

Abstract
Patients with an apparently balanced translocation and an abnormal phenotype may carry a cryptic deletion/duplication at their translocation breakpoints that may explain their abnormalities. Using microarray CGH (aCGH) and gene expression arrays we studied a child with t(15;22)(q26.1;q11.2), developmental delay and mild dysmorphic features. A high density aCGH study with 244,000 oligo probes demonstrated a 3.3 Mb deletion immediately adjacent to the 15q breakpoint. Gene expression studies with 44,000 oligos displayed an approximately 50% reduction of the expression of IGF1R gene that was translocated to the der(22). There are 18 known or hypothetical protein coding genes within the deleted region according to UniProt, RefSeq, and GenBank mRNA (UCSC HG17, May 2004). Although two of these genes, RGMA and ST8SIA2, play an important role in neural development, the mild phenotype of our patient indicates that loss of one copy of these genes may not be critical developmentally. The 50% reduction of IGF1R expression could be responsible for the growth deficiency in the patient. Reviewing the few 15q26 microdeletion cases that have been characterized by aCGH, we discovered that deletion of the segment including distal 15q26.2 to the proximal part of 15q26.3 is associated with severe phenotypes. Our experience demonstrates that high-density oligonucleotide-based aCGH is a quick and precise way to identify cryptic copy number changes in “balanced translocations.” Expression studies can also add valuable information regarding gene expression changes due to a chromosomal rearrangement. Both approaches can assist in the elucidation of the etiology of unexplained phenotypic differences in cases such as this one.
Keywords: 15q26 microdeletion, chromosome microdeletion, developmental delay, microarray CGH
INTRODUCTION
Chromosome rearrangements involving the distal band of the long arm of chromosome 15 (15q26) are a relatively common cause of multiple congenital abnormalities (MCA) and mental retardation. Most of these cases present themselves as ring chromosome 15 [Tumer et al., 2004; Glass et al., 2006], terminal deletion [Bhakta et al., 2005; Tonnies et al., 2007], or unbalanced translocation involving the terminal part of chromosome 15q [Kato et al., 2001; Mercer et al., 2005]. Common features of ring chromosome 15 syndrome include short stature, microcephaly, triangular facies, cardiac anomalies and mental retardation [Jacobsen, 1966]. Only a few 15q terminal deletion cases have been reported, and most share a similar phenotype with the ring chromosome 15 syndrome. The phenotype in patients with an unbalanced translocation varies significantly depending on the partner chromosome and the size of the segments involved in the translocation. In contrast, interstitial deletion of 15q26 appears to be rare. There have been a few 15q interstitial deletion cases reported, and the majority of them are cytogenetically visible deletions of various sizes in the 15q21.1–26.1 region [Martin et al., 1990; Spruijt et al., 2004]. We present a child with developmental delay, mild dysmorphic features and an apparently balanced translocation t(15;22)(q26.1;q11.2). With high density oligo aCGH and gene expression arrays a cryptic 3.3 Mb deletion within 15q26 was identified and further characterized.
CLINICAL REPORT
The study was approved by the Institutional Review Board of Tulane University Medical School. The patient is a Hispanic male adopted from Guatemala. He was born full-term to a 21-year-old woman with a history of syphilis. Little is known about the pregnancy history or his biological father. His birth weight was 2,449 g. He was rapid plasma reagin (RPR) positive at birth, and has since been fluorescent treponemal antibody (FTA) negative without therapy. He was put in a foster family after birth and was adopted at the age of 81/2 months by an American family. His history was notable for developmental delay. He started to sit and roll over at 81/2 months of age, stand at 18 months, and walk at 20 months. His height and head circumference have been consistently at or below the 3rd centile; his weight has been at or below the 5th centile. Language delay was apparent. Speech therapy was implemented at 18 months of age. Although some improvement was achieved, at 51/2 years of age his language development was at the 3-year-old level. His bone age at 18 months of age was at the 6-month-old level, and at 36 months, the 18-month-old level. He was diagnosed with developmental motor coordination disorder at 23/4 years of age. His fine motor coordination has been greatly improved through continuous physical therapy and occupational therapy. He had two episodes of febrile seizures. Left eye amblyopia was surgically corrected at age 2. The patient showed some mild dysmorphic features including anteverted nares, unilateral auricular pit, fingertip pads, a low posterior hairline, and hirsutism at his back. Echocardiogram, head CT scan, brain MRI, thyroid hormone level, renal function, plasma amino acids, and urine organic acids are all within normal limits.
MATERIALS AND METHODS
Cytogenetics and FISH Studies
A peripheral blood sample was collected from the patient and cultured for chromosome analysis. Metaphase chromosomes were prepared using conventional GTW methods. DNA probes used in FISH studies included a triple probe mix of D15Z1 at 15p11.2, SNRPN at 15q11.2, and PML at 15q22 (Vysis, Inc., Downers Grove, IL) and a bacterial clone (BAC) RP11-262P8 containing the DNA sequence of the IGF1R gene (http://bacpac.chori.org/). FISH studies were performed according to the manufacturer’s suggested protocols with minor modification. FISH images were captured and analyzed using CytoVision ChromoFluor™ (Applied Imaging Corp., San Jose, CA).
Molecular Studies
DNA was extracted from the patient’s blood. Methylation-specific PCR (M-PCR) was performed after Sodium Bisulfite treatment of the DNA. PCR primers specific for the methylated (maternal) and unmethylated (paternal) alleles of the CpG island of the SNRPN gene were used to detect the presence or absence of maternal and paternal copies of the gene [Kubota et al., 1996].
Translocation Breakpoint Mapping
A lymphoblastoid cell line was established from the blood sample using EB virus infection. Chromosome preparations from the cell line were used for translocation breakpoint mapping by FISH. Cosmid probes flanking the chromosome 22 breakpoint were isolated from the LL22NCO3 library as previously described [Budarf et al., 1995]. BAC clones flanking the chromosome 15 breakpoint were selected based on their map location in the UCSC database (http://genome.ucsc.edu/) and purchased from BACPAC resources (http://bacpac.chori.org/). FISH studies were carried out as previously described with minor modifications [Nimmakayalu et al., 2000].
Array CGH and Gene Expression Arrays
aCGH
Agilent Human Genome CGH 1×244K microarrays with an average spatial resolution of 6.4 kb were used in the study (Agilent Technologies, Santa Clara, CA). Genomic DNA from the patient and pooled normal female reference DNA (Promega Corporation, Madison, WI) were digested with AluI and RsaI (Promega Corporation) and labeled using Agilent Genomic DNA Labeling Kit according to manufacturer’s instructions. Patient and reference DNA were labeled with Cy3 and Cy5 respectively and vice versa. Labeled patient DNA and reference DNA were co-hybridized to arrays for 40 hr at 65°C in a rotating oven (Agilent Technologies) at 20 rpm. The arrays were then washed with Agilent wash buffer and scanned by using an Agilent Microarray Scanner.
Expression arrays
The Whole Human Genome Oligo Microarray 4×44K arrays were used in the study. Two-group experiment, diseased versus normal was carried out. cDNA was synthesized from RNA samples from the patient and three normal controls and then used to synthesize fluorescent cRNA. cRNA samples were labeled with a Low Input Linear Amplification Kit (Agilent Technologies) as per the recommended protocol. Labeled cRNA samples were hybridized to 2 4×44K slides using an Agilent Gene Expression Hybridization Kit. Hybridizations were carried out in Agilent’s SureHyb Hybridization Chambers for 17 hr at 65°C in a rotating oven (Agilent Technologies) at 4 rpm. The arrays were then washed using the Agilent Gene Expression Wash Pack and scanned using an Agilent Microarray Scanner.
Data analysis
Scanned data were extracted using Feature Extraction 9.1 software (Agilent Technologies). Extracted data from aCGH were analyzed using CGH Analytics 3.4 software (Agilent Technologies). Genomic copy number changes were identified with the assistance of the Aberration Detection Method 1 algorithm. Extracted data from the expression arrays were analyzed using Gene-Spring GX 7.3 software (Agilent Technologies). Fold change and t-test were utilized for statistical analyses of the expression arrays.
RESULTS
Cytogenetics, FISH, and Molecular Studies
Conventional cytogenetic studies revealed an apparently balanced translocation 46,XY,t(15;22) (q26.1;q11.2) (Fig. 1). A FISH study using three chromosome 15 probes (D15Z1/SNRPN/PML, Vysis, Inc.) resulted in a normal disomic signal pattern. FISH with the probe RP11-262P8 showed two signals, one on the normal 15, and the other, on the der(22) (Fig. 2). M-PCR displayed the presence of a 174 bp methylated allele and a 100 bp unmethylated allele of the SNRPN gene, indicating biparental inheritance of the gene.
Fig. 1.
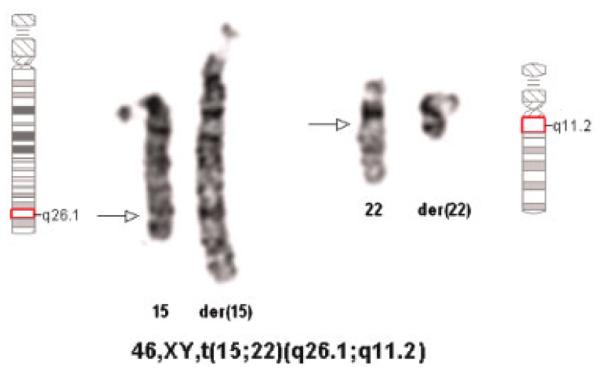
Partial karyotype showing the balanced translocation t(15;22). Arrows indicate the breakpoints. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Fig. 2.
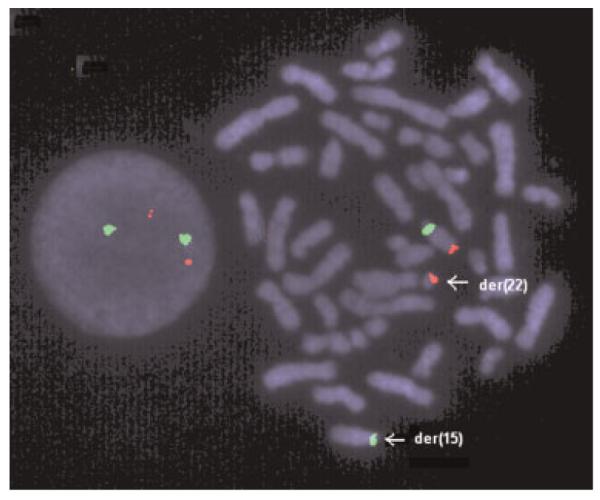
FISH with the probes RP11-262P8 (red) and D15Z1 (green). Signals from the BAC RP11-262P8 (red) were present on the normal 15 and the der(22), indicating that the IGF1R gene is translocated to the der(22). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Translocation Breakpoint Mapping
The cosmid probes for 22q11.2 loci D22S788 (N41) and ZNF74 were used. These probes correspond to regions just proximal and distal, respectively, of LCR-B. LCR-B is the common breakpoint found in the recurrent t(11;22) and a constitutional t(17;22) [Shaikh et al., 1999; Kurahashi et al., 2003]. The signal for c68a1 (D22S788) was seen on the normal and the der(22), whereas that of c87f9 (ZNF74) was detected on the normal 22 and the der(15), positioning the breakpoint of the patient within LCR-B (data not shown).
Additional FISH analyses were performed in order to localize the chromosome 15 breakpoint. A total of 15 fully sequenced BAC clones that were separated by approximately 1 Mb of genomic sequence or less on 15q (Fig. 3) were selected and used as probes. In each experiment, two probes were labeled separately with either spectrum red or spectrum green and cohybridized to metaphase chromosomes. This dual color FISH approach rapidly led to the identification of a significant deletion on the der(15) (Fig. 4). Of the 15 clones used in the analysis, 11 clones were deleted (Fig. 3). The clone bordering the deleted region on the centromeric end (405a15) mapped to the normal 15 and the der(15), whereas the clone (349o9) on the telomeric end of the deletion mapped to the normal 15 and the der(22). These data suggested that the deletion involved in this rearrangement was at least ≈2.7 Mb in size.
Fig. 3.

Diagrammatic representation of the BAC clones used in mapping the t(15;22). A total of 11 clones spanning from 2o19 to 76e17 are deleted from the der(15) suggesting that at least ≈2.7 Mb is missing as a result of this rearrangement.
Fig. 4.

FISH mapping of BAC clones at 15q26. A: Signals from clones 2o19 (green) and 354m14 (red) seen only on the normal chromosome 15 suggesting a deletion involved in this rearrangement. B: Signals from clones 405a15 (red) and 349o9 (green) which represent the clones flanking the deleted region. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Array CGH and Gene Expression Arrays
Array CGH revealed a deletion of 316 contiguous oligo probes covering 3.3 Mb immediately adjacent to the breakpoint on the der(15) (Fig. 5). The average log 2 ratio of the 316 probes was −0.96, consistent with the expected log 2 ratio =−1 for a single copy loss in a diploid genome. The proximal breakpoint of the deletion was between probes A_16_P03083328 and A_16_P20345216 (89,184,954-89,185,013 and 89,197,342-89,197,401, UCSC HG17, May 2004). The distal breakpoint of the deletion was between probes A_14_P117027 and A_16_P20353086 (92,489, 582-92,489,641 and 92,510,939-92,510,998, UCSC HG17, May 2004). Based on the known sequence position of the probes, the deletion extends from the middle part of 15q26.1 to the proximal part of 15q26.2. Gene expression arrays showed a close to 50% reduction of IGF1R expression (normalized intensity for the patient was 0.685 vs. normal 1.153).
Fig. 5.

Microarray CGH study. Two color aCGH with dye swap using 244K oligo probes demonstrated a 3.3 Mb heterozygous deletion of chromosome 15q26.1-q26.2. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
We have studied a patient with an apparently balanced translocation, t(15;22)(q26.1;q11.2), short stature, developmental delay and mild dysmorphic features. Using translocation breakpoint mapping, and aCGH, we identified a cryptic deletion of 3.3 Mb immediately adjacent to the chromosome 15 breakpoint. Gene expression analysis demonstrated a reduction of IGF1R gene expression in the patient. An extensive literature search revealed three patients with microdeletions within 15q26 characterized by aCGH. All three patients were ascertained by congenital diaphragmatic hernia (CDH). Slavotinek et al. [2005] described two cases of 15q26 microdeletion and CDH in great detail. Both patients had severe phenotypes with cardiovascular anomalies, cleft palate, and died soon after birth. Cytogenetic analysis revealed a 46,XX karyotype in both patients. aCGH and microsatellite marker analyses demonstrated an 8–11 Mb deletion in Patient 1, and a 1–2 Mb deletion in Patient 2. While the proximal part of the deletion in Patient 1 largely overlaps with the deletion in our patient, the entire deletion in Patient 2 is distal to the deletion in our case (Fig. 6). The third patient reported by Klaassens et al. [2005] had CDH, genital anomalies, IUGR, and an interstitial deletion of 6.3 Mb determined by aCGH and confirmed by FISH with BAC clones within and flanking the deleted region. The deletion overlaps the deletion in our patient by approximately 3 Mb (Fig. 6). The phenotype and genotype information of these four patients is summarized in Table I. In an attempt to investigate phenotype/genotype correlation, we also included in Table I two patients (Patients 4 and 5) with ring chromosome 15 in which the 15q breakpoints were confirmed to be within 15q26 by aCGH [Glass et al., 2006]. The deletion found in Patient 5 is discontinuous and may be the result of a more complex rearrangement. In most deletion syndromes, the size of deletion usually correlates with the severity of the phenotype. However, we discovered that deletion size does not always correlate with phenotypic severity in 15q26 microdeletion patients. Our patient with a 3.3 Mb deletion showed a relatively mild phenotype, while Patient 2 with a <2 Mb deletion died soon after birth due to severe MCA. Likewise, Patients 1 and 2 had smaller deletions than Patient 4. They died soon after birth while Patient 4 survived, presumably due to a less severe phenotype. Patients 1, 2, 3, and 4 who showed severe phenotypes share a common deleted region from the distal part of 15q26.2 to the proximal part of 15q26.3 (minimum overlap extends from 95, 883,860 to 96,766,282, UCSC, HG17, May 2004). On the other hand, the present case and case 5 whose deletions did not involve this region showed much milder phenotypes. Therefore, it is reasonable to propose that the deleted segment of 15q26 extending from 95,883,860 to 96,766,282 is associated with a severe phenotype. Within this segment, there is only one known gene ARRDC4 based on the UCSC genome browser. Although mutations of this gene have been identified in patients with CDH [Slavotinek et al., 2006], none of these mutations appeared to be unambiguously causal for CDH.
Fig. 6.

Comparison of deleted segments of four 15q26 microdeletion cases and two ring chromosomes 15 involving breakpoints in 15q26. Solid bars indicate deleted regions confirmed by FISH or microsatellite markers; hatched bars indicate possible deletions that were not confirmed due to uninformative microsatellite markers. The blue rectangle indicates the segment associated with a severe phenotype. *Slavotinek et al. [2005], **Klaassens et al. [2005], and ***Glass et al. [2006]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
TABLE I.
Phenotype and Genotype Correlation of 15q26 Microdeletion and Ring Chromosome 15
|
Slavotinek et al. [2005] |
Klaassens et al. [2005] |
Glass et al. [2006] |
||||
|---|---|---|---|---|---|---|
| Features | Present case | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
| Growth | ||||||
| IUGR | NR | + | − | + | + | − |
| Short stature | + | NA | NA | NR | + | + |
| Delayed bone age | + | NR | NR | NR | NR | NR |
| Developmental delay | + | NA | NA | NR | + | + |
| Speech delay | + | NA | NA | NR | + | + |
| Pulmonary hypoplasia | − | + | + | NR | − | − |
| Cardiovascular | ||||||
| Translocation of arteries | − | − | + | NR | − | − |
| Mitral or aortic valve defects | − | + | + | NR | + | − |
| Septal defects | − | − | + | NR | + | − |
| Hypoplastic left heart | − | + | − | NR | − | − |
| Musculoskeletal | ||||||
| Anomalies of extremities | − | − | + | NR | + | + |
| Talipes or rocker bottom feet | − | + | + | NR | − | − |
| Urogenital | ||||||
| Renal anomalies | − | − | + | NR | + | − |
| Double uterus/vagina | NA | − | + | NA | − | − |
| Genital anomaly | − | − | − | + | − | − |
| Craniofacial | ||||||
| Facial dysmorphism | Mild | + | + | NR | + | NR |
| Cleft/high arched palate | − | + | + | NR | + | − |
| Skin | ||||||
| Café au lait | − | − | − | NR | + | − |
| Hirsutism | + | − | + | NR | − | − |
| Other | ||||||
| CDH | − | + | + | + | − | − |
| Single umbilical artery | NR | + | + | NR | NR | NR |
| Seizure | + | NA | NA | NR | + | − |
| Karyotype | 46,XY, t(15;22) (q26.1;q11.2) |
46,XX | 46,XX | 46,XY, t(1;14), inv(6),del(15)(q26) |
46,XX, r(15)(q26) | 46,XX, r(15)(q26) |
| Deletion size | 3.3 Mb | 8–11 Mb | 1–2 Mb | 6.3 Mb | 8–11 Mb | <3 Mb |
| UCSC | 89,197,342 –92,489,641 |
89,274,936 –99,149,459 |
95,883,860 –96,766,282 |
90,475,196 –97,234,642 |
89,275,717 or 92,238,363– |
89,224,936–a |
NR, not reported; NA, not applicable; +, present; −, absent.
Discontinuous deletion.
There are 18 known or hypothetical protein coding genes within the 3.3 Mb deleted region in the present case (UCSC HG17, May 2004). Among the known genes, RGMA and ST8SIA2 appear to be important in neural development. RGMA is a cell-survival factor that suppresses the proapoptotic activity of its receptor neogenin. A recent study has shown that RGMA overexpression promotes neuronal differentiation in chick embryos, whereas RGMA small interference RNA represses it [Matsunaga et al., 2006]. Thus, haploinsufficiency of RGMA may be responsible for the delayed neuromuscular development in our patient. ST8SIA2 codes for a type II membrane protein involved in the production of polysialic acid, a modulator of the adhesive properties of neural cell adhesion molecule (NCAM1). There have been no reports to demonstrate that ST8SIA2 is dosage sensitive. A mutation has been identified in ST8SIA2 in a patient with CDH [Slavotinek et al., 2006]; however, a causal relationship between the mutation and CDH could not be established. Another gene worth mentioning is VPS33B. VPS33B plays an important role in segregation of intracellular molecules into distinct organelles. A homozygous mutation of VPS33B causes arthrogryposis-renal dysfunction-cholestasis syndrome (ARC) [Gissen et al., 2004]. Deletion of one copy of VPS33B presumably renders our patient a heterozygous carrier of an ARC deletion. Nonetheless, the relatively mild phenotype of our patient indicates that few genes within the 3.3 Mb deleted region are developmentally critical.
The IGF1R gene is located in 15q26.3 and is dosage sensitive [Butler et al., 1988; Faivre et al., 2002]. Patients 1, 3, and 4 have lost a copy of their IGF1R gene due to the deletion. IUGR was seen in all three patients; postnatal growth retardation was seen in Patient 4. Consistent with the role of IGF1R in prenatal and postnatal growth, the deletions in Patients 2 and 5 spare the IGF1R gene and both patients show normal prenatal growth. The postnatal growth failure in Patient 5 is likely to be multifactorial. Though our patient carries two copies of the IGF1R gene, the reduced level of IGF1R expression may be responsible for his growth defects and developmental delay. The IGF1R gene is located about 4.5 Mb distal to the 15q breakpoint and was translocated to the der(22). The mechanism responsible for the reduction of IGF1R expression is not clear. It may be caused by a position effect as a result of the translocation or haploinsufficiency of a gene or genes within the deleted region that modifies IGF1R expression. Our experience with this case demonstrates that high-density aCGH is a quick and accurate way to identify cryptic copy number changes in apparently “balanced” translocations. Expression studies can add valuable information regarding gene expression changes due to a chromosomal rearrangement. Both approaches can assist in the elucidation of the etiology of unexplained phenotypic differences in cases such as the one described in this report.
ACKNOWLEDGMENTS
These studies were supported in part by funds from grants CA39926 and HD26979 from the National Institutes of Health and the Charles E.H. Upham Chair of Pediatrics (BSE).
Grant sponsor: National Institutes of Health; Grant numbers: CA39926, HD26979.
REFERENCES
- Bhakta KY, Marlin SJ, Shen JJ, Fernandes CJ. Terminal deletion of chromosome 15q26.1: Case report and brief literature review. J Perinatol. 2005;25:429–432. doi: 10.1038/sj.jp.7211301. [DOI] [PubMed] [Google Scholar]
- Budarf ML, Collins J, Gong W, Roe B, Wang Z, Bailey LC, Sellinger B, Michaud D, Driscoll DA, Emanuel BS. Cloning a balanced translocation associated with DiGeorge syndrome and identification of a disrupted candidate gene. Nat Genet. 1995;10:269–278. doi: 10.1038/ng0795-269. [DOI] [PubMed] [Google Scholar]
- Butler MG, Fogo AB, Fuchs DA, Collins FS, Dev VG, Phillips JA., III Two patients with ring chromosome 15 syndrome. Am J Med Genet. 1988;29:149–154. doi: 10.1002/ajmg.1320290119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Gosset P, Cormier-Daire V, Odent S, Amiel J, Giurgea I, Nassogne M, Pasquier L, Munnich A, Romana S, Prieur M, Vekemans M, de Blois M, Turleau C. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: Report of two families and review of the literature. Eur J Hum Genet. 2002;10:699–706. doi: 10.1038/sj.ejhg.5200879. [DOI] [PubMed] [Google Scholar]
- Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, Cooper WN, McKiernan PJ, Klomp LW, Morris AA, Wraith JE, McClean P, Lynch SA, Thompson RJ, Lo B, Quarrell OW, Di Rocco M, Trembath RC, Mandel H, Wali S, Karet FE, Knisely AS, Houwen RH, Kelly DA, Maher ER. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- Glass IA, Rauen KA, Chen E, Parkes J, Alberston DG, Pinkel D, Cotter PD. Ring chromosome 15: Characterization by array CGH. Hum Genet. 2006;118:611–617. doi: 10.1007/s00439-005-0030-z. [DOI] [PubMed] [Google Scholar]
- Jacobsen P. A ring chromosome in the 13-15group associated with microcephalic dwarfism, mental retardation and emotional immaturity. Hereditas. 1966;55:188–191. [Google Scholar]
- Kato R, Kishibayashi J, Shimokawa O, Harada N, Niikawa N, Matsumoto N. Congenital glaucoma and Silver-Russell phenotype associated with partial trisomy 7q and monosomy 15q. Am J Med Genet. 2001;104:319–322. [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: Determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, Horsthemke B, Beaudet AL, Ledbetter DH. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet. 1996;66:77–80. doi: 10.1002/(SICI)1096-8628(19961202)66:1<77::AID-AJMG18>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Shaikh T, Takata M, Toda T, Emanuel BS. The constitutional t(17;22): Another translocation mediated by palindromic AT-rich repeats. Am J Hum Genet. 2003;72:733–738. doi: 10.1086/368062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Platt J, Tawn EJ, Burn J. A de novo interstitial deletion of 15(q21.2q22.1) in a moderately retarded adult male. J Med Genet. 1990;27:637–639. doi: 10.1136/jmg.27.10.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E, Nakamura H, Chedotal A. Repulsive guidance molecule plays multiple roles in neuronal differentiation and axon guidance. J Neurosci. 2006;26:6082–6088. doi: 10.1523/JNEUROSCI.4556-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer D, Dvorak C, Techakittiroj C, Andersson H, Dise D, Li MM. Molecular cytogenetic study of a 45, X male with cryptic Y/15 translocation and deletion of IGF1R gene. The American Society of Human Genetics, 55th Annual Meeting A835.2005. [Google Scholar]
- Nimmakayalu M, Henegariu O, Ward DC, Bray-Ward P. Simple method for preparation of fluor/hapten-labeled dUTP. Biotechniques. 2000;28:518–522. doi: 10.2144/00283st11. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet. 1999;65:1595–1607. doi: 10.1086/302666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek A, Lee SS, Davis R, Shrit A, Leppig KA, Rhim J, Jasnosz K, Albertson D, Pinkel D. Fryns syndrome phenotype caused by chromosome microdeletions at 15q26.2 and 8p23.1. J Med Genet. 2005;42:730–736. doi: 10.1136/jmg.2004.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, Shaw GM, James B, Ptacek L, Pennacchio LA. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: Mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26.2. Eur J Hum Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- Spruijt L, Engelen JJ, Bruinen-Smeijsters IP, Albrechts JC, Schrander J, Schrander-Stumpel CT. A patient with a de novo 15q24q26.1 interstitial deletion, developmental delay, mild dysmorphism, and very blue irises. Am J Med Genet Part A. 2004;129A:312–315. doi: 10.1002/ajmg.a.30185. [DOI] [PubMed] [Google Scholar]
- Tonnies H, Schulze I, Hennies H, Neumann LM, Keitzer R, Neitzel H. De novo terminal deletion of chromosome 15q26.1 characterised by comparative genomic hybridisation and FISH with locus specific probes. J Med Genet. 2007;38:617–621. doi: 10.1136/jmg.38.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumer Z, Harboe TL, Blennow E, Kalscheuer VM, Tommerup N, Brondum-Nielsen K. Molecular cytogenetic characterization of ring chromosome 15 in three unrelated patients. Am J Med Genet Part A. 2004;130A:340–344. doi: 10.1002/ajmg.a.30035. [DOI] [PubMed] [Google Scholar]