


Abstract
XRCC1 plays a central role in mammalian single-strand break repair. Although it has no enzymatic activity of its own, it stimulates the activities of polynucleotide kinase/phosphatase (PNKP), and this function is enhanced by protein kinase CK2 mediated phosphorylation of XRCC1. We have previously shown that non-phosphorylated XRCC1 stimulates the kinase activity of PNKP by increasing the turnover of PNKP. Here we extend our analysis of the XRCC1-PNKP interaction taking into account the phosphorylation of XRCC1. We demonstrate that phosphorylated and non-phosphorylated XRCC1 interact with different regions of PNKP. Phosphorylated XRCC1 binds with high affinity (Kd = 3.5 nM and 1 : 1 stoichiometry) to the forkhead associated (FHA) domain, while non-phosphorylated XRCC1 binds to the catalytic domain of PNKP with lower affinity (Kd = 43.0 nM and 1 : 1 stoichiometry). Under conditions of limited enzyme concentration both forms of XRCC1 enhance the activities of PNKP, but the effect is more pronounced with phosphorylated XRCC1, particularly for the kinase activity of PNKP. The stimulatory effect of phosphorylated XRCC1 on PNKP can be totally inhibited by the presence of excess FHA domain polypeptide, but non-phosphorylated XRCC1 is not susceptible to competition by the FHA domain. Thus, XRCC1 can stimulate PNKP by two independent mechanisms.
INTRODUCTION
DNA single-strand breaks (SSB) can be directly induced by oxidative attack at the sugar-phosphate backbone or indirectly during the course of repair of damaged bases generated by both internal and external DNA damaging agents such as oxygen free radicals, ionizing radiation and alkylating agents (1). If left unrepaired, SSBs can be converted into potentially lethal double-strand breaks (DSBs) (1). SSBs often bear non-ligatable termini, such as 3′-phosphate and 5′-hydroxyl termini (2,3), which are required to be converted respectively to 3′-hydroxyl and 5′-phosphate termini so as to facilitate downstream DNA synthesis and ligation by DNA polymerase β and DNA ligase III (1,4).
Human polynucleotide kinase (PNKP) is a DNA repair enzyme that processes DNA termini (5,6). It contains three domains: a forkhead-associated (FHA) domain, a kinase domain and a phosphatase domain (5–7). The latter two domains are responsible for the enzyme’s dual cellular functions of phosphorylating 5′-termini and dephosphorylating 3′-termini. PNKP has been shown to play a critical role in several repair pathways including base excision/single-strand break repair (BER/SSBR) (4,8,9) and the nonhomologous end-joining (NHEJ) repair pathway for DSB repair (10–12). Decreased expression of PNKP in A549 cell lines using siRNA silencing gave rise to cells sensitized to a range of genotoxic agents, most notably ionizing radiation and the topoisomerase I inhibitor, camptothecin, a slower repair rate of DNA strand breaks, and increased spontaneous mutation frequency (13).
Unlike the phosphatase and kinase domains in PNKP, the FHA domain of PNKP has no enzymatic activity. FHA domains are generally regarded to be phospho-protein recognition motifs that are often found in regulatory proteins in the nucleus including kinases, phosphatases and transcription factors (14,15). The PNKP FHA domain has been shown to bind to the DNA repair proteins XRCC1 and XRCC4 following phosphorylation of the proteins by protein kinase CK2 (11,16).
XRCC1 is considered to be a scaffold protein primarily involved in SSB repair (1,17,18), but may also play a role in a newly-described DSB repair pathway (19,20). The protein can bind to PNKP, DNA polymerase β and DNA ligase III. Binding of XRCC1 to PNKP stimulates PNKP’s enzymatic activities (8), and we have shown that this is most likely due to enhanced protein turnover (21). XRCC1 is rich in serine (Ser: 9.8%) and threonine (Thr: 4.6%) residues, which are the potential phosphorylation sites in proteins. Loizou et al. (16) originally demonstrated that XRCC1 can interact with the PNKP FHA domain through a peptide sequence in XRCC1 phosphorylated by CK2, and this interaction stimulates PNKP enzyme activity even more than the interaction between PNKP and non-phosphorylated XRCC1. Recently, Ali et al. (22) synthesized a series of phospho-peptides based on an XRCC1 phosphorylated segment (Tyr515–Glu526), and demonstrated, using isothermal titration calorimetry and co-crystallization methods, that there are two binding sites in this peptide for the FHA domain of PNKP, and that the phosphorylated peptide can bind two separate FHA domains in tandem. Since there are potentially more phosphorylation motifs for CK2 on XRCC1 the situation may be more complicated for the interaction of the full-length proteins.
Therefore, in order to gain more insight into the XRCC1-PNKP interaction, we examined XRCC1 phosphorylation by CK2 in vitro, its interaction with the FHA domain of PNKP, and compared the influence of phosphorylated versus non-phosphorylated XRCC1 on the phosphatase and kinase activities of PNKP.
MATERIALS AND METHODS
Expression and purification of proteins
XRCC1 and PNKP-XRCC1 cDNA cloned in pET16BXH vector and PNKP cDNA cloned in pET16b were transformed in Escherichia coli BL21 (DE3) for expression and affinity purification following previously reported procedures (6,23). The fractions containing high concentration of each protein after affinity purification on Ni–NTA beads were pooled and dialyzed against buffer containing 50 mM Tris (pH 7.5), 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT and quantified using established extinction coefficients, 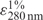 of 7.9 for XRCC1 (23) and
of 7.9 for XRCC1 (23) and 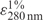 of 12.2 for PNKP (24). The proteins were stored at −80°C.
of 12.2 for PNKP (24). The proteins were stored at −80°C.
FHA and catalytic domains of PNKP—Human PNKP cDNA with a 10× His tag was previously cloned between Xba I and Bam HI restriction sites in the BlueScript plasmid (21). To generate a construct encoding the FHA domain, a stop codon and BamH1 restriction site were introduced at the end of codon 130 using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The modified construct was then digested with Bam HI and Xba I, and the 497 bp fragment corresponding to the FHA domain was ligated into Bam HI/Xba I cleaved pET16b (Novagen, Gibbstown, NJ). The resultant plasmid encoding the FHA domain (from Met1 to Lys130 with a 10× His tag at the N-terminus) was transformed into E. coli DE3 (BL21) pLysS (Novagen) for protein expression following the protocol recommended by Novagen. The protein was purified from the E. coli lysate using Ni–NTA agarose beads (Qiagen, Mississauga, ON) as directed by the manufacturer. The recovered protein was dialyzed and concentrated using a Millipore microconcentrator (Millipore, Etibicoke, ON) and stored in buffer containing 50 mM Tris pH 7.5, 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT at −80°C. The 16.5 kDa protein was quantified by the Bradford assay (25), and shown to be detectable by both mono and polyclonal anti-PNKP antibodies (Supplementary Figure S1). The C-terminal (CT) catalytic kinase/phosphatase domain (K141-G521) of PNKP was subcloned and expressed in E. coli in a similar manner.
CK2—The cDNA of CK2α previously cloned in pGEX-3X vector with an N-terminal GST tag (26) was transformed into E. coli BL21 (DE3) for protein expression. The cells were first cultured at 37°C with continuous shaking until the OD600 reached 0.6. Protein expression was then induced by IPTG at 30°C for 3 h. The cells were then harvested at 8000 rpm and 4°C for 15 min, washed with cold PBS, suspended in PBS containing 50 µM PMSF and subjected to sonication on ice for 5 × 30 s. Triton X-100 was added to the mixture to make the final concentration 1% Triton X-100, and the mixture was incubated on ice with shaking for 15 min followed by centrifugation at 4°C to obtain the soluble supernatant. The GST-tagged CK2α was then bound to glutathione–Sepharose 4B beads (GE Healthcare, Baie d’Urfe, PQ) by incubating the lysate with GSH beads for 1 h on ice. The beads were then washed with PBS containing 50 µM PMSF, and the protein was eluted with Tris–HCl buffer (50 mM, pH 8.0) containing 10 mM GSH. The fractions with the highest concentration of CK2α were dialyzed against buffer containing 50 mM Tris–HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA and 50% glycerol three times at 4°C, quantitated by Bradford assay, and stored at −80°C for further use.
In vitro phosphorylation of XRCC1 by CK2
Phosphorylation of XRCC1 by CK2 was carried out in kinase buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM MgCl2 and 0.1 mM ATP. To follow the phosphorylation, 1.5 µg XRCC1 and 3.3 pmol of 32P labeled ATP were added to the vial containing kinase buffer. Then 0.1 µg CK2 (diluted immediately from the stock using cold dilution buffer containing 1 mg/ml BSA, 5 mM MOPS pH 7.0 and 200 mM NaCl) was added to the vial. The reaction was then incubated at 30°C for up to 90 min. Aliquots of the reaction mixture were sampled at 0, 30, 60 and 90 min, mixed with Laemmli buffer, and heated at 95°C for 5 min. The samples were then loaded on an SDS–PAGE gel to monitor the phosphorylation of XRCC1.
To prepare phosphorylated XRCC1 (pXRCC1) for further use, radio-labeled ATP was omitted while maintaining the same ratio of XRCC1 and CK2 in the reaction mixture. In this case, an elemental detector, i.e. inductively coupled plasma mass spectrometer, was applied to quantify the phosphate incorporated in the proteins. After phosphorylation, the resultant reaction mixture was incubated with 100 µl GSH-Sepharose beads at 4°C for 30 min to remove the GST-tagged CK2α, and the supernatant was collected for further use. To test the level of phosphorylation, 50 µl of the supernatant was desalted into HPLC grade water using a BioSpin column (Bio-Rad Laboratory, Mississauga, ON). One portion of the resultant filtrate was analyzed by inductively coupled plasma mass spectrometry using scandium as an internal calibrator and free ATP as an external calibrator. The protein in the other portion of filtrate was quantified using the Bradford assay. The molar ratio of phosphate to protein was then calculated based on the above measurements.
Trypsin digestion of XRCC1
Phosphorylated XRCC1 was purified by 10% SDS–PAGE, and stained with 0.1% Coomassie blue. After destaining, the band with an apparent MW of ∼80 kDa was extracted and digested at 37°C overnight using proteomics grade trypsin (Promega, Madison, WI) with a mass ratio of trypsin to protein = 1 : 50, according to the protocol for in-gel digestion of protein previously described (27). The resultant peptides were then mixed with 2,5-dihydroxybenzoic acid (25 mg/ml in 70% acetonitrile and 1% H3PO4) in 1 : 1 ratio (v/v). One microliter of the resultant peptide mixture was loaded onto the MALDI plate for peptide analysis.
MALDI-TOF measurements of proteins and peptides
An Autoflex III MALDI-TOF mass spectrometer (Bruker, Fremont, CA) was employed to analyze the molecular weight of XRCC1 before and after phosphorylation by CK2, and also for analyzing the trypsin digestion products combined with peptide mapping. For analysis of the full-length proteins, XRCC1 and the phosphorylation reaction mixtures (containing both pXRCC1 and CK2α) were first desalted using C8 ziptips (Millipore, Billerica, MA), mixed with sinapinic acid, and loaded onto the MALDI probe for molecular weight analysis. The instrument was calibrated using standard BSA and its dimers. The molecular weight accuracy was within 100 ppm. For peptide analysis, a standard peptide from Bruker was used for calibration of the instrument, and the mass accuracy was generally within 20 ppm. Proteomics software such as PeptideMass (http://www.expasy.org/) was applied to predict the theoretical peptides generated by trypsin digestion.
Co-immunoprecipitation of XRCC1 with PNKP and its truncated domains
Non-phosphorylated XRCC1 (npXRCC1) or pXRCC1 (1.5 µg) were separately mixed with the FHA domain of PNKP at different molar ratios in buffer containing 50 mM Tris–HCl (pH 7.5), 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT, and incubated on ice for 30 min. Anti-XRCC1 polyclonal antibody (0.6 µg, Santa Cruz Biotech, Santa Cruz, CA) was added to the reaction vial, and incubated on ice with continuous shaking for 2 h. Protein A Sepharose beads (10 µl, GE Healthcare) were then added to the reaction mixture, and the reaction mixture was incubated on ice for another 2 h. The beads were washed three times with Tris buffer containing 0.1% Tween-20 and the protein remaining bound to the beads was eluted by boiling the beads in Laemmili buffer containing 0.125 M Tris–HCl, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol and 0.004% bromphenol blue. The resultant supernatant solution was subjected to 10% SDS–PAGE and western blotting analysis using a monoclonal anti-PNKP antibody (28), horseradish peroxidase-conjugated goat anti-mouse IgG antibody, and the ECL Plus western blot detection system (GE Healthcare).
Steady-state fluorescence spectroscopy
The interaction between p/npXRCC1 and PNKP and its FHA and CT catalytic domains was studied using acrylodan-labeled PNKP as described previously for studying PNKP-XRCC1 interactions (21,23). The interaction between pXRCC1 and single-stranded DNA was studied using the intrinsic fluorescence due to tryptophan residues as described previously for npXRCC1 (23). GraphPad Prism software (GraphPad Software Inc. La Jolla, CA) was used for the analysis of binding data.
Circular dichroism spectroscopy
Far-UV CD measurements were performed with an Olis DSM 17 CD spectropolarimeter (Bogart, GA) as previously described (23).
Substrate preparation
HPLC purified single-strand oligonucleotides (Oligos 1–6) with the sequences shown in Table 1 were synthesized by Integrated DNA Technology (Coralville, IA). The oligonucleotides were then annealed following the scheme shown in Table 2. The annealed products were purified on 8% native PAGE gels followed by ethanol precipitation. The resultant double-stranded substrates were resuspended in distilled water before use.
Table 1.
Sequences of the oligonucleotides synthesized
| Name | Length | Sequence |
|---|---|---|
| Oligo1 | 18mer | 5′-(6-FAM)TAGCATCGATCAGTCCTCp-3′ |
| Oligo2 | 21mer | 5′-GAGGTCTAGCATCGTTAGTCA(TAMRA)-3′ |
| Oligo3 | 39mer | 5′-TGACTAACGATGCTAGACCTCGAGGACTGATCGATGCTA-3′ |
| Oligo4 | 40mer | 5′-TGACTAACGATGCTAGACCTCTGAGGACTGATCGATGCTA-3′ |
| Oligo5 | 39mer | 5′-TGACTAACGATGCTAGACCTCATCCGTTCAGTACGTAGG-3′ |
| Oligo6 | 24mer | 5′-GGCGCCCACCACCACTAGCTGGCC-3′ |
Table 2.
Schematic representation of model substrates
 |
PNKP kinase and phosphatase assay using fluorescent substrates
We followed the protocol of Dobson and Allinson (29) with minor modifications to simultaneously monitor the kinase and phosphatase activities of PNKP. Briefly, substrate (80 nM) was added to a buffer containing 80 mM succinate (pH 5.5), 10 mM MgCl2, 1 mM DTT and 0.1 mM ATP followed by addition of 4 nM PNKP. The reaction mixture was then incubated at 37°C. At each time point (0, 2, 5, 10 and 20 min), 3 µl aliquots were withdrawn and mixed with 2× denaturing loading buffer, incubated at 95°C for 5 min and analyzed on a polyacrylamide/urea gel. The gel was visualized using a Typhoon 9400 or Typhoon Trio laser scanning system (GE Healthcare). The image was then analyzed using ImageQuant 5.2 or ImageQuant TL software (GE Healthcare), and the data obtained were plotted using Igor Pro software (WaveMetrics, Portland, OR).
Conventional PNKP kinase activity assay using 32P labeling
To examine the kinase activity of PNKP using 32P ATP labeling assay, we applied the method reported previously (21). Briefly, 0.2 nmol 24mer single-strand oligonucleotide (Oligo 6, Table 1), cold ATP (0.4 nmol) and 3.3 pmol [γ-32P] labeled ATP were added to 50 µl reaction buffer, followed by the addition of 0.5 µg PNKP. The reaction was then incubated at 37°C. At various times aliquots were withdrawn, mixed with 2× denaturing loading buffer, denatured, electrophoresed and visualized using the Typhoon 9400 laser scanning system as described above.
Influence of npXRCC1 and pXRCC1 on the dual activities of PNKP
To test the impact of XRCC1 phosphorylation on the PNKP activity, PNKP was first used to process the substrate for 20 min as described above. After 20 min, 5-fold molar excess of npXRCC1, pXRCC1, or BSA was added to the reaction, and the reaction was allowed to proceed for another 20 min. The reaction mixture was sampled at various times. Each aliquot was mixed with 2× denaturing loading buffer and denatured for SDS–PAGE analysis and laser scanning.
Effect of competitive binding of FHA to pXRCC1 on the dual activities of PNKP
PNKP, pXRCC1 or npXRCC1, and FHA in Tris buffer were mixed at a molar ratio of 1 : 5 : 20 on ice for 15 min. The protein mixture was then added to the reaction vials containing substrate in 80 mM succinate (pH 5.5), 10 mM MgCl2, 1 mM DTT and 0.1 mM ATP and incubated at 37°C for 20 min. An equal volume of 2× loading buffer was added and the DNA was heat denatured at 95°C for 5 min before urea–PAGE analysis and fluorescence imaging scanning.
RESULTS AND DISCUSSION
Characterization of XRCC1 phosphorylation and identification of phosphorylation sites of XRCC1 by mass spectrometry
Using 32P-labeled ATP, we observed the phosphorylation of XRCC1 by CK2 (Figure 1A) and calculated that the degree of phosphorylation varied between experiments with the molar ratio ranging between 4.5 and 6 phosphates per XRCC1. Measurement by inductively coupled plasma mass spectrometry (ICPMS) to selectively monitor phosphorus present in the protein yielded a similar molar ratio.
Figure 1.

In vitro phosphorylation of XRCC1. (A) 32P-labeling demonstrating the phosphorylation of XRCC1 by CK2. (B) Linear MALDI-TOFMS analysis demonstrating the molecular weight increase of XRCC1 after phosphorylation. The upper trace shows the spectrum for pXRCC1 and the lower trace is for the npXRCC1. The peak with an m/z value of 66 477 shows the BSA calibration standard. (C) Linear MALDI-TOFMS analysis of the trypsin digestion products indicating multiple phosphorylation within peptide segments P1 (459–494) and P2 (503–546). Upper trace pXRCC1, lower trace npXRCC1. (D) Reflective MALDI-TOFMS analysis of the trypsin digest indicating multiple phosphorylation of peptide segment P3 (401–427). Upper trace pXRCC1, lower trace npXRCC1.
The phosphorylation of XRCC1 results in a molecular mass increase in the protein by 80 Da for the addition of each phosphate group to the protein. Using MALDI-TOF/MS to measure the molecular mass of the protein following its phosphorylation, we observed that the molecular mass of XRCC1 increased to 71.2 kDa, which is ∼400 Da larger than non-phosphorylated XRCC1 (70.8 kDa) (Figure 1B), indicating that an average of five residues were phosphorylated by CK2, which was consistent with the quantitative assay using ICPMS analysis.
To further identify the sites of phosphorylation in the protein, trypsin digestion followed by MALDI-TOF peptide mapping was applied. We compared the mass spectra obtained before and after phosphorylation by CK2 (Figure 1C and D). With the MALDI linear TOFMS mode, the m/z range of 3500–9000 was monitored, in which peptides P1 (459–494) and P2 (503–546) were found to shift to higher m/z after CK2 phosphorylation. Peptide P1 can be triply phosphorylated with m/z increases of 80, 160 and 240, indicating that 3 of 4 potential sites (S461, S475, S485 and T488) have been phosphorylated, among which only T488 is the conventional motif for CK2 phosphorylation (16,30,31). Peptide P2, can be tetraphosphorylated with m/z increases of 80, 160, 240 and 320, indicating that four potential sites (S518, T519, T523 and S525) can all be phosphorylated, though only S518 and T523 are typical CK2 sites. With reflective TOFMS mode, we also monitored the mass spectrum at the low m/z range, and found another peptide, P3 (401–427), was phosphorylated with a weak signal at m/z of 3133 and a much more intense peak at 3149 (arising from oxidation of the methionine), which were 480 m/z higher than the unphosphorylated peptide P1 (m/z = 2652) and its oxidized form at m/z of 2668 (Figure 1D), indicating that six serine sites on this segment were fully phosphorylated, with S408, S409, S410 and S421 as the CK2 consensus sites, while S416 and S418 are atypical sites for CK2. Under our conditions, we observed singly phosphorylated P1 and triply phosphorylated P2 to be the most abundant modifications. Our data showing multiple sites of phosphorylation at the three peptide sites are in broad agreement with those of Loizou et al. (16). In their analysis of the phosphorylation of three peptides with amino acid sequences corresponding to P1, P2 and P3, these authors observed that the P2 peptide underwent the most rapid phosphorylation by CK2. Furthermore, XRCC1 immunoprecipitated from HeLa cells displayed multiple phosphorylation (up to four residues) in the P1 peptide sequence and similarly up to four sites of phosphorylation in the P2 peptide region, with the largest signal arising from triple phosphorylation (16). Thus our in vitro phosphorylation of XRCC1 appears to closely reflect the in vivo phosphorylation profile.
FHA domain of PNKP binds to pXRCC1 specifically
The interaction of the FHA domain of PNKP with non-phosphorylated XRCC1 (npXRCC1) and CK2-phosphorylated XRCC1 (pXRCC1) in vitro was first examined by immunoprecipitation. The complex formed between the FHA domain and XRCC1 was immunoprecipitated using an anti-XRCC1 antibody and probed with an antibody to PNKP that recognizes an epitope in the FHA domain (28). As shown in Figure 2A, a band at 16 kDa was observed following coincubation of the FHA domain and pXRCC1, indicating tight binding of the FHA to pXRCC1. In contrast, no FHA signal was detected following immunoprecipitation of the mixture of npXRCC1 with FHA, indicating that there is no discernable binding between FHA and npXRCC1. When human A549 cell lysate was incubated with increasing amounts of the FHA domain and immunoprecipitated with anti-XRCC1 antibody, the FHA peptide was co-immunoprecipitated (Figure 2B), indicating the presence of phosphorylated XRCC1 in the A549 cells with similar properties to XRCC1 phosphorylated in vitro.
Figure 2.

The FHA domain of PNKP can interact with XRCC1 phosphorylated in vitro by CK2 and in A549 cell lines. (A) Phosphorylated XRCC1 (pXRCC1) and non-phosphorylated XRCC1 (npXRCC1) was incubated with the FHA domain in different molar ratios and then immunoprecipitated. The figure shows the results of western blots to determine if the FHA domain co-precipitated with the XRCC1 proteins. (B) Co-immunoprecipitation of the FHA domain after incubation with A549 cell lysate and immunoprecipitation with anti-XRCC1 antibodies, indicating the presence of phosphorylated XRC1 in A549 cells.
Evidence for different PNKP binding sites for npXRCC1 and pXRCC1
In order to obtain quantitative data for the interaction between the FHA domain and pXRCC1, we labeled the single Cys residue (Cys-46) in the FHA domain with acrylodan (AC). When FHA-AC was excited at 380 nm, the emission maximum occurred at 495 nm, and addition of pXRCC1 resulted in ∼50% AC fluorescence quenching. Titration of FHA-AC with pXRCC1 yielded the relative fluorescence intensity curve shown in Figure 3A inset. The maximum quenching observed at saturating dose of pXRCC1 was taken as 1, and the quenching at different concentrations of pXRCC1 was plotted as the fraction bound versus pXRCC1 concentration (Figure 3A). Non-linear regression analysis of this binding data yielded a Kd value for FHA-AC binding to pXRCC1 of 3.5 ± 0.5 nM. In Figure 3B, log(F0 − F/F − F∞) is plotted against log[pXRCC1] to obtain the stoichiometric relationship between the two proteins, where F0, F and F∞ are the fluorescence intensities of solutions of FHA-AC alone, FHA-AC in the presence of various concentrations of pXRCC1, and FHA-AC saturated with pXRCC1, respectively (32). This plot yielded a slope of 1.08, indicating a 1 : 1 interaction between pXRCC1 and FHA-AC domain. In contrast, npXRCC1 addition had no significant effect on AC fluorescence (∼3% quenching), thus providing no evidence for its binding to the FHA domain (data not shown).
Figure 3.

Interaction of XRCC1 with different domains of PNKP. (A) Fluorescence titration of the AC labeled FHA domain versus pXRCC1. FHA-AC (0.065 µM) was excited at 380 nm and the relative fluorescence (Rel.Fluor) intensities were monitored at 480 nm (see inset). The fraction bound versus pXRCC1 concentration is plotted. (B) Sample plot of fluorescence data from titration with pXRCC1. F0, F and F∞ are the relative fluorescence intensities at 480 nm of FHA-AC alone, FHA-AC in the presence of a given concentration of pXRCC1, and FHA-AC saturated with pXRCC1, respectively. The plot is according to Chipman et al. (32). (C) Fluorescence titration of the AC labeled PNKP CT-domain versus npXRCC1. CT-AC (0.3 μM) was excited at 380 nm and the relative fluorescence (Rel. Fluor) intensities were monitored at 480 nm (see inset). The fraction bound versus npXRCC1 concentration is plotted. (D) Analysis of the fluorescence titration data of CT-AC with npXRCC1 as described for FHA-AC in panel B.
The binding constant we obtained for the pXRCC1/FHA domain interaction indicates considerably tighter binding between the full length XRCC1 and FHA domain than seen with short phosphorylated XRCC1-based peptides and the FHA domain (22). For example, the Kd value for binding of the triphosphorylated peptide YAGSTDENTDSE (phosphorylated amino acids underlined) to the FHA domain was 190 nM. This implies that other components of XRCC1 contribute to the stabilization of the protein–protein interaction. In our hands, we determined a 1 : 1 stoichiometry between the two proteins in contrast to Ali et al. (22), who observed a 1 : 2 stoichiometry for the phospho-peptide:FHA domain complex. It is not clear if this difference in stoichiometry is due to the different species examined, i.e. full length XRCC1 versus phospho-peptide or to the different modes of analysis, i.e. fluorescence quenching versus isothermal calorimetry.
The CT catalytic kinase/phosphatase domain (K141-G521) of PNKP was similarly subcloned, expressed and labeled with AC. The labeled protein was found to be functionally active, retaining ∼90% of its kinase activity (data not shown). When the labeled protein CT-AC was excited at 380 nm, the emission maximum was centered at 485 nm. Addition of npXRCC1 resulted in partial quenching of AC fluorescence and amounted to ∼30%, when the two proteins were mixed in a 1 : 1 molar ratio. Fluorescence titration (Figure 3C) yielded a Kd value for the binding of npXRCC1 to the CT domain of 43.0 ± 3.0 nM, in close agreement with the Kd value obtained with the AC-labeled full length PNKP (PNKP-AC) (21). The slope of the plot of log(F0 − F/F − F∞) versus log[pXRCC1] was 1.1 (Figure 3D) indicative of a 1 : 1 stoichiometric interaction between the proteins. In contrast to the addition of npXRCC1, pXRCC1 induced quenching of AC fluorescence of only ∼8%, signifying modest interaction between the two proteins. This level of quenching was not sufficient to allow us to determine a Kd value. Collectively, our data therefore indicate that there appear to be two major modes of interaction between PNKP and XRCC1: a high affinity FHA and pXRCC1 mediated interaction and another, lower affinity, interaction that is not dependent on phosphorylation and involves XRCC1 binding to the CT catalytic domain of PNKP.
To further confirm binding of npXRCC1 to the PNKP catalytic domain, we analyzed the PNKP-CT/npXRCC1 interaction by far-UV circular dichroism. Typical CD spectra of the two individual proteins are shown in Figure 4A and B. Both proteins exhibited two negative CD bands centered ∼209 and 218 nm, indicating the presence of α-helical organization. Analysis of the CD spectra according to Chen et al. (33), revealed PNKP-CT to possess ∼30% α-helix and ∼30% β-structure, with the remaining ∼40% representing random structure, while XRCC1 possesses ∼35% α-helix and ∼30% β-structure and ∼35% random structure. CD measurements can be applied to study protein–protein interactions. Since the ellipticity is an additive parameter, one can generate the theoretical CD spectrum for a mixture of non-interacting proteins by adding together the spectra of individual proteins and this spectrum can be compared to that observed experimentally to see if the interaction has induced any conformational change. The far-UV CD spectrum obtained for PNKP-CT:npXRCC1 complex (1 : 1 molar ratio) is shown in Figure 4C along with the theoretical spectrum. The experimentally observed ellipticity values deviate from the theoretical values particularly in the 209 and 218 nm wavelength regions, demonstrating that an interaction between these two proteins has produced a conformational change. For instance, the difference between the observed and the theoretical ellipticity value at 218 nm was ∼2600 deg cm2/dmol, while the experimental error in these measurements is only ±300 deg cm2/dmol. Analysis of the CD spectrum of the PNKP-CT:npXRCC1 complex yielded ∼23% α-helix and ∼32% β-structure and random structure accounted for ∼45%. Comparison of the secondary structure of the complex with the secondary structure of the individual proteins suggests that the interaction between PNKP-CT and XRCC1 resulted in an increase in the random structure at the expense of the α-helical structure.
Figure 4.

Protein analysis by circular dichroism showing interaction of the catalytic domain of PNKP with non-phosphorylated XRCC1. Far-UV CD spectra of npXRCC1 (A) and PNKP-CT (B) in 50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 5 mM MgCl2. (C) Experimentally observed (filled square) and theoretical (filled triangle) CD spectra of the PNKP-CT:XRCC1 complex. For the theoretical spectrum the proteins are assumed to be non-interacting.
Binding of pXRCC1 to DNA
We have previously obtained the Kd value for npXRCC1 binding to single-stranded DNA (45mer oligonucleotide) by measuring the quenching of the intrinsic Trp fluorescence of the protein at 340 nm following excitation at 295 nm as a function of DNA concentration (21,23). As shown in Figure 5, we used the same approach to determine if phosphorylation of XRCC1 alters its affinity for DNA. A representative plot of relative fluorescence intensity versus the concentration of DNA is shown (inset). Non-linear regression analysis of the binding data revealed unimodal binding with a Kd value of 0.80 ± 0.10 μM, which represents ∼3-fold lower affinity compared with the binding of npXRCC1 (Kd = 0.26 ± 0.02 μM) (23). The DNA binding domain of XRCC1 has been mapped to the N-terminal domain (17,34). It remains to be determined if phosphorylation of the CT domain reduces the affinity of XRCC1 for DNA due to altered protein conformation or possibly charge repulsion between the multiply phosphorylated protein and DNA backbone.
Figure 5.

Binding of pXRCC1 to DNA. Fluorescence titration of pXRCC1 with a 45mer single-stranded oligonucleotide. pXRCC1 (0.4 μM) in 50 mM Tris–HCl (pH 7.5), 100 mM NaCl and 5 mM MgCl2 was excited at 295 nm, and the intrinsic fluorescence intensity was monitored as a function of DNA concentration at 340 nm (see inset). The fraction bound (i.e. relative fluorescence quenching) versus ligand concentration is plotted.
Analysis of the dual activities of PNKP on nicked and gapped substrates
We adopted an assay recently developed by Dobson and Allinson (29) that utilizes a double-fluorophore labeled substrate to measure the kinase and phosphatase activity of PNKP simultaneously. In agreement with Dobson and Allinson, we observed that PNKP showed a faster reaction rate in processing the 3′-phosphate terminus than processing the 5′-OH terminus in a substrate with a nick (Figure 6A and B). Examination of a series of different substrates indicated a marked variation in the efficiency of processing of the termini (Figure 7A–D). With the molar ratio of PNKP to substrate set at 1 : 15 and reducing the quantity of enzyme 4-fold from the previous experiment in order to monitor the reaction over a longer period of time, we observed that PNKP removed the 3′-terminal phosphate from the 1-nucleotide (1-nt) gap substrate more efficiently than from the nicked substrate. After 5 min incubation, the phosphatase reactions reached equilibrium, with more than 95% of the 1-nt gap substrate dephosphorylated, but only 30% removal of the phosphate from the nick substrate. In contrast, the kinase reaction tended to reach equilibrium after 10 min reaction. Up to 60% of the 1-nt gap substrate was phosphorylated, while processing of the nick substrate was <5%, making it difficult to quantify. Thus, although PNKP has previously been shown to preferentially phosphorylate SSB termini rather than sterically more accessible blunt-ended DSB termini (7,35,36), steric factors are probably responsible for the higher reactivity towards a 1-nt gapped substrate than a substrate with a nick. Interestingly, however, for the substrate with an extended recess, the level of 5′-phosphorylation showed intermediate activity reaching ∼25% under the same conditions, implying that the PNKP kinase activity is enhanced by the presence of a DNA strand downstream to the site of phosphorylation.
Figure 6.

Simultaneous monitoring of kinase and phosphatase activities of PNKP using the fluorescence-based assay of Dobson and Allinson (29). A nicked DNA substrate (80 nM) bearing a 3′-phosphate (P) and 5′-hydroxyl group (OH) was incubated with 0.1 mM ATP and 20 nM PNKP. The products of the reaction, i.e. FAM-labeled 18mer (3′-phosphatase product) and TAMRA-labeled p21mer (5′-kinase product) were monitored by gel electrophoresis (A) and quantified (B) over time.
Figure 7.

Comparison of PNKP activity with different substrates. Substrates (80 nM) bearing a nick (A), recessed 5′-terminus (B) and 1-nucleotide gap (C) were incubated with 4 nM PNKP and 0.1 mM ATP. (D) Plot of percentage of product accumulated over time. (open circle) 3′-dephosphorylation product from the nick substrate; (open square) 3′-dephosphorylation product from the gap substrate; (filled square) 5′-phosphorylation product from the gap substrate; (filled triangle) 5′-phosphorylation product from the recessed substrate. The data points represent the mean of three individual determinations and the error bars show the standard deviation from the mean.
Stimulation of PNKP enzyme activity by npXRCC1 and pXRCC1
The influence of XRCC1 on phosphatase activity of PNKP was examined using the nick substrate. The rate of product accumulation decreased over time and reached a plateau at ∼10 min (Figure 8A). When 5-fold molar excess of npXRCC1 or pXRCC1 was added to the reaction after 20 min incubation, it led to the restimulation of the phosphatase activity and dephosphorylated product continued to accumulate to another plateau. pXRCC1 appeared to enhance the PNKP phosphatase to a greater extent than npXRCC1. Addition of BSA failed to stimulate PNKP, indicating that the stimulatory effect of pXRCC1 and npXRCC1 was not the result of non-specific enzyme stabilization.
Figure 8.

Stimulation of PNKP end-processing activities by pXRCC1 and npXRCC1. (A) The effect of pXRCC1 and npXRCC1 on PNKP phosphatase activity. The fluorescently labeled nick substrate (80 nM) was incubated with 4 nM PNKP and 0.1 mM ATP for 20 min and then divided into four aliquots. Three of the aliquots were supplemented with 20 nM pXRCC1, npXRCC1 or BSA and the fourth received buffer only. The reactions were followed for a further 20 min. (B) The effect of both pXRCC1 and npXRCC1 on the kinase activity of PNKP acting on the 5′OH group of a single-stranded 24-mer, using the same conditions as described above. The data were obtained from three individual determinations and the error bars represent the standard deviation from the mean.
The effect of XRCC1 phosphorylation on PNKP kinase activity was tested under limited enzyme concentration using a single-stranded 24mer oligonucleotide containing a 5′-OH group (Figure 8B). Addition of npXRCC1 increased the product yield by a further 40%, which is consistent with our previous report (21). Addition of pXRCC1, on the other hand, more than doubled the level of phosphorylated oligonucleotide. The results above indicated that both npXRCC1 and pXRCC1 can re-activate and enhance the turnover rate of PNKP. Moreover, pXRCC1 can enhance PNKP turnover rate more efficiently than npXRCC1.
Inhibition of the pXRCC1 stimulation of PNKP by purified FHA domain polypeptide
We made use of the tight binding of pXRCC1 to the FHA domain to determine whether the stimulation of PNKP activity by pXRCC1 is dependent on interaction with the FHA domain. PNKP was preincubated with npXRCC1 or pXRCC1 at 4°C for 15 min, in the presence or absence of the FHA domain, and was then added to the nick substrate using a molar ratio of PNKP to substrate of 1 : 10. As shown in Figure 9, in the presence of pXRCC1, but absence of the FHA domain, kinase activity was enhanced from 3.6 to 17.5%, and phosphatase activity was enhanced from 45 to 67% compared to PNKP alone. However, pre-incubation of PNKP with pXRCC1 in the presence of the FHA domain almost completely inhibited pXRCC1 stimulation of both the kinase and phosphatase activity of PNKP. In contrast, co-incubation of the PNKP-npXRCC1 complex with the FHA domain did not reduce the stimulatory action of npXRCC1.
Figure 9.

Competitive binding of the FHA domain to pXRCC1 abrogates the stimulation of PNKP kinase and phosphatase by pXRCC1. PNKP (8 nM) was incubated with either npXRCC1 or pXRCC1 (40 nM) in the absence or presence of FHA domain polypeptide (200 nM) at 4°C for 15 min and then allowed to react with the nick substrate (80 nM) at 37°C for 20 min and the kinase (dark shade) and phosphatase (light shade) activities were measured. The data points represent the mean of three individual determinations and the error bars show the standard deviation from the mean.
CONCLUSION
XRCC1 is known to stimulate the activity of PNKP (8). One potential explanation has been that XRCC1, in its capacity as a scaffold protein, recruits PNKP to the site of damage although there is evidence to suggest that the opposite may be the case, i.e. that PNKP recruits XRCC1 (37). As an alternative explanation, we have previously shown that XRCC1 stimulates the dissociation of PNKP from 5′-phosphorylated DNA, thereby increasing PNKP turnover. Here we have shown that XRCC1 similarly stimulates the PNKP phosphatase activity and that an elevation of the rate of enzyme turnover is at least partly responsible for the increased stimulation afforded by CK2 phosphorylated XRCC1. However, even though phosphorylated and non-phosphorylated XRCC1 both appear to stimulate PNKP by a similar mechanism, we have demonstrated that the interaction between pXRCC1 and PNKP differs substantially from that between npXRCC1 and PNKP. The former is totally dependent on XRCC1 binding to the FHA domain while the latter involves XRCC1 interaction with the catalytic domain. Although the bulk of XRCC1 in human cells appears to be phosphorylated, it is not clear if the key site for interaction with the FHA domain is fully phosphorylated (16). Furthermore, there appears to be a requirement for additional phosphorylation of XRCC1 by CK2 for rapid repair of SSBs immediately after cellular exposure to oxidative damaging agents such as H2O2 (4,16). It is therefore possible that the enhanced CK2 phosphorylation of XRCC1 is required for cells to cope in a timely fashion with a major challenge to the repair machinery (4), while under normal circumstances unstressed cells make use of non-phosphorylated XRCC1 or XRCC1 with a limited level of phosphorylation to stimulate PNKP activity by the alternative mode of interaction to handle the comparatively low level of DNA damage produced endogenously.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research (grant number 15385 to M.W.); a post-doctoral fellowship from the Alberta Heritage Foundation for Medical Research (to M.L.). Funding for open access charge: Canadian Institutes of Health Research (grant number 15385 to M.W.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Sarah Allinson (Lancaster University, UK) for her assistance in establishing the fluorescence-based assay of PNKP activity.
REFERENCES
- 1.Friedberg ECW, Siede GC, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: ASM Press; 2006. [Google Scholar]
- 2.Lennartz M, Coquerelle T, Bopp A, Hagen U. Oxygen – effect on strand breaks and specific end-groups in DNA of irradiated thymocytes. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1975;27:577–587. doi: 10.1080/09553007514550611. [DOI] [PubMed] [Google Scholar]
- 3.Henner WD, Rodriguez LO, Hecht SM, Haseltine WA. gamma Ray induced deoxyribonucleic acid strand breaks. 3′ Glycolate termini. J. Biol. Chem. 1983;258:711–713. [PubMed] [Google Scholar]
- 4.Breslin C, Caldecott KW. DNA 3′-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol. Cell. Biol. 2009;29:4653–4662. doi: 10.1128/MCB.00677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, Lasko DD. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3'-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- 6.Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJ, Sgouros J, Miller GG, Fakhrai H, Davis EM, Le Beau MM, et al. Molecular characterization of a human DNA kinase. J. Biol. Chem. 1999;274:24187–24194. doi: 10.1074/jbc.274.34.24187. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, Mani RS, Galicia S, Koch CA, Cass CE, et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell. 2005;17:657–670. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 9.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koch CA, Agyei R, Galicia S, Metalnikov P, O'D;onnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004;23:3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res. 2007;67:6619–6625. doi: 10.1158/0008-5472.CAN-07-0480. [DOI] [PubMed] [Google Scholar]
- 13.Rasouli-Nia A, Karimi-Busheri F, Weinfeld M. Stable down-regulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc. Natl Acad. Sci. USA. 2004;101:6905–6910. doi: 10.1073/pnas.0400099101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hofmann K, Bucher P. The FHA domain: a putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem. Sci. 1995;20:347–349. doi: 10.1016/s0968-0004(00)89072-6. [DOI] [PubMed] [Google Scholar]
- 15.Durocher D, Jackson SP. The FHA domain. FEBS Lett. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- 16.Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, Sarno S, Meggio F, Pinna LA, Caldecott KW. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 17.Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular 'nick-sensor' in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 19.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 20.Audebert M, Salles B, Weinfeld M, Calsou P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J. Mol. Biol. 2006;356:257–265. doi: 10.1016/j.jmb.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 21.Mani RS, Fanta M, Karimi-Busheri F, Silver E, Virgen CA, Caldecott KW, Cass CE, Weinfeld M. XRCC1 stimulates polynucleotide kinase by enhancing its damage discrimination and displacement from DNA repair intermediates. J. Biol. Chem. 2007;282:28004–28013. doi: 10.1074/jbc.M704867200. [DOI] [PubMed] [Google Scholar]
- 22.Ali AA, Jukes RM, Pearl LH, Oliver AW. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009;37:1701–1712. doi: 10.1093/nar/gkn1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mani RS, Karimi-Busheri F, Fanta M, Caldecott KW, Cass CE, Weinfeld M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry. 2004;43:16505–16514. doi: 10.1021/bi048615m. [DOI] [PubMed] [Google Scholar]
- 24.Mani RS, Karimi-Busheri F, Cass CE, Weinfeld M. Physical properties of human polynucleotide kinase: hydrodynamic and spectroscopic studies. Biochemistry. 2001;40:12967–12973. doi: 10.1021/bi011383w. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Bosc DG, Graham KC, Saulnier RB, Zhang C, Prober D, Gietz RD, Litchfield DW. Identification and characterization of CKIP-1, a novel pleckstrin homology domain-containing protein that interacts with protein kinase CK2. J. Biol. Chem. 2000;275:14295–14306. doi: 10.1074/jbc.275.19.14295. [DOI] [PubMed] [Google Scholar]
- 27.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 28.Fanta M, Zhang H, Bernstein N, Glover M, Karimi-Busheri F, Weinfeld M. Production, characterization, and epitope mapping of monoclonal antibodies against human polydeoxyribonucleotide kinase. Hybridoma. 2001;20:237–242. doi: 10.1089/027245701753179811. [DOI] [PubMed] [Google Scholar]
- 29.Dobson CJ, Allinson SL. The phosphatase activity of mammalian polynucleotide kinase takes precedence over its kinase activity in repair of single strand breaks. Nucleic Acids Res. 2006;34:2230–2237. doi: 10.1093/nar/gkl275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinna LA, Meggio F. Protein kinase CK2 (“casein kinase-2”) and its implication in cell division and proliferation. Prog. Cell Cycle Res. 1997;3:77–97. doi: 10.1007/978-1-4615-5371-7_7. [DOI] [PubMed] [Google Scholar]
- 31.Pinna LA. Protein kinase CK2: a challenge to canons. J. Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 32.Chipman DM, Grisaro V, Sharon N. The binding of oligosaccharides containing N-acetylglucosamine and N-acetylmuramic acid to lysozyme. The specificity of binding subsites. J. Biol. Chem. 1967;242:4388–4394. [PubMed] [Google Scholar]
- 33.Chen YH, Yang JT, Chau KH. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 34.Marintchev A, Mullen MA, Maciejewski MW, Pan B, Gryk MR, Mullen GP. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol. 1999;6:884–893. doi: 10.1038/12347. [DOI] [PubMed] [Google Scholar]
- 35.Karimi-Busheri F, Weinfeld M. Purification and substrate specificity of polydeoxyribonucleotide kinases isolated from calf thymus and rat liver. J. Cell. Biochem. 1997;64:258–272. [PubMed] [Google Scholar]
- 36.Bernstein NK, Hammel M, Mani RS, Weinfeld M, Pelikan M, Tainer JA, Glover JN. Mechanism of DNA substrate recognition by the mammalian DNA repair enzyme, Polynucleotide Kinase. Nucleic Acids Res. 2009;37:6161–6173. doi: 10.1093/nar/gkp597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons JL, Dianova II, Boswell E, Weinfeld M, Dianov GL. End-damage-specific proteins facilitate recruitment or stability of X-ray cross-complementing protein 1 at the sites of DNA single-strand break repair. FEBS J. 2005;272:5753–5763. doi: 10.1111/j.1742-4658.2005.04962.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.