


Abstract
The HIV-1 viral infectivity factor (Vif) allows productive infection of non-permissive cells (including most natural HIV-1 targets) by counteracting the cellular cytosine deaminases APOBEC-3G (hA3G) and hA3F. The Vif-induced degradation of these restriction factors by the proteasome has been extensively studied, but little is known about the translational repression of hA3G and hA3F by Vif, which has also been proposed to participate in Vif function. Here, we studied Vif binding to hA3G mRNA and its role in translational repression. Filter binding assays and fluorescence titration curves revealed that Vif tightly binds to hA3G mRNA. Vif overall binding affinity was higher for the 3′UTR than for the 5′UTR, even though this region contained at least one high affinity Vif binding site (apparent Kd = 27 ± 6 nM). Several Vif binding sites were identified in 5′ and 3′UTRs using RNase footprinting. In vitro translation evidenced that Vif inhibited hA3G translation by two mechanisms: a main time-independent process requiring the 5′UTR and an additional time-dependent, UTR-independent process. Results using a Vif protein mutated in the multimerization domain suggested that the molecular mechanism of translational control is more complicated than a simple physical blockage of scanning ribosomes.
INTRODUCTION
The human immunodeficiency virus-type 1 (HIV-1) infects primary T-cells, macrophages and monocytes, ultimately leading to the destruction of the immune system, infection by opportunistic pathogens, and death, if its replication cannot be inhibited. However, these cell lineages possess an innate defense system directed against retroviruses, retrotransposons and retroelements and HIV-1 had to develop a countermeasure against this protective system in order to be able to infect them. Primary T-cells, macrophages and monocytes, as well as some lymphocyte-derived cell lines, collectively named non-permissive cells, express two related cytosine deaminases, APOBEC3G (hA3G) and APOBEC3F (hA3F) that restrict HIV-1 replication (1,2). However, these antiviral factors are counteracted by the HIV-1 viral infectivity factor (Vif), a basic 23 kDa protein required for HIV-1 propagation in vivo and pathogenesis (1–5).
The N- and C-terminal domains of hA3G and hA3F possess significant similarity to APOBEC1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme. The APOBEC family includes the cellular cytidine deaminases APOBEC1, APOBEC2, APOBEC3, APOBEC4 and the activation-induced deaminase (AID). In humans, there are seven apobec3 genes coding for hA3A, hA3B, hA3C, hA3DE, hA3F, hA3G and hA3H (3–6). Among these proteins, hA3G has the most potent anti-HIV-1 activity, while the activity of hA3F is weaker but significant, and the anti-HIV-1 activity of hA3DE is very weak (3,4). Other APOBEC3 members display potent activities against various viruses and retroelements (3). In the absence of Vif (i.e. in HIV-1 Δvif), hA3G and hA3F are incorporated into HIV-1 and they catalyze cytosine deamination during (−) strand DNA synthesis, ultimately resulting in (i) degradation of the viral DNA and/or (ii) lethal hypermutagenesis (1,2,7–12). Notably, accumulating evidence shows that catalytically inactive hA3G and hA3F mutants also significantly inhibit HIV-1 DNA synthesis and replication (3–5), and some of the antiviral effects of these proteins might not require packaging into the viral particles.
HIV-1 Vif decreases the intracellular concentration of hA3G and hA3F and prevents their incorporation into HIV-1 particles (7–9,13–16). HIV-1 Vif suppresses hA3G and hA3F antiviral functions by hijacking a cellular E3 ubiquitin ligase and inducing their degradation by the 26S proteasome (8,10–12,16–18). Even though the Vif induced-degradation of hA3G by the 26S proteasome has been extensively studied, it is not the only mechanism, and possibly not the main mechanism, by which Vif counteracts hA3G and hA3F. Indeed, Vif has been shown to be able to inhibit packaging and antiviral activity of a degradation resistant hA3G variant (19). In one of the first publications in the field, Landau and coworkers reported a 4.6-fold reduction of hAG3 synthesis in the presence of Vif and concluded that Vif-induced degradation of hA3G does not appear to be the primary mechanism by which it blocked hA3G encapsidation (7). It is possible that the Vif-induced, proteasome-dependent, degradation of Vif was underestimated in this study (8,10), but the effect of Vif on hA3G synthesis was confirmed by several independent groups (10,13). The mechanism by which Vif downregulates hA3G translation has never been studied.
Recently, we showed that Vif binds the 5′-region of the HIV-1 genomic RNA with high affinity and moderate cooperativity (20,21), and that Vif has an RNA chaperone activity (22). In addition, mutations reducing the affinity of Vif for RNA have been shown to diminish viral replication in non-permissive cells (23), suggesting that RNA binding plays a central role in Vif function. We therefore studied binding of HIV-1 Vif to hA3G mRNA by biochemical and biophysical methods. As the 5′ and 3′UTRs of mRNAs are often involved in negative translational control by proteins (24,25), we compared Vif binding to the full-length mRNA and RNA fragments corresponding to the 5′ and 3′UTRs and to the hA3G coding region. Our results showed that wild-type Vif has a high affinity for the hA3G mRNA, and especially for its 3′ UTR. Using chemical and enzymatic footprinting, we identified several Vif binding sites not only in the 3′UTR, but also in the 5′UTR of hA3G mRNA. Finally, we compared the effect of Vif on the in vitro translation of full-length hA3G mRNA and mRNAs lacking the 5′ or/and 3′UTR(s) regions. Vif had two negative effects on hA3G translation: a time-dependent, UTR-independent effect and a more important time-independent effect that required the 5′UTR. Our results suggest that Vif binding to hA3G mRNA, and particularly to its 5′UTR might be crucial for Vif function, by downregulating hA3G translation.
MATERIALS AND METHODS
Plasmid construction
The DNA sequence corresponding to hA3G was obtained after polyA+ mRNA isolation from H9 cells (PolyATtract® mRNA Isolation Systems—Promega) and RT–PCR analysis (Superscript®—Invitrogen) using a sense primer corresponding to the major transcription site defined by Muckenfuss et al. (26) and an antisense primer corresponding to the 3′-end of the 3′UTR (Table 1). Amplified PCR products containing the entire human hA3G mRNA sequence were digested by EcoRI and XbaI and ligated into pCMV6-XL5 previously digested with the same restriction enzymes. Fragments corresponding to the 5′UTR, the 3′UTR or the coding region ± the 5′ and/or the 3′UTR of hA3G mRNA were PCR amplified using primers listed in Table 1 and cloned into pCR TOPO 2.1 (Invitrogen), for in vitro run off transcription, and pCMV6-XL5, for in vitro translation assays. The resulting plasmids pCMV-hA3G, pCMV-hA3GΔUTR, pCMV-hA3GΔ5′UTR and pCMV-hA3GΔ3′UTR contain nucleotides 1–1771, 298–1452, 298–1771 and 1–1432 of hA3G, respectively and pTOPO-hA3G-5′UTR and pTOPO-hA3G-3′UTR contain nucleotides 1–297 and 1454–1771, respectively. The inserts of all plasmids were checked by sequence analysis. All nucleotide positions refer to the transcription start site of the major isoform corresponding to hA3G mRNA in human T-cell lines A3.01 (26).
Table 1.
Oligonucleotides used in this study
| RNA | Primers | |
|---|---|---|
| hA3G | pS 1–21 | EcoRI-TCTTTCCCTTTGCAATTGCC |
| pAS 1750–1771 | XbaI-AAGATTTAGTATTTCATTT | |
| hA3G-ΔUTR | pS 298–321 | EcoRI-GGATGAAGCCTCACTTCAGAAACAC |
| pAS 1432–1452 | XbaI-TCAGTTTTCCTGATTCTGGAG | |
| hA3G-Δ5′UTR | pS 298–321 | EcoRI-GGATGAAGCCTCACTTCAGAAACAC |
| pAS 1750–1771 | XbaI-AAGATTTAGTATTTCATTT | |
| hA3G-Δ3′UTR | pS 1–21 EcoRI | EcoRI-TCTTTCCCTTTGCAATTGCC |
| pAS 1432–1452 | XbaI-TCAGTTTTCCTGATTCTGGAG | |
| hA3G-5′UTR | pST7 1–21 | EcoRI-T7-TCTTTCCCTTTGCAATTGCC |
| pAS 277–297 | StuI-TGGCCGGCTAGTCCCGAC | |
| hA3G-3′UTR | pST7 1454–1478 | EcoRI-T7-GGATGGGCCTCAGTCTCTAAGGAAG |
| pAS 1750–1771 | XbaI-AAGATTTAGTATTTCATTT | |
| Vif AALA | pSAALA hVif | CCAAAACAGATAAAGGCAGCATTGGCAAGTGTTAGGAAACTG |
| pASAALA hVif | CAGTTTCCTAACACTTGCCAATGCTGCCTTTATCTGTTTTGG | |
Plasmid pD10 WT-Vif containing a 6His-tag fused at the N-terminal domain of Vif lacking the methionine initiation codon was used to perform site-directed mutagenesis (QuickChange mutagenesis—Stratagene) to obtain a Vif protein mutated in the multimerization domain 161PPLP164. The proline residues were replaced by alanines using oligonucleotides vif-M1 et vif-M2 as indicated in Table 1. The presence of the mutation was checked in the resulting plasmid, pD10 AALA-Vif, by DNA sequencing.
RNA synthesis and labeling
After linearization of plasmids pCMV-hA3G, pCMV-hA3GΔUTR, pCMV-hA3GΔ5′UTR, pCMV-hA3GΔ3′UTR, pCRTOPO-hA3G5′UTR or pCRTOPO-hA3G3′UTR by StuI or XbaI, in vitro transcription was performed with bacteriophage T7 RNA polymerase in presence of α-32P ATP (Amersham) as described earlier (27). Internally labeled RNA was purified on denaturing polyacrylamide gels or on agarose gels.
Recombinant Vif proteins
Expression plasmids pD10WTVif or pD10AALAVif were used to transform Escherichia coli BL21 cells as described (28). Detailed protocols of Vif expression and purification, as well as a biophysical characterization of the wild-type and mutant Vif proteins will be published elsewhere (S. Bernacchi, et al., manuscript in preparation). Briefly, Vif proteins were purified under denaturing conditions using a Ni-NTA column. After elution, proteins were renaturated by slow dialysis against buffers with decreasing guadinium chloride concentration, and finally against a buffer containing 50 mM MOPS pH 6.5, 150 mM NaCl, 10% glycerol. Vif proteins were stored in small aliquots at −80°C and kept at 4°C after thawing. As both wild-type and AALA Vif tend to aggregate, the protein stock solutions were centrifuged at 100 000g for 30 min at 4°C immediately prior use. The protein concentration in the supernatant was determined spectrophotometrically using an extinction coefficient of 43 040 cm−1M−1 at 280 nm. UV spectroscopy revealed that purified Vif proteins were not contaminated by nucleic acids.
Filter binding assays
Internally 32P-labeled full-length or truncated hA3G mRNA (20 000 c.p.m., final concentration <2 nM) and 1.5 µg of unlabeled E. coli total tRNA in 5 µl of Milli-Q (Millipore) water were heated for 2 min at 90°C and chilled on ice for 2 min. After addition of 10-fold concentrated binding buffer [final concentrations: 30 mM Tris–HCl (pH 7.5), 300 mM NaCl, 2.5 mM MgCl2], 1.4 mM β-mercaptoethanol and 5 Us of RNasin (Promega), RNA was renatured for 15 min at 37°C. In parallel, Vif proteins were renatured for 15 min at 37°C in binding buffer supplemented with 0.02% (w/v) bovine serum albumin, and added to RNA in a final volume of 60 µl. After incubation for 30 min at 37°C and for 30 min at 4°C in the presence of 0.01% Triton X-100, RNA–protein complexes were loaded onto 0.45 µm pore size cellulose filters (MultiscreenTM 96-well plate, Millipore), presoaked with 100 µl of binding buffer. After three washes with 100 µl of ice-cold binding buffer, the filters were air-dried and the radioactivity remaining on the filters was determined by liquid scintillation counting.
Quantitative analysis of filter-binding assays
The hypothesis underlying filter binding assays is that all RNA–protein complexes are retained on the filter. As we previously showed that Vif binds RNA cooperatively (20,21), filter binding data can be analyzed using the Hill equation for cooperative binding (21). Curve fitting of the experimental data was performed with Origin7 or Prism5 softwares using equation:
| 1 |
where f corresponds to the RNA fraction retained on the filter, [L] is the protein concentration, αH is the Hill constant and is an index to the cooperativity and Kd is the ligand concentration at which 50% of the RNA is retained on the filter.
Steady-state fluorescence measurements
Fluorescence experiments were recorded in quartz cells at 20 ± 0.5°C on a Fluoromax-4 fluorometer (HORIBA Jobin–Yvon). The excitation wavelength was 295 nm, the emission wavelength was scanned from 310 to 450 nm, the integration time was 0.1 s, and the excitation and emission bandwidths were 5 nm. Fluorescence titrations were performed by adding increasing amounts of nucleic acid to 100 nM or 50 nM Vif in 30 mM Tris–HCl (pH 7.5), 200 mM NaCl and 10 mM MgCl2.
Determination of Vif/RNA binding parameters
To determine the binding parameters of Vif to the hA3G mRNA fragments, we measured the decrease of the fluorescence intensity, I, at a fixed concentration of protein in presence of increasing RNA concentrations. The fluorescence intensity was then converted into the intrinsic fluorescence quenching Qobs in Equation 2:
| 2 |
with I0 corresponding to the protein fluorescence intensity in absence of nucleic acid. The number of consecutive nucleotides occluded upon binding of one ligand, n, the observed affinity, Kobs and the cooperativity parameter, ω, were recovered using the model of McGhee and von Hippel for cooperative binding (29), in Equation 3:
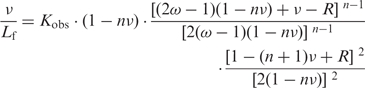 |
3 |
with R = {[1 − (n + 1)v]2 + 4v(1 − nv)}1/2.
Because the McGhee and von Hippel (30) model was derived with the simplifying assumption of an infinite lattice of binding sites, we introduced a correction factor taking into account the finite lattice size, N. The correction merely amounts to multiplying Equation 3 by the factor (N−n+1)/(N)
For ω = 1, Equation 3 simplifies to Equation 4, which corresponds to the Scatchard formulation for non-interacting ligands:
| 4 |
where ν corresponds to the binding density (mol of bound protein/mol of nucleotide), and Lf to the concentration of free protein. These quantities are linked by Equations 5–7 as described in (31):
| 5 |
| 6 |
| 7 |
where Dt is the total nucleotide concentration, Lt and Lb are the total and the bound protein concentrations, respectively, and Qmax corresponds to the maximal fluorescence quenching when all protein molecules are bound to nucleic acids. Fitting of experimental data with those equations was performed with an algorithm written with Mathematica (Wolfram Research) (20).
Chemical modifications of RNA and enzymatic footprinting
Chemical modification experiments were performed on the full-length hA3G mRNA and on the hA3G-5′UTR and hA3G-3′UTR RNAs. RNAs were modified with DMS (Fluka) or CMCT (Merck) after a renaturation procedure in a high ionic strength buffer in the presence of 2 µg of E. coli total tRNA. For DMS modification, the reaction was carried out in buffer D1 (50 mM sodium cacodylate, pH 7.5, 300 mM KCl, 5 mM MgCl2) for 4 and 8 min at 37°C with DMS freshly diluted 1/20 (v/v) in ethanol. For CMCT, modifications were performed at 25°C in buffer D2 (100 mM sodium borate pH 8.0, 300 mM KCl and 5 mM MgCl2) with 5 µl of freshly dissolved CMCT (42 mg/ml in water) for 15, 30 and 45 min. Reactions were stopped by precipitation in ethanol. Modified/protected bases were detected by extension of a 5′ 32P-labeled primer with avian myeloblastosis virus reverse transcriptase (MP Bio-medicals) as described earlier (21) and analyzed on denaturing 8% polyacrylamide gels.
Enzymatic footprinting experiments were performed on hA3G-5′UTR and hA3G-3′UTR RNAs in the presence of increasing concentrations of Vif using ribonuclease (RNase) V1, T1 and A (Ambion). Briefly, RNA (1 µM) was renatured for 15 min in the presence of 2 µg of E. coli total tRNA in the buffer provided by the manufacturer. After addition of Vif protein (0, 0.5, 1.0, 2.0 and 3.0 µM), Vif–RNA complexes were allowed to form for 30 min at 37°C and 30 min at 4°C. RNases V1, T1 or A were then added and incubation was continued for 15 min at room temperature. Reactions were stopped by phenol/chloroform extraction and ethanol precipitation. Footprints were detected by primer extension as described earlier (21).
In vitro translation assays
The wild-type and mutant Vif proteins, and control AspRS were transcribed and translated for 90 min at 30°C using the Promega TNT T7-coupled reticulocyte lysate system according to the manufacturer’s instructions. The synthesized Vif, Vif AALA or AspRS were added on ice to a new transcription/translation mix containing either pCMV-hA3G, hA3GΔUTR, hA3GΔ5′UTR or hA3GΔ3′UTR. The reaction media were then incubated for 15, 30, 45 or 60 min at 30°C and stopped on ice by the addition of SDS–PAGE loading buffer. Proteins were then separated on 12.5% SDS–PAGE and analyzed by autoradiography.
RESULTS
Characterization of wild-type and AALA Vif proteins
Due to the tendency of wild-type and AALA Vif proteins to aggregate, they were centrifuged at 100 000g prior each experiments and only the soluble fraction of the proteins was used. The soluble proteins were further characterized by size-exclusion chromatography and dynamic light scattering. Both techniques indicated that wild-type Vif self-associates into homogenous oligomers containing 6–10 proteins, while Vif AALA formed dimers or/and trimers (S. Bernacchi et al., manuscript in preparation). Thus, even though the PPLP motif is important for Vif multimerization, its mutation does not completely abolish Vif self-association.
Filter binding assays
In order to detect and analyze binding of wild type (WT) HIV-1 Vif to hA3G mRNA, we first performed filter binding assays using either full-length hA3G mRNA, mRNAs deleted of the 5′ or the 3′UTR or RNA fragments corresponding to the 5′UTR, the hA3G coding region or the 3′UTR, respectively (Figure 1A). All binding curves displayed a sigmoid shape suggesting that binding of Vif to the hA3G mRNA and the fragments thereof is cooperative (Figure 2A). The experimental data were adequately fitted with the Hill equation (Figure 2A), and the apparent dissociation constants (Kd) and the Hill constant (αH), which reflect cooperativity, extracted from this analysis are summarized in Table 2. No cooperativity corresponds to αH = 1, while αH > 1 indicates positive cooperativity. In addition, αH gives a lower limit of the number of Vif binding sites on RNA. For this analysis and all other quantitative analyses in the next sections, the Vif concentrations were expressed as concentrations of Vif monomers, even though characterization of wild-type and AALA Vif proteins indicated that they are oligomeric (see above). Indeed, as we do not know how many RNA binding sites are present per Vif oligomer, it would be very speculative to use more complex models. All dissociation constants must therefore be considered as apparent dissociation constants, which are nevertheless very useful to compare Vif binding to different RNA species.
Figure 1.

Schematic representation of the RNA and Vif proteins used in this study. (A) Representation of the full-length hA3G mRNA and the truncated RNAs. The coding region is indicated by a black box. (B) Scheme of the Vif protein. The name of the viral and cellular partners that interact with different regions of Vif are indicated. CUL5: Cullin 5; ELOC: Elongin C; GAG: HIV-1 Pr55Gag precursor. The sequence of the multimerization domain of the WT protein and of the Vif AALA mutant are indicated.
Figure 2.

Titration curves of full-length and truncated hA3G mRNA by Vif determined by filter binding. Radiolabeled full-length hA3G mRNA or hA3G mRNA fragments were incubated with increasing concentration of WT Vif (A) or Vif AALA (B), and filtered through a cellulose membrane. The fraction of bound radioactivity is plotted versus the concentration of Vif. Curves correspond to the best fit of equation (1) to the experimental data. The data points represent the mean ± SD of >10 experiments after exclusion of outliers falling outside of the 95% confidence interval.
Table 2.
Binding parameters derived from the filter binding assays
| WT Vif |
Vif AALA |
|||
|---|---|---|---|---|
| RNA | Kda(nM) | αH | Kd (nM) | αH |
| hA3G | 120 ± 10b | 2.0 ± 0.3 | 276 ± 23 | 2.4 ± 0.2 |
| hA3G-Δ5′UTR | 112 ± 5 | 1.9 ± 0.1 | 231 ± 14 | 3.0 ± 0.3 |
| hA3G-Δ3′UTR | 148 ± 11 | 1.9 ± 0.2 | 202 ± 7 | 3.4 ± 0.3 |
| hA3G-ΔUTR | 145 ± 8 | 2.3 ± 0.3 | 261 ± 11 | 2.7 ± 0.2 |
| hA3G-5′UTR | 115 ± 4 | 2.6 ± 0.2 | 292 ± 18 | 3.0 ± 0.3 |
| hA3G-3′UTR | 57 ± 3 | 2.3 ± 0.2 | 204 ± 8 | 3.3 ± 0.3 |
aKd and αH were obtained by fitting the experimental data to the Hill equation (see ‘Materials and Methods’ section).
bMean ± SD.
Comparison of hA3G-ΔUTR, hA3G-5′UTR, and hA3G-3′UTR RNAs showed that WT Vif bound to these three regions with significant affinity, especially as the binding buffer contained high salt concentration in order to reduced unspecific electrostatic interactions. However, Vif displayed a higher affinity for the 3′UTR of hA3G mRNA, while binding to the coding region was slightly weaker than to the 5′UTR (Figure 2A and Table 2). These experiments suggested that the 3′UTR of hA3G mRNA contains at least one high affinity Vif binding site. In keeping with this interpretation, WT Vif binds tighter to hA3G and hA3G-Δ5′UTR RNAs, which contain the 3′UTR, than to hA3G-Δ3′UTR (Table 2). WT Vif appeared to bind all RNA fragments with moderate cooperativity.
Unexpectedly, Vif bound hA3G and hA3G-Δ5′UTR RNAs weaker than hA3G-3′UTR RNA, even though these three RNAs contain the same high affinity Vif binding site(s). This result revealed that the assumption that all RNA–protein complexes are retained with the same efficiency on the cellulose membrane was not fulfilled. Instead, retention of the RNA–Vif complexes on the filter probably increased as an increasing number of proteins bound to the same RNA molecule. Even though a theoretical framework has been developed to determine the retention efficiency of RNA–protein complexes in filter binding experiments (32), it could not be applied to our experiments, which involved an unkown number of RNA–protein complexes with different retention efficiencies. Thus, the Kd values in Table 2 do not correspond to the binding dissociation constant of Vif from the highest affinity binding site present in each RNA fragment, but correspond to a weighted average of the binding dissociation constants of all binding sites present on each RNA.
As Vif multimerization is required to counteract the antiviral activity of hA3G (4,33–35), and as it would also help to explain the cooperative binding of this protein to hA3G mRNA, we mutated the Vif multimerization domain, which has been mapped to the 161PPLP164 motif (Figure 1) (36). Detailed characterization of the WT and mutant Vif proteins will be described elsewhere (S. Bernacchi et al., manuscript in preparation). Interestingly, we observed that mutation of the PPLP motif did decrease the oligomerization of Vif but did not totally prevent it, as Vif AALA behaved as dimers or/and trimers (see above and S. Bernacchi et al., manuscript in preparation). As compared to WT Vif, the mutant Vif AALA protein displayed very little preference for any of the RNA fragments we tested (Figure 2 and Table 2). While the mutant Vif had a slightly stronger affinity for the isolated 3′UTR than for the 5′UTR, it also displayed a slightly better affinity for the hA3G-Δ3′UTR RNA than for the hA3G-Δ5′UTR. Thus, it seems that the mutant Vif did not display any global preference for one of the UTR or for the coding region of the hA3G mRNA. However the PPLP motif was not required for cooperative binding, since αH values were consistently higher for Vif AALA than for WT Vif (Table 2).
Fluorescence spectroscopy
In order to reinforce the conclusions drawn from the filter binding experiments, we studied Vif binding to the hA3G mRNA by fluorescence spectroscopy. HIV-1 Vif protein contains eight Trp residues, including seven that are located in the N-terminal region of Vif, which corresponds to the RNA binding domain (4,36). Thus, Vif displays a high intrinsic fluorescence that is quenched upon binding to nucleic acids (20). While increasing amounts of protein were used to titrate a negligible amount of RNA in the filter binding assays, a constant and non negligible amount (50 or 100 nM) of protein was titrated with increasing amount of nucleic acids in the fluorescence experiments. Thus, the same formalism could not be used to analyze the two kinds of binding curves, and we analyzed the fluorescence binding curves using the McGhee and von Hippel model for cooperative binding (see ‘Materials and Methods’ section).
Compared to the filter binding assays, fluorescence titrations consistently yielded lower Kobs (Table 3), as expected if several Vif proteins have to bind to a same RNA molecule to obtain complete retention of the complexes on the filters. Alternatively, this difference might also indicate that some Vif–RNA complexes dissociate during washing of the filters. However, both techniques indicated that WT Vif bound all RNA fragments with moderate cooperativity. Fluorescence titration curves revealed that WT Vif had a high affinity for hA3G-3′UTR RNA, in agreement with filter binding assays, but also for hA3G-ΔUTR RNA, a feature not observed in the previous experiments (Table 3). As a result, Vif also demonstrated a high affinity for all RNA fragments containing either the 3′UTR or the coding region of hA3G mRNA. Note that as binding cooperativity is not extremely high, the Kobs values derived from the fluorescence titrations are mean values of all binding events quenching the Vif fluorescence, whereas, as previously pointed out, the Kd values derived from the filter binding assay are affected by the differential retention of the Vif–RNA complexes on the filters. Thus, differences in the Kd and Kobs values obtained by these two techniques are not completely unexpected.
Table 3.
Binding parameters derived from analysis of the fluorescence titration curves using the model of McGhee and von Hippel
| RNA | Kobs (nM)a | ωa | Stoichiometryc | RNA length (nt) |
|---|---|---|---|---|
| hA3G | 36 ± 6b | 108 | ∼6–7 vif/RNA | 1773 |
| hA3G-Δ5′UTR | 42 ± 5 | 78 | ∼2–4 vif/RNA | 1476 |
| hA3G-Δ3′UTR | 40 ± 2 | 109 | ∼2–3 vif/RNA | 1452 |
| hA3G-ΔUTR | 46 ± 7 | 76 | ∼2–3 vif/RNA | 1155 |
| hA3G-5′UTR | 120 ± 16 | 254 | ∼3–4 vif/RNA | 297 |
| hA3G-3′UTR | 49 ± 11 | 125 | ∼3–4 vif/RNA | 321 |
aKobs and ω values were obtained by fitting the experimental data to equation 3 (see ‘Materials and Methods’ section).
bMean ± SD of at least three experiments.
cThe binding stoichiometry was determined graphically from the fluorescence binding curves (see text).
The fluorescence binding curves also allowed determination of the binding stoichiometry of Vif to the various RNA fragments from the intersection of the initial slope with the fluorescence plateau (Table 3 and data not shown) (20). Indeed, only a limited number of Vif monomers bound to the full-length hA3G mRNA, with as few as 2–4 Vif molecules binding to the coding region, and the 3′ and 5′UTRs (Table 3). Indeed, this low stoichiometry indicates that there are several RNA binding sites per wild-type Vif oligomer (corresponding to ∼9 Vif monomers). In addition, the Vif binding density is significantly higher (≤1 protein/100 nucleotides) in the UTRs than in the hA3G coding region (≤1/400 nucleotides) (Table 3).
In an attempt to get a better estimation of the binding affinity of Vif for the highest affinity binding site present in each of the hA3G-5′UTR, hA3G-ΔUTR and hA3G-3′UTR RNAs, we analyzed the fluorescence binding data using the Scatchard equation (see ‘Materials and Methods’ section). This analysis is justified because these RNAs bound only a few Vif molecules (Table 3). When plotting the fraction of the bound to unbound protein as a function of the fraction of bound protein, the fluorescence binding data was linear over a wide range of RNA concentration (Figure 3A), indicating that these data can indeed be used to determine the Kd of Vif for the highest affinity binding site present in each RNA (KSca). Fitting the data from several experiments with Equation 4 yielded KSca values of 27 ± 6, 24 ± 5 and 37 ± 4 nM for hA3G-5′UTR, hA3G-3′UTR and hA3G-ΔUTR RNAs, respectively (Table 4). Expectedly, these values are lower than the ‘mean values’ obtained from the same data with the McGhee and von Hippel equation (Table 3). They reveal that the highest affinity binding sites present in the 5′ and 3′UTRs of hA3G mRNA bind Vif equally well, whereas binding to the highest affinity binding site located in the coding region is significantly weaker.
Figure 3.

Scatchard representation of the fluorescence binding data of wild-type Vif (A) and Vif AALA (B) to hA3G-5′UTR, hA3G-3′UTR, and hA3G-ΔUTR RNAs. The ratio of bound to unbound Vif is plotted as a function of the fraction of bound protein.
Table 4.
Determination of the apparent dissociation constant (KSca) of the highest affinity Vif binding site using the Scatchard equation
| RNA |
KSca (nM)a |
|
|---|---|---|
| WT Vif | Vif AALA | |
| hA3G-5′UTR | 27 ± 6b | 9 ± 2 |
| hA3G-3′UTR | 24 ± 5 | 11 ± 1 |
| hA3G-ΔUTR | 37 ± 4 | 20 ± 6 |
aKSca values were obtained by fitting the experimental data to Equation 4 (see ‘Materials and Methods’ section).
bMean ± SD of at least three experiments.
Filter binding experiments indicated that the mutant Vif AALA has a significantly reduced affinity for A3G mRNA (Figure 2B and Table 2). Fluorescence titration and Scatchard analysis using this protein (Figure 3B and Table 4) yielded unexpected results. Indeed, the affinity of Vif AALA for the highest affinity binding sites present in hA3G-5′UTR, hA3G-3′UTR and hA3G-ΔUTR RNAs was not decreased. On the contrary, the KSca values determined for the mutant protein were reproducibly 2- to 3-fold lower than for the wild-type protein (Table 4). This difference in the KSca values may not reflect a higher affinity of the mutant protein for these sites, but may be the consequence of the lower oligomerization state of the mutant Vif protein. Indeed, the KSca values likely reflect binding of the first Vif oligomer to a single RNA binding site, and thus it would be appropriate to express the Vif concentrations as concentration of Vif oligomers for this analysis. We choose not to do so for a matter of consistency throughout the present study. Nevertheless, the important point is that although mutation of the PPLP motif decreases the overall affinity of Vif for hA3G mRNA and fragments thereof, it does not reduce the affinity for the highest affinity binding sites present in hA3G-5′UTR, hA3G-3′UTR and hA3G-ΔUTR RNAs.
RNA probing and footprinting of Vif
To further investigate Vif binding to hA3G mRNA and its possible role in regulating hA3G translation, we next used chemical and enzymatic probing to determine the secondary structure of this mRNA and enzymatic footprinting to identify Vif binding sites. As the 5′ and 3′UTRs play an important role in the translational regulation of many mRNAs (24,25), we focused our structural analysis on these regions.
To test whether the 5′ and 3′UTRs fold independently or whether they interact with each other or with the hA3G coding region, we first incubated hA3G-5′UTR RNA with hA3G-Δ5′UTR RNA and hA3G-3′UTR RNA with hA3G-Δ3′UTR in a high salt and Mg2+ buffer. Agarose gel electrophoresis performed at 4°C revealed no complex, strongly suggesting that no long range interaction takes place between the 5′UTR, the coding region, and the 3′UTR of the hA3G mRNA (data not shown) (37). Accordingly, no significant difference was found when chemical probing was performed on full-length hA3G mRNA or on hA3G-5′UTR and hA3G-3′UTR RNAs (Figures 4 and 5, and data not shown).
Figure 4.

Chemical and enzymatic probing of the 5′UTR of hA3G mRNA and RNase footprinting of Vif. (A–C) Representative gels of structure probing with DMS (A) and RNAse V1 footprinting (B and C). Nucleotides modified by DMS (A) and RNase V1 cleavages protected by Vif (B and C) are indicated by red bars or dots. (D) Secondary structure model of the 5′-UTR of hA3G mRNA summarizing the experimental data.
Figure 5.

Chemical and enzymatic probing of the 3′UTR of hA3G mRNA and RNase footprinting of Vif. (A–C) Representative gels of structure probing with CMCT (A) and footprinting using RNase A (B) or RNAse V1 (C). Nucleotides modified by CMCT (A) and RNase cleavages protected by Vif (B and C) are indicated by red bars or dots. (D) Secondary structure model of the 3′UTR of hA3G mRNA summarizing the experimental data.
The 5′UTR of hA3G mRNA
As indicated by probing with DMS, CMCT, RNAse A and RNase T1, two chemicals and two RNases that specifically modify or cleave unpaired nucleotides, and RNase V1, which cuts paired and stacked residues, the 5′UTR of hA3G mRNA folds into three structured domains separated by single-stranded junctions (Figure 4). Most nucleotides in the 5′-J12 and 5′-J23 junctions were slightly to highly reactive towards chemical and 5′-J23 was also cleaved by RNase A (Figure 4A and D). The structure of the structured domains (5′-D1 to 5′-D3) was also unambiguous. Most nucleotides in the stems were not reactive, except some moderate reactivity close to some helix ends. In addition, the existence of several stems in 5′-D1 and 5′-D3 was confirmed by RNase V1 cuts (Figure 4B–D). On the other hand, as expected, most nucleotides that are single-stranded in our model were strongly modified by chemicals or/and cut by RNases A and T1 (Figure 4A and D). The only ambiguous region in the 5′-UTR of hA3G mRNA is the 5′-strand of the upper internal loop of 5′-D1 (52–55 nt). This loop was simultaneously cleaved by RNases A and V1, while theses nucleotides were reactive to CMCT and DMS, suggesting that these nucleotides are unpaired but stacked.
The RNAse V1 cleavages in this region increased upon addition of increasing Vif concentration, except at the highest Vif concentration (3 µM) at which they almost completely disappear. It is plausible that Vif binds first to the adjacent unstable stem (46–50 nt), increasing stacking of the internal loop, then binds to the loop itself at 3 µM Vif. However, this internal loop must be regarded as a weak Vif binding site, considering the Vif concentration at which protection was observed. No Vif footprint was detected in 5′-D2, while several were detected in 5′-D3 (Figure 4C and D). RNase V1 cleavages at nt 212–218 were strongly inhibited at ≥1 µM Vif, reflecting Vif binding to the lower helix of 5′-D3. Similarly, RNAse V1 cleavages at 257–262 nt and RNAse V1 and RNase A cleavages at 265–267 nt were strongly protected at 2 µM Vif (Figure 4C and D). These two regions may either represent a unique Vif binding site or two contiguous binding sites.
The 3′UTR of hA3G mRNA
Chemical and enzymatic probing of the 3′-UTR of hA3G mRNA revealed that it also consists in three independent structural domains separated by two single stranded junctions (Figure 5). Junction 3′-J23 was highly reactive towards chemical probes, while most nucleotides in 3′-J12 were only slightly reactive (Figure 5A and D). Indeed, the 3′-J12 3′ half may form an unstable helix with the very 3′-end of the 3′UTR (Figure 5D). The model structure of 3′-D1 is well supported by experimental data: nucleotides involved in helices were not reactive towards chemicals, except at the end of some helices, and the existence of most helices was supported by RNase V1 cleavages (Figure 5C and D). The proposed structure of domain 3′-D2 is in good agreement with experimental data, except for a weak RNase A cut after nt U1621, which is located in the middle of a helix, as supported by the lack of reactivity of the neighboring nucleotides (Figure 5A, B and D). The short apical stem of 3′-D2 consisting only of A–U and G–U base pairs is very unstable and might not exist at all (Figure 5A and D). The secondary structure of 3′-D3 is in good agreement with the probing data, except for the lower stem of this domain (Figure 5D). Thus, this stem might not exist, or might adopt alternative conformations, as suggested by cleavage by both RNase A and RNase V1 between nucleotides 1643 and 1648 (Figure 5B–D).
Vif footprints were observed in the three structured domains (Figure 5B–D). In 3′-D1, Vif-induced protections were split in several short regions but without a detailed knowledge of its 3D structure, it is difficult to estimate how many Vif molecules bind to this flexible domain. In 3′-D2, the two protected regions located in a continuous helical domain might be to far away to be produced by a single Vif protein. Similarly, it is unclear whether the protections observed in the 11 nt stretch encompassing the 3′-end of 3′-J23 and the 5′-region of 3′-D3 correspond to a single or two contiguous Vif binding site(s). To note, inhibition of the RNAse V1 cleavages in this region was already observed at 0.3 µM Vif, while protections in the other domains required higher protein concentrations.
In vitro translation
As Vif was found to bind with high affinity to hA3G mRNA, including to the 5′ and 3′ UTRs which are often involved in translational regulation (24,25), we studied the effects of WT Vif and Vif AALA on in vitro translation of full length and truncated versions of hA3G mRNA (Supplementary Figure S1 and Figure 6).
Figure 6.

Effect of Vif on the in vitro translation of hA3G. The amount of hA3G synthesized from full-length hA3G, hA3G-ΔUTR, hA3G-Δ5′UTR and hA3G-Δ3′UTR RNAs in the presence of WT Vif (A) or Vif AALA (B) was compared to that obtained in the presence of human AspRS. Data are the mean values of four independent experiments ±SDs. (C) The amount of hA3G synthesized from full-length hA3G and hA3G-ΔUTR RNAs in the presence of WT Vif in the absence or in the presence of MG132, a proteasome inhibitor, was compared to that obtained in the presence of human AspRS.
In order to ensure maximal activity of the WT and mutant Vif proteins and to avoid the inhibitory effect of the Vif storage buffer on in vitro translation, these proteins or human AspRS, which was used as a reference, were synthesized by in vitro coupled transcription/translation in rabbit reticulocyte lysate. These proteins were then added to new transcription/translation reaction mixtures including expression plasmids allowing translation of hA3G from full-length hA3G, hA3G-Δ5′UTR, hA3G-Δ3′UTR or hA3G-ΔUTR mRNAs. As a control, we also checked the effects of WT Vif and AspRS on the translation of luciferase. Radiolabeled proteins were then analyzed by SDS–PAGE and quantified (Supplementary Figure S1 and Figure 6).
When hA3G was translated from the full-length hA3G mRNA for 15 min, the amount of protein synthesized in the presence of WT Vif was 45% of that obtained in the presence of AspRS, indicating that Vif strongly repress translation from this mRNA (Supplementary Figure S1A and Figure 6A). Inhibition of translation from hA3G mRNA by WT Vif further increased at longer incubation times, and a ∼3-fold reduction of hA3G was observed at 45 min. Importantly, at 15 min, WT Vif did not significantly reduce hA3G synthesis from hA3G-ΔUTR RNA, indicating that the UTRs of hA3G mRNA play a crucial role in the translational repression mediated by WT Vif (Supplementary Figure S1B and Figure 6A). Further analysis showed that translation from hA3G-Δ3′UTR RNA was repressed at the same level as the full-length hA3G mRNA, whereas hA3G-Δ5′UTR RNA behaved as hA3G-ΔUTR RNA (Supplementary Figure S1B–D and Figure 6A). These results demonstrated that the 5′UTR was necessary and sufficient to allow Vif-mediated translational repression of hA3G. As the incubation time increased to 45 min, WT Vif gradually repressed hA3G synthesis from all RNAs, including hA3G-ΔUTR and hA3G-Δ5′UTR RNAs. However the amount of hA3G synthesized from these RNAs was always twice that obtained from full-length and hA3G-Δ3′UTR RNAs (Figure 6A). To further ensure that the Vif-mediated translational repression of hA3G was specific, we checked that WT-Vif had not effect on translation of luciferase (Supplementary Figure S1E). In addition, adding MG132, a proteasome inhibitor, in the rabbit reticulocyte lysate did not affect the results (Figure 6C), indicating that we were really looking at differences in protein synthesis, and not at protein degradation.
Finally, we tested whether the Vif multimerization domain played a role in the inhibition of translation. Indeed, the results we obtained with Vif AALA were almost identical to (and do not show any statistically significant difference with) those obtained with WT Vif (Figure 6B). They demonstrated that the Vif multimerization domain is not required for Vif-mediated repression of hA3G translation, and confirmed the role of the 5′UTR of hA3G mRNA in this process.
DISCUSSION
In this work, we first analyzed Vif binding to hA3G mRNA using filter binding and fluorescence assays (Figures 2 and 3 and Tables 2 to 4). Even though both techniques gave rather divergent dissociation constant values (120 and 36 nM, respectively, which will be discussed below) they allow to conclude that Vif binds strongly to hA3G mRNA, especially as we used buffers containing high concentrations of monovalent and divalent cations to reduce unspecific electrostatic interactions. We previously showed that Vif binds with high affinity and moderate cooperativity to the 5′-region of HIV-1 genomic RNA, while it binds very weakly or does not bind at all to the central and 3′-regions of this RNA, to E. coli tRNAs and 5S RNA, and to the 3′-region of Drosophila melangaster bicoid mRNA, indicating that Vif is not an unspecific RNA binding protein (20,21). Indeed, the Kd (from filter binding assays) and Kobs (from fluorescence titration curves) values we determined here for Vif binding to hA3G mRNA compare favorably with the Kd (45–79 nM) and Kobs (42–70 nM) values for Vif binding to 500 nt long RNA fragments derived from the HIV-1 genomic RNA (20,21).
There are however two noticeable differences between Vif binding to HIV-1 genomic RNA and to hA3G mRNA. First, with the latter RNA, we obtained rather divergent Kd and Kobs values (compare Tables 2 and 3), whereas Kd and Kobs values were very close with the former RNA (20,21). Second, the binding density of Vif to the hA3G mRNA fragments [one Vif molecule per ≥100 nt in the UTRs, and per ≥400 nt in the hA3G coding region (Table 3)] was much lower than the one we observed with the HIV-1 RNA fragments [one Vif molecule per ∼10 nt for RNA fragments derived from the 5′-region of HIV-1 genomic RNA (20)]. This lower binding density to hA3G mRNA was confirmed by our RNase footprinting experiments, which showed much more limited protections upon Vif binding to 5′ and 3′UTRs of hA3G mRNA (Figures 4 and 5), than to the 5′-region of HIV-1 genomic RNA (21).
Interestingly, this low binding density might explain the divergences between Kd and Kobs values observed with hA3G mRNA fragments. As explained in the ‘Results’ section, our filter binding data imply that not all complexes formed between Vif and hA3G mRNA fragments were retained with the same efficiency on the cellulose filters. Indeed, retention of the Vif/RNA complexes probably increased with the Vif binding density up to a threshold value above which retention was complete. If the Vif binding density is high, as for the HIV-1 genomic RNA fragments, most Vif–RNA complexes are completely retained on the filters. However, if the binding density is low, a bias is introduced in the filter binding experiments that artificially increases the Kd. If our assumption is correct the differences in the Kd and Kobs values should be smaller for the shorter RNAs, which have the higher Vif binding density (Table 3), than for the longer ones. This hypothesis is indeed verified, since the two techniques yielded very similar Kd and Kobs values for the short hA3G-5′UTR and hA3G-3′UTR RNAs, while divergent values were obtained for the longer RNAs. To summarize, (i) both techniques demonstrated that Vif has a stronger affinity for the 3′UTR than for the 5′UTR of hA3G mRNA (Tables 2 and 3) and (ii) for longer RNAs, Kobs derived from fluorescence titrations are more reliable than Kd values derived from filter binding assays. Interestingly, the fluorescence data showed that Vif has also a high affinity for the hA3G coding region (Table 3).
However, the Kobs values extracted from the fluorescence titration curves using the McGhee and von Hippel model (29) are also mean values of the individual Vif binding sites. As shown in Figure 3, it was also possible to analyze these data using the Scatchard equation in order to extract the KSca value for the binding site having the highest affinity for Vif in hA3G-5′UTR, hA3G-3′UTR and hA3G-ΔUTR. This analysis revealed that the highest affinity Vif binding sites present in the 5′ and 3′UTRs of hA3G mRNA bind Vif with similar affinity (KSca = 27 ± 6 and 24 ± 5 nM, respectively), even though the overall binding of Vif to the 5′UTR is significantly weaker than to the 3′UTR (Tables 2 and 3). Thus, the secondary Vif binding sites located in the 5′UTR must have considerably weaker affinity for this protein than those located in the 3′UTR of hA3G mRNA. In contrast, the KSca of the highest affinity Vif binding site in hA3G-ΔUTR (KSca = 37 ± 4 nM), is identical, within experimental errors to Kobs (46 ± 7 nM), indicating that all binding sites present in the coding region of hA3G mRNA have a similar affinity for Vif.
We showed that mutation of the Vif multimerization motif reduced the Vif oligomerization state but did totally abolished it, suggesting that an additional region is involved in Vif multimerization. Interestingly, this mutation decreased the overall affinity of the Vif AALA protein for hA3G mRNA, but it did not decrease its affinity for the RNA sites with the highest affinity for Vif (Figure 3).
RNase footprinting revealed 3 to 4 Vif binding sites in the 5′UTR (Figure 4) and 3 to 6 Vif binding sites in the 3′UTR (Figure 5), in agreement with the fluorescence data (Table 3). In addition, while most protections in the 3′UTR were obvious at 1.5 µM Vif, only one Vif-binding site, located in the lower stem of domain 5′-D3, was clearly detected at 1 µM Vif. This observation is in keeping with the overall higher affinity of Vif for the 3′UTR of hA3G mRNA, even though the protein concentrations required to observe a footprint could not be directly compared to those used in titration experiments.
In the last part of this study, we analyzed the effect of Vif on the in vitro translation of hA3G from full-length hA3G mRNA and mRNAs devoid of the 5′UTR or/and the 3′UTR. Vif induced a 2-fold reduction of translation that was strictly dependent on the 5′UTR, while the 3′UTR had no influence on translation (Figures 6 and S1). Hence, there is no correlation between the relative binding affinity of Vif for the 5′ and 3′UTRs and their role in Vif-mediated translational repression. The same conclusion holds true when comparing WT Vif and Vif AALA: mutation of the Vif multimerization domain reduced the overall affinity and specificity of Vif binding to hA3G mRNA fragments (Table 2), but it didn’t significantly affect translation (Figure 6). The fact that wild-type Vif and Vif AALA have similar affinities for the highest affinity Vif binding site located in the 5′UTR of hA3G RNA (Figure 3 and Table 4), may explain why they have similar effects on hA3G mRNA translation. These results suggest that Vif does not simply physically prevent scanning of the ribosomes through the 5′UTR of hA3G mRNA, but raise the possibility of a specific mechanism of Vif-mediated translational repression. Mutation of the individual Vif binding sites we identified in the 5′UTR of hA3G mRNA (Figure 4) should prove very useful in order to decipher this mechanism. Numerous examples in the literature showed that both the 5′ and the 3′UTRs can be involved in translational regulation (24,25). However, despite Vif binding to both UTRs of hA3G mRNA, our data clearly show that the 3′UTR is dispensable for the translational regulation by Vif. Otherwise, several lines of evidence indicated that mRNAs ‘circularize’ during translation, and this phenomenon is also involved in some translational control mechanisms (24,25). The experiments we performed in the present study do not allow to rule out this possibility, as our hA3G-Δ3′UTR expression construct, although lacking the authentic 3′-UTR of the hA3G mRNA, contains the 3′-UTR present in the expression vector.
The 5′UTR-dependent translational repression of hA3G by Vif was observed at all incubation times (Figure 6). In addition, a general repression of hA3G synthesis by Vif was gradually observed at increasing incubation times, irrespective of the mRNA used for its synthesis (Figure 6). This phenomenon ultimately resulted in a 30–40% reduction in hA3G synthesis. Several groups previously reported a negative impact of Vif on hA3G translation. Pulse-chase experiments in cells transfected with a tagged hA3G indicated a 30–78% decrease in hA3G synthesis in the presence of Vif (7,10,38), while Vif impaired hA3G translation by 70–75% in an in vitro-coupled transcription/translation system (10). Importantly, none of these studies used expression vectors with the authentic 5′UTR of hA3G mRNA (26). Thus, these authors very likely only observed the time-dependent, UTR-independent secondary effect, while they missed the main 5′UTR-dependent, time-independent effect. Even though we cannot dismiss that possibility, we think it is unlikely that the time-dependent inhibition of translation is due to Vif aggregation, because (i) in all these experiments (including ours) Vif was directly synthesized in the in vitro translation system and (ii) one would expect a decrease of Vif activity upon aggregation (i.e. aggregation of Vif would likely decrease inhibition of translation over time).
Since the seminal publication by Sheehy et al. (1), it has become increasingly clear that the function of Vif is to counteract the restriction factors hA3G and hA3F. Even though several mechanisms have been proposed for Vif function, the proteasome-mediated, Vif-induced degradation of these factors has been by far the most studied. In agreement with previous studies suggesting that the RNA binding properties of Vif are essential for its function (23), our present work indicates that Vif binding to hA3G mRNA, and particularly to its 5′UTR might be crucial for Vif function, by downregulating hA3G translation. Even though other authors already reported a negative effect of Vif on hA3G translational (7,8,10,38), our present work suggest that these authors missed most of the effect by working with expression vectors lacking the authentic 5′UTR of hA3G mRNA, which had not been identified at that time. We are presently performing experiments to confirm that Vif binding to the 5′UTR of hA3G mRNA inhibits hA3G synthesis in cells, and to evaluate the contribution of this effect to HIV-1 replication in the presence of hA3G expression (G. Mercenne et al., manuscript in preparation).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French ‘Agence Nationale de Recherches contre le SIDA’ (ANRS to J.C.P. and R.M.); fellowships of the French Ministry of Higher Education and Research and of SIDACTION (to G.M.); SIDACTION (to S.H.). Funding for open access charge: Agence Nationale de Recherches sur le SIDA (ANRS).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Philippe Dumas, Philippe Wolff and Magali Frugier (all from the Architecture et Réactivité de l’ARN, Université de Strasbourg, CNRS) for their help with curve fitting in Mathematica and HPLC chromatography, and for the gift of the human AspRS expression plasmid, respectively.
REFERENCES
- 1.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 2.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu YL, Greene WC. The APOBEC3 Cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 4.Henriet S, Mercenne G, Bernacchi S, Paillart JC, Marquet R. Tumultuous relationship between the human immunodeficiency virus type 1 viral infectivity factor (Vif) and the human APOBEC-3G and APOBEC-3F restriction factors. Microbiol. Mol. Biol. Rev. 2009;73:211–232. doi: 10.1128/MMBR.00040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 7.Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 8.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 9.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 10.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 11.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 12.Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Potash MJ, Volsky DJ. Functional domains of APOBEC3G required for antiviral activity. J. Cell Biochem. 2004;92:560–572. doi: 10.1002/jcb.20082. [DOI] [PubMed] [Google Scholar]
- 15.Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J. Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 17.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 18.Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 2007;81:8236–8246. doi: 10.1128/JVI.02694-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernacchi S, Henriet S, Dumas P, Paillart JC, Marquet R. RNA and DNA binding properties of HIV-1 Vif protein: a fluorescence study. J. Biol. Chem. 2007;282:26361–26368. doi: 10.1074/jbc.M703122200. [DOI] [PubMed] [Google Scholar]
- 21.Henriet S, Richer D, Bernacchi S, Decroly E, Vigne R, Ehresmann B, Ehresmann C, Paillart JC, Marquet R. Cooperative and specific binding of Vif to the 5′ region of HIV-1 genomic RNA. J. Mol. Biol. 2005;354:55–72. doi: 10.1016/j.jmb.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 22.Henriet S, Sinck L, Bec G, Gorelick RJ, Marquet R, Paillart JC. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 2007;35:5141–5153. doi: 10.1093/nar/gkm542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Pomerantz RJ, Dornadula G, Sun Y. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J. Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee S, Pal JK. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell. 2009;101:251–262. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- 25.Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 2003;28:182–188. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- 26.Muckenfuss H, Kaiser JK, Krebil E, Battenberg M, Schwer C, Cichutek K, Munk C, Flory E. Sp1 and Sp3 regulate basal transcription of the human APOBEC3G gene. Nucleic Acids Res. 2007;35:3784–3796. doi: 10.1093/nar/gkm340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skripkin E, Isel C, Marquet R, Ehresmann B, Ehresmann C. Psoralen crosslinking between human immunodeficiency virus type 1 RNA and primer tRNA3(Lys) Nucleic Acids Res. 1996;24:509–514. doi: 10.1093/nar/24.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Goncalves J, Gabuzda D. Phosphorylation of Vif and its role in HIV-1 replication. J. Biol. Chem. 1996;271:10121–10129. doi: 10.1074/jbc.271.17.10121. [DOI] [PubMed] [Google Scholar]
- 29.McGhee JD, von Hippel PH. Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 30.Tsodikov OV, Holbrook JA, Shkel IA, Record MT., Jr Analytic binding isotherms describing competitive interactions of a protein ligand with specific and nonspecific sites on the same DNA oligomer. Biophys. J. 2001;81:1960–1969. doi: 10.1016/S0006-3495(01)75847-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mely Y, de Rocquigny H, Sorinas-Jimeno M, Keith G, Roques BP, Marquet R, Gerard D. Binding of the HIV-1 nucleocapsid protein to the primer tRNA(3Lys), in vitro, is essentially not specific. J. Biol. Chem. 1995;270:1650–1656. doi: 10.1074/jbc.270.4.1650. [DOI] [PubMed] [Google Scholar]
- 32.Woodbury CP, Jr, von Hippel PH. On the determination of deoxyribonucleic acid-protein interaction parameters using the nitrocellulose filter-binding assay. Biochemistry. 1983;22:4730–4737. doi: 10.1021/bi00289a018. [DOI] [PubMed] [Google Scholar]
- 33.Miller JH, Presnyak V, Smith HC. The dimerization domain of HIV-1 viral infectivity factor Vif is required to block APOBEC3G incorporation with virions. Retrovirology. 2007;4:81–92. doi: 10.1186/1742-4690-4-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang B, Gao L, Li L, Lu Z, Fan X, Patel CA, Pomerantz RJ, DuBois GC, Zhang H. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J. Biol. Chem. 2003;278:6596–6602. doi: 10.1074/jbc.M210164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S, Sun Y, Zhang H. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J. Biol. Chem. 2001;276:4889–4893. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barraud P, Paillart JC, Marquet R, Tisne C. Advances in the structural understanding of Vif proteins. Curr. HIV Res. 2008;6:91–99. doi: 10.2174/157016208783885056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R. In vitro evidence for a long range pseudoknot in the 5′-untranslated and matrix coding regions of HIV-1 genomic RNA. J. Biol. Chem. 2002;277:5995–6004. doi: 10.1074/jbc.M108972200. [DOI] [PubMed] [Google Scholar]
- 38.Kao S, Akari H, Khan MA, Dettenhofer M, Yu XF, Strebel K. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J. Virol. 2003;77:1131–1140. doi: 10.1128/JVI.77.2.1131-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.