients and their parents in order to identify familial deletions. Thus, the deletions enumerated (Fig. 3) occurred de novo in the proband or, in the rare familial cases, only a single member of a family is reported. Figure 3 depicts the relevant 22q11.2 region including the low copy repeats (LCRs) and relative location of markers and probes used for FISH. Probes adjacent to LCRs A and D (deletion endpoints of the standard 3 Mb deletion) and within that interval were used to determine the extent of the deletion. We found that in a group of 300 patients, 259 of the deletions (86.3%) span the same 3 Mb region from LCR A to LCR D. From the same cohort of 300, we found that 22 (7.3%) had a smaller approximately 1.5 Mb deletion extending from LCR-A to LCR-B (Fig. 3). Smaller numbers of individuals had other recurrent deletions (i.e., from A-C and from a small block internal to A or A′-D in Fig. 3). Further, there were nine deletions with atypical endpoints. These latter cases required numerous additional experiments using probes different from the standard deletion-sizing probe set. These probes are the same clones derived from the cosmid contig that was used to generate the sequence of the chromosome 22q11.2 region [Dunham et al., 1999].
Fig. 3.

Diagrammatic representation of the TDR on 22q11.2q and deletion size in 300 patients. The orientation of the region is centromere (left) to telomere (right). LCRs-A, -B, -C, and -D are indicated as filled boxes. The small, hatched box between markers S22S36 and D22S75 indicates the proximal deletion endpoint (A′) of several variant deletions not deleted for cosmid c103a2. The cosmids used as FISH probes for deletion sizing are indicated with asterisks. Above the line are the markers from chromosome 22q contained within the cosmid clones.
This mechanism of deletion, involving meiotic homologous recombination after misalignment of low copy repeats (LCRs), should result in one deleted chromosome, and one chromosome containing a duplication of the deleted sequence. Although individuals with 22q11 duplications have been identified, they seem to be fewer than anticipated based upon the prevalence of the deletion. Nonetheless, they often seem to share some phenotypic similarities with patients who have the 22q11.2 deletion [Lindsay et al., 1995b; Edelmann et al., 1999a; Ensenauer et al., 2003; Meins et al., 2003; Hassed et al., 2004; Portnoi et al., 2005; Yobb et al., 2005; Vorstman et al., 2006; de La Rochebrochard et al., 2006; Mukaddes and Herguner, 2007; Alberti et al., 2007; Jalali et al., 2007], suggesting that they might be amongst the individuals who are clinically considered to be nondeleted patients. This indicates that a better diagnostic modality to determine variation in 22q11.2 DNA copy number is indicated for such individuals. Further, numerous individuals with the clinical phenotype who do not appear to harbor detectable deletions or mutations have been described, suggesting that they would also benefit from better diagnostic tools. Limited numbers of mutations in genes within the TDR have been identified in such patients suggesting that another, more comprehensive diagnostic approach is likely to be required [Gong et al., 2001; Yagi et al., 2003; Paylor et al., 2006].
DIAGNOSTIC APPROACHES TO DELETION DETECTION AND DELETION ENDPOINT IDENTIFICATION
The diagnostic procedure most often used for detection of deletions and duplications at 22q11.2 is chromosomal analysis coupled with FISH using commercially available probes located between LCRs A and B. For deletion size analysis multiple FISH experiments or characterization with polymorphic markers have been the preferred methodologies employed [Carlson et al., 1997; Saitta et al., 1999; Shaikh et al., 2000]. Recently, we as well as others have shown that the existing 22q11.2DS MLPA kit (P023; MRC-Holland, Amsterdam) is a cost-effective, rapid, and sensitive method for the detection of the typical recurrent deletions and duplications in proximal 22q11 extending to LCR-D, [Fernandez et al., 2005; Vorstman et al., 2006]. However, copy number changes at 22q11.2 have been identified that would not have been detected by the current commercially available diagnostic FISH probes or the existing MLPA P023 kit [Vorstman et al., 2006; Sivertsen et al., 2007]. The detection and analysis of these genomic copy number alterations at 22q11.2 is significant, because, to date, little information is available with regard to their prevalence and whether there are consistently associated phenotypic differences. Indeed, identification of these variant cases is of particular interest since it may provide insight into which genes or genomic regions are crucial for specific phenotypic manifestations and are likely to assist in the quest to determine deletion and duplication mechanisms.
Therefore, in collaboration with MRC-Holland, a high-density MLPA (HDMLPA) probe set has been developed [Jalali et al., 2007]. The new HDMLPA probe set incorporates probes starting proximal to LCR-A and covers the region flanked by the four LCRs distal to LCR-D. LCR-D is the distal boundary of the typical 22q11.2 deletions (Fig. 4). The HDMLPA kit has been designed to contain 37 loci located on the long arm of chromosome 22. Most of the probe sets are located within genes, of which 15 loci are located between LCR-A and LCR-D. Three unique loci are located within LCR-D designed to detect differences in deletion endpoints. The HDMLPA kit provides more extensive coverage of the TDR within 22q11.2 allowing for the identification and characterization of atypical deletions. It also includes 10 probe sets that are located distal to LCR-D in the interval between LCR-D and LCR-H. The ability of this new high-density (HD) probe set to detect copy number alterations on numerous samples with copy number changes at 22q11.2 has been successfully tested [Jalali et al., 2007].
Fig. 4.

Graphical representation of HDMLPA data. Shown above the representation of chromosome 22 are the FISH probes (cosmids) used in the laboratory designated either by cosmid address or locus name. The MLPA probes (orange rectangles, existing MLPA PO23 loci; yellow rectangles, new MLPA loci) are shown in map order based upon data derived from the UCSC genome browser. All MLPA loci were tested in all samples. Copy number for MLPA probes is indicated by color (grey = disomic, 2 copies; red = hemizygous deletion). The 22q11.2 deletions studied are shown as samples 1–10: (1,2) CH99-023 and CH02-125 with 3 Mb LCR-A to LCR-D deletions. Patient 2 has an extended deletion. (3,4) CH06-198, DO6-231 with LCR-B to LCR-D deletions. (5,6) 06-006997 and 07000363 with a distal variant of LCR-B to LCR-D deletions. (7) 06-006837 an atypical deletion. (8) 06-004115 a LCR-C to LCR-D deletion. (9) CH98-018 a LCR-D to LCR-E deletion, (10) 07-0000553 a LCR-D to LCR-F deletion.
Because the new assay is highly accurate with excellent sensitivity and specificity values, we have adopted HDMLPA as the method of choice for detection of copy number differences in 22q11.2. The new MLPA probe set correctly detects gains or losses of genomic material, accurately indicates the number of allelic copies (from n = 0 to n = 4) and is able to delineate the extent of the region involved in the rearrangement. Using the HDMLPA probe set, it has been demonstrated that the group of patients with a 3 Mb deletion is not a homogeneous group. Several patients clearly showed a difference in deletion size. Examination of 77 patients with typical deletions of LCR-A to LCR-D showed that 6.5% of these patients had deletions that extended to include the HIC2 locus. The signals of the HIC2 probe in normal control samples were found to cluster closely around 1.0 (n = 2), which indicated a good performance for this particular probe. It is of interest that although the proximal part of the HIC2 region is within a reported CNV region, the MLPA probe covering HIC2 is outside the copy number variant region [Wong et al., 2007, http://projects.tcag.ca/variation/]. This variation in deletion endpoint might eventually be of utility in explaining differences in the mechanism of the rearrangement in affected patients as well in finding explanations for the extreme variability in the spectrum of the phenotype.
Several groups have also evaluated the use of array-based comparative genomic hybridization (aCGH) as another efficient tool for identifying chromosomal imbalances such as deletions and duplications of 22q11.2 [Krepischi-Santos et al., 2006; Urban et al., 2006; Stanczak et al., 2007; Tokuyasu et al., 2007]. Similar to MLPA, aCGH allows simultaneous interrogation of numerous DNA probes. Thus, it offers an efficient and high-throughput alternative for detecting microdeletions and duplications. For example, a clone-based platform was applied to 44 patients with a phenotype of the 22q11.2 deletion syndrome. Twenty-five patients had the deletion of chromosome 22 characteristic of this syndrome as determined by FISH. This array contained 2464 BAC, PAC, and P1 clones that provide coverage with an average 1.4 Mb spacing across the human genome. Array measurements were in complete concordance with FISH analysis, supporting the diagnostic utility of the approach [Tokuyasu et al., 2007]. Another clone-based platform was able to identify three patients with overlapping deletions of 22q11.21, who were ascertained by an atypical clinical presentation [Krepischi-Santos et al., 2006]. Their features, which were not typical or specific enough to allow diagnosis before detection of chromosome 22q11.2 imbalances, included a heart defect, uterovaginal aplasia, and mental retardation associated with psychotic disease. The results of this study demonstrated that ascertainment through whole-genome screening of syndromic patients by array-CGH can lead to the recognition of a broader spectrum of features for the 22q11.2DS. Thus, aCGH may uncover 22q11.2 deletions that were not suspected on the basis of phenotype.
Several oligonucleotide-based platforms have recently been used to provide proof of principle that the technology could be successfully applied for detection of the hemizygous loss of DNA in the 22q11.2DS, including the Affymetrix 50K array [Stanczak et al., 2007]. In addition, arrays with an 85-bp tiling path of chromosome 22 were used to analyze DNA from patients heterozygous deletions and duplications as well as partial triploidies and partial tetraploidies of chromosome 22q11.2. The alterations were mapped with high resolution (typically up to 200 bp) in each patient, and the precise breakpoints of two deletions were confirmed by DNA sequencing [Urban et al., 2006]. In this study, it was demonstrated that high resolution CGH allows prediction of the extent of deletions that stretch into LCR regions, showing differences in deletion endpoints. These findings further demonstrate that differences in gene complement might ultimately allow for substantial progress in explaining differences in phenotype between patients with the 22q11.2DS whose deletions involve what appears to be the same region. Thus, the ultimate choice for diagnostic purposes will depend upon the level of suspicion of a 22q11.2 anomaly and the relative cost to the patient. MLPA should be considered as a cost-effective diagnostic screen, whereas oligo-based aCGH will be definitive as a final test as it will be able to detect 22q11 copy number changes in patients with nonclassic phenotypes.
It is clear that newer higher resolution techniques such as MLPA, BAC or oligonucleotide based microarrays are now capable of providing more sensitive and rapid breakpoint localization without the need for reiterative FISH or PCR-based experiments [Vorstman et al., 2006; Urban et al., 2006; Jalali et al., 2007]. Application of these techniques has permitted the identification of numerous distal deletions, interstitial duplications and has also facilitated breakpoint differences in several proximal 22q11.2 deletions as well [Urban et al., 2006; Jalali et al., 2007]. In this manner, investigations could be performed to determine whether the BCRL modules serve as “hotspots” for aberrant recombination mediating the DGS/VCFS deletions, as the data from cloned distal deletions suggests. The ability to localize, clone, and sequence breakpoints within the LCRs of 22q11.2, has also allowed the identification of structural variation within the genome that may predispose a given chromosome to NAHR or translocation. This not only has implications for the deletion mechanisms of chromosome 22q11.2 in particular, but also provides important insights related to the role of genomic architecture in chromosomal rearrangements, chromosome evolution, and in human disease.
Acknowledgments
National Organization for Rare Disorders (NORD); Grant numbers: HL74731, HL84410, CA39926.
The authors research efforts are also supported by funds from the Charles E. H. Upham endowed chair in Pediatrics. The author gratefully acknowledges Dr. Tamim Shaikh for kindly providing the figure showing the organization of the LCRs on chromosome 22 and Dr. G. Reza Jalali for providing the HDMLPA figure graphic.
References
- Alberti A, Romano C, Falco M, et al. 1.5 Mb de novo 22q11.21 microduplication in a patient with cognitive deficits and dysmorphic facial features. Clin Genet. 2007;71:177–182. doi: 10.1111/j.1399-0004.2007.00750.x. [DOI] [PubMed] [Google Scholar]
- Amati F, Conti E, Novelli A, et al. Atypical deletions suggest five 22q11.2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur J Hum Genet. 1999;7:903–909. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- Baumer A, Dutly F, Balmer D, et al. High level of unequal meiotic crossovers at the origin of the 22q11. 2 and 7q11.23 deletions. Hum Mol Genet. 1998;7:887–894. doi: 10.1093/hmg/7.5.887. [DOI] [PubMed] [Google Scholar]
- Baumer A, Riegel M, Schinzel A. Non-random asynchronous replication at 22q11.2 favours unequal meiotic crossovers leading to the human 22q11.2 deletion. J Med Genet. 2004;41:413–420. doi: 10.1136/jmg.2003.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn J, Takao A, Wilson D, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22. J Med Genet. 1993;30:822–824. doi: 10.1136/jmg.30.10.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AH, Kelly D, Halford S, et al. Molecular genetic study of the frequency of monosomy 22q11 in DiGeorge syndrome. Am J Hum Genet. 1992;5:964–970. [PMC free article] [PubMed] [Google Scholar]
- Carlson C, Sirotkin H, Pandita R, et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet. 1997;61:620–929. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Chapelle A, Herva R, Koivisto M, et al. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57:253–256. doi: 10.1007/BF00278938. [DOI] [PubMed] [Google Scholar]
- de La Rochebrochard C, Joly-Helas G, Goldenberg A, et al. The intrafamilial variability of the 22q11.2 microduplication encompasses a spectrum from minor cognitive deficits to severe congenital anomalies. Am J Med Genet A. 2006;140:1608–1613. doi: 10.1002/ajmg.a.31227. [DOI] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992a;50:924–933. [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Spinner NB, Budarf ML, et al. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet. 1992b;44:261–268. doi: 10.1002/ajmg.1320440237. [DOI] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counseling and pre-natal diagnosis. J Med Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Shimizu N, Roe BA, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet. 1999a;64:1076–1086. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Spiteri E, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999b;8:1157–1167. doi: 10.1093/hmg/8.7.1157. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Spiteri E, Koren K, et al. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am J Hum Genet. 2001;68:1–13. doi: 10.1086/316952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensenauer RE, Adeyinka A, Flynn HC, et al. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am J Hum Genet. 2003;73:1027–1040. doi: 10.1086/378818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez L, Lapunzina P, Arjona D, et al. Comparative study of three diagnostic approaches (FISH, STRs and MLPA) in 30 patients with 22q11.2 deletion syndrome. Clin Genet. 2005;68:373–378. doi: 10.1111/j.1399-0004.2005.00493.x. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, et al. The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet. 1980;56:2l–5l. doi: 10.1007/BF00281567. [DOI] [PubMed] [Google Scholar]
- Gong W, Gottlieb S, Collins J, et al. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. doi: 10.1136/jmg.38.12.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Nimmakayalu MA, Jalali GR, et al. A palindrome-driven complex rearrangement of 22q11.2 and 8q24.1 elucidated using novel technologies. Genome Res. 2007;17:470–481. doi: 10.1101/gr.6130907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Shaikh TH, Budarf ML, et al. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum Mol Genet. 2004;13:103–115. doi: 10.1093/hmg/ddh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassed SJ, Hopcus-Niccum D, Zhang L, et al. A new genomic duplication syndrome complementary to the velocardiofacial (22q11 deletion) syndrome. Clin Genet. 2004;65:400–404. doi: 10.1111/j.0009-9163.2004.0212.x. [DOI] [PubMed] [Google Scholar]
- Hurles ME, Willey D, Matthews L, et al. Origins of chromosomal rearrangement hotspots in the human genome: evidence from the AZFa deletion hotspots. Genome Biol. 2004;5:R55. doi: 10.1186/gb-2004-5-8-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iselius L, Lindsten J, Aurias A, et al. The 11q;22q translocation: a collaborative study of 20 new cases and analysis of 110 families. Hum Genet. 1983;64:343–355. doi: 10.1007/BF00292366. [DOI] [PubMed] [Google Scholar]
- Jalali GR, Vorstman JA, Errami A, et al. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum Mutat. 2007 doi: 10.1002/humu.20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Haussler J, Krone W, et al. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum Genet. 1997;99:237–247. doi: 10.1007/s004390050346. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Zackai EH, Emanuel BS, et al. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. Disabil Rehabil. 1982;101:197–200. doi: 10.1016/s0022-3476(82)80116-9. [DOI] [PubMed] [Google Scholar]
- Kerstjens-Frederikse WS, Kurahashi H, Driscoll DA, et al. Microdeletion 22q11.2: clinical data and deletion size. J Med Genet. 1999;36:721–723. [PMC free article] [PubMed] [Google Scholar]
- Krepischi-Santos AC, Vianna-Morgante AM, Jehee FS, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res. 2006;115:254–261. doi: 10.1159/000095922. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Hu P, et al. Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22) Hum Mol Genet. 2000a;9:1665–1670. doi: 10.1093/hmg/9.11.1665. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh TH, Zackai EH, et al. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22) Am J Hum Genet. 2000b;67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh T, Takata M, et al. The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am J Hum Genet. 2003;72:733–738. doi: 10.1086/368062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Hosoba E, et al. Molecular cloning of a translocation breakpoint hotspot in 22q11. Genome Res. 2007;17:461–469. doi: 10.1101/gr.5769507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leana-Cox J, Pangkanon S, Eanet KR, et al. Familial DiGeorge/velocardiofacial syndrome with deletions of chromosome area 22q11: report of five families with a review of the literature. Am J Med Genet. 1996;65:309–316. doi: 10.1002/(SICI)1096-8628(19961111)65:4<309::AID-AJMG12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Greenberg F, Shaffer LG, et al. Submicroscopic deletions at 22q11.2: variability of the clinical picture and delineation of a commonly deleted region. Am J Med Genet. 1995a;56:191–197. doi: 10.1002/ajmg.1320560216. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Shaffer LG, Carrozzo R, et al. De novo tandem duplication of chromosome segment 22q11-q12: clinical, cytogenetic, and molecular characterization. Am J Med Genet. 1995b;56:296–299. doi: 10.1002/ajmg.1320560316. [DOI] [PubMed] [Google Scholar]
- Li M, Budarf ML, Chien P, et al. Clustering of DiGeorge/velocardiofacial-associated translocations suggestive of a translocation “hotspot”. Am J Hum Genet. 1995;57(Suppl):A119. [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. (Review) [DOI] [PubMed] [Google Scholar]
- Matsuoka R, Kimura M, Scambler PJ, et al. Molecular and clinical study of 183 patients with conotruncal anomaly face syndrome. Hum Genet. 1998;103:70–80. doi: 10.1007/s004390050786. [DOI] [PubMed] [Google Scholar]
- McDermid HE, Duncan AMV, Brasch KR, et al. Characterization of the supernumerary chromosome in cat eye syndrome. Science. 1986;232:646–648. doi: 10.1126/science.3961499. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, LaRossa D, et al. The 22q11.2 deletion: screening for deletion, diagnostic workup, and outcome of results. Report on 181 patients. Genet Test. 1997;1:99–108. doi: 10.1089/gte.1997.1.99. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen ML, et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net. Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- Meins M, Burfeind P, Motsch S, et al. Partial trisomy of chromosome 22 resulting from an interstitial duplication of 22q11.2 in a child with typical cat eye syndrome. J Med Genet. 2003;40:e62. doi: 10.1136/jmg.40.5.e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaddes NM, Herguner S. Autistic disorder and 22q11.2 duplication. World J Biol Psychiatry. 2007;8:127–130. doi: 10.1080/15622970601026701. [DOI] [PubMed] [Google Scholar]
- Nimmakayalu MA, Gotter AL, Shaikh TH, et al. A novel sequence-based approach to localize translocation breakpoints identifies the molecular basis of a t(4;22) Hum Mol Genet. 2003;12:2817–2825. doi: 10.1093/hmg/ddg301. [DOI] [PubMed] [Google Scholar]
- Pavlicek A, House R, Gentles AJ, et al. Traffic of genetic information between segmental duplications flanking the typical 22q11.2 deletion in velo-cardio-facial syndrome/DiGeorge syndrome. Genome Res. 2005;15:1487–1495. doi: 10.1101/gr.4281205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoi MF, Lebas F, Gruchy N, et al. 22q11.2 duplication syndrome: two new familial cases with some overlapping features with DiGeorge/velocardiofacial syndromes. Am J Med Genet A. 2005;137:47–51. doi: 10.1002/ajmg.a.30847. (Review) [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta SC, Harris SE, Gaeth AP, et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum Mol Genet. 2004;13:417–428. doi: 10.1093/hmg/ddh041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta SC, McGrath JM, Mensch H, et al. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Genet. 1999;65:562–566. doi: 10.1086/302514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet. 2000;34:297–329. doi: 10.1146/annurev.genet.34.1.297. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Emanuel BS. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet Med. 2001;3:6–13. doi: 10.1097/00125817-200101000-00003. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O’ Connor RJ, Pierpont ME, et al. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivertsen A, Lie RT, Wilcox AJ, et al. Prevalence of duplications and deletions of the 22q11 DiGeorge syndrome region in a population-based sample of infants with cleft palate. Am J Med Genet A. 2007;143:129–134. doi: 10.1002/ajmg.a.31445. [DOI] [PubMed] [Google Scholar]
- Spiteri E, Babcock M, Kashork CD, et al. Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22s) and telomeric bands of partner chromosomes. Hum Mol Genet. 2003;12:1823–1837. doi: 10.1093/hmg/ddg203. [DOI] [PubMed] [Google Scholar]
- Stanczak CM, Chen Z, Nelson SF, et al. Representational oligonucleotide microarray analysis (ROMA) and comparison of binning and change-point methods of analysis: application to detection of del22q11.2 (DiGeorge) syndrome. Hum Mutat. 2007;29:176–181. doi: 10.1002/humu.20593. [DOI] [PubMed] [Google Scholar]
- Summar ML, Dasouki MJ, Butler MJ, et al. Recurring de novo unbalanced translocations involving 22q11.2, a hot spot for rearrangement? Am J Hum Genet. 1995;57:A128. [Google Scholar]
- Tapia-Páez I, O’Brien KP, Kost-Alimova M, et al. Fine mapping of the constitutional translocation t(11;22)(q23;q11) Hum Genet. 2000;106:506–516. doi: 10.1007/s004390000287. [DOI] [PubMed] [Google Scholar]
- Tokuyasu TA, Cotter PD, Segraves R, et al. Detection of single clone deletions using array CGH: identification of submicroscopic deletions in the 22q11.2 deletion syndrome as a model system. Am J Med Genet A. 2007;143:925–932. doi: 10.1002/ajmg.a.31662. [DOI] [PubMed] [Google Scholar]
- Urban AE, Korbel JO, Selzer R, et al. High resolution mapping of DNA copy alterations in human chromosome 22 using high density tiling oligonucleotide arrays. Proc Natl Acad Sci USA. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Jalali GR, Rappaport EF, et al. MLPA: a rapid, reliable, and sensitive method for detection and analysis of abnormalities of 22q. Hum Mutat. 2006;27:814–821. doi: 10.1002/humu.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksberg R, Stachon AC, Squire JA, et al. Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum Genet. 2007;120:837–845. doi: 10.1007/s00439-006-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, deLeeuw RJ, Dosanjh NS, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yobb TM, Somerville MJ, Willatt L, et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am J Hum Genet. 2005;76:865–876. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackai EH, Emanuel BS. Site-specific reciprocal translocation, t(11;22)(q23;q11) in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet. 1980;7:507–52l. doi: 10.1002/ajmg.1320070412. [DOI] [PubMed] [Google Scholar]