

Abstract
Purpose
Smad3, a mediator of TGF-β signaling has been shown to be involved in the epithelial-to-mesenchymal transformation (EMT) of lens epithelial cells in a lens injury model. In this study, the role of Smad3 in anterior subcapsular cataract (ASC) formation was investigated in a transgenic TGF-β/Smad3 knockout mouse model.
Methods
TGF-β1 transgenic mice (containing a human TGF-β1 cDNA construct expressed under the αA-crystallin promoter) were bred with mice on a Smad3-null background to generate mice with the following genotypes: TGF-β1/Smad3-/- (null), TGF-β1/Smad3+/-, TGF-β1/Smad3+/+, and nontransgenic/Smad3+/+. Lenses from mice of each genotype were dissected and prepared for histologic or optical analyses.
Results
All transgenic TGF-β1 lenses demonstrated subcapsular plaque formation and EMT as indicated by the expression of α-smooth muscle actin. However, the sizes of the plaques were reduced in the TGF-β1/Smad3-/- lenses, as was the level of type IV collagen deposition when compared with TGF-β1/Smad3+/- and TGF-β1/Smad3+/+ lenses. An increased number of apoptotic figures was also observed in the plaques of the TGF-β1/Smad3-/- lenses compared with TGF-β1/Smad3+/+ littermates.
Conclusions
Lens-specific expression of TGF-β1 induced ASC formation in the absence of the Smad3 signaling mediator, suggests that alternative TGF-β-signaling pathways participate in this ocular fibrotic model.
Transforming growth factor (TGF)-β is a secreted polypeptide that is involved in various cellular processes, including cell proliferation, differentiation, apoptosis, migration, and extracellular matrix (ECM) formation.1-5 TGF-β has been shown to promote wound healing by stimulating the production and deposition of ECM, which is essential to normal tissue repair after injury.6 The profibrotic actions of TGF-β have also been implicated in multiple fibrotic diseases, including those of the eye, such as glaucoma and anterior subcapsular cataract (ASC).7-10
The ocular lens is a transparent structure that provides part of the refractive power needed to focus images on the retina of the eye. A cataract involves a reduction in transparency of the lens, which can lead to vision loss. Specifically, ASC can occur after ocular trauma, ocular surgery or in conjunction with diseases such as atopic dermatitis and retinitis pigmentosa.11,12 The development of ASC involves the transformation and proliferation of the anterior epithelial cells of the lens into plaques of large “spindle shaped” cells, or myofibroblasts, through a phenomenon known as epithelial-to-mesenchymal transition (EMT).13-15 The appearance of the myofibroblasts promotes lens capsule wrinkling and an aberrant deposition of extracellular matrix (ECM), both of which are features of ASC.1,7,16,17 Similarly, in secondary cataract (also known as posterior capsular opacification [PCO]), a complication that develops after cataract surgery, lens epithelial cells that remain within the capsule are triggered to proliferate and migrate to the posterior lens capsule where they transition into myofibroblasts through EMT.18,19
Under physiological conditions, TGF-β in the lens and ocular media mainly exists in its latent form, whereas biologically active TGF-β has been detected in the ocular media of patients with ASC.20,21 There are three functionally and structurally related species of TGF-β: TGF-β1, -β2, and -β3.22 Studies using in vitro rat lens cultures and lens epithelial explants have shown that all three TGF-β isoforms can induce cataractous changes similar to those observed in humans. However, TGF-β1 is 10 times less potent than TGF-β2 and -β3.23 The in vivo expression of self-activating TGF-β1 in a transgenic mouse model has been useful for examining the morphologic and molecular changes involved in ASC formation which closely resemble those in humans.1,7,24 However, the TGF-β-mediated signaling pathways regulating fibrosis including those governing ASC formation have not been clearly defined.
Smad proteins are intracellular molecules involved in TGF-β signal transduction from the receptors to the target genes in the nucleus.22,25,26 On ligand binding and TGF-β receptor activation, phosphorylation of receptor-regulated Smad2 and -3 occurs, which leads to the formation of hetero-oligomeric complexes with Smad4 (co-Smad).22,25,26 The Smad complexes then translocate into the nucleus, where they regulate target gene expression in collaboration with other coactivators and corepressors. The Smad3-knockout mouse model has been useful in determining TGF-β mediated fibrotic events requiring Smad3 signaling.27-30 An important finding is that ablation of Smad3 in mice prevents the EMT of lens epithelial cells that occurs on injury to the lens capsule,31 suggesting that EMT in the lens may be entirely Smad3 dependent. However, the effect of TGF-β on ASC formation in the absence of Smad3 has not been directly tested.
In the present study, we directly determined the requirement for Smad3 in ASC formation using an in vivo transgenic TGF-β1/Smad3 knockout mouse model. Evidence of ASC formation in TGF-β1/Smad3-/- mice and their TGF-β1/Smad wild-type littermates was evaluated histologically and immunohistochemically using the EMT marker α-smooth muscle actin (SMA), as well as fibrotic markers, fibronectin and collagen types I and IV. Formation of ASC in mice was also evaluated by using a quantitative measure of lens optical quality. The results showed that EMT and the formation of ASC plaques occurred in the TGF-β1/Smad3-/- mice, accompanied by a significant decrease in optical quality of the lens. However, the plaques in the TGF-β1/Smad3-/- mice were smaller, contained a greater number of apoptotic cells and substantially less collagen than did their Smad3 heterozygote and wild-type littermates. Together these findings demonstrate that while Smad3 signaling contributes to some aspects of the TGF-β1-induced ASC phenotype, it is not necessary for ASC formation, and additional Smad3-independent pathways are involved in this ocular fibrotic disease.
Materials and Methods
Generation of Transgenic TGF-β1/Smad3 Knockout Mice
All animal studies were performed according to the Canadian Council on Animal Care Guidelines and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The transgenic TGF-β1 mice contain a human TGF-β1 cDNA construct with an αA-crystallin promoter designed for lens-specific expression of active TGF-β11 on a FVB/N/C57BL/6J background. The Smad3-null mutant was generated by the deletion of exon 8 of the Smad3 gene, which truncated 89 amino acids from the C-terminal end.32 Exon 8 contains an SSVS consensus phosphorylation site and an L3 loop, which are essential for interaction with the TGF-β receptor.2 TGF-β1 transgenic mice were bred with Smad3-null and heterozygous mice on a SvEv/C57BL background to generate mice with the following genotypes: TGF-β1/Smad3-/- (null), TGF-β1/Smad3+/- (heterozygous), TGF-β1/Smad3+/+, Smad3-/-, and nontransgenic/Smad+/+ (wild-type). Wild-type littermates were used to ensure that results were not due to the various differences between the strains of mice.
Genotype Analysis
DNA extraction and purification from mouse ear tissue was performed with a kit (DNeasy; Qiagen Inc., Toronto, ON, Canada). Genotypes were determined by polymerase chain reaction (PCR) analysis. The TGF-β1 transgene was identified by using primers specific for the simian virus 40 (SV40) sequences in the transgene: the sense primer (5′-GTGAAGGAACCTTACTTCTGTGGTG-3′) and the antisense primer (5′-GTCCTTGGGGTCTTCTACCTTTCTC-3′) yield a 300-bp fragment in the PCR reactions.33 PCR reactions were performed for 36 cycles using the following conditions: initial heating for 3 minutes at 94°C (only for cycle 1); denaturation for 30 seconds at 94°C; annealing for 1 minute at 57°C; and extension for 1 minute at 72°C. A final extension was performed for 2 minutes at 72°C. Agarose gel electrophoresis (1.5% agarose) with ethidium bromide (EtBr) detection was used to visualize the PCR reaction products.
The Smad3 wild-type and knockout alleles were detected using three primers. The Smad3 wild-type allele were detected by using primers 1 (5′-CCACTTCATTGCCATATGCCCTG-3′) and 2(5′-CCCGAACAGTTGGATTCACACA-3′). Primer 1 (located 5′ to the deletion) and primer 2 (located within the deletion) amplify a 400-bp fragment from wild-type and heterozygous knockout mice.32 The Smad3-knockout allele was detected by using primers 1 and 3 (5′CCAGACTGCCTTGGGAAAAGC-3′; located in the pLoxpneo) to yield a 250-bp fragment, which is detected in both the heterozygous and homozygous Smad3 knockout mice.32 PCR reactions were performed for 31 cycles in the following conditions: initial heating for 2 minutes at 94°C (only for cycle 1); denaturation for 30 seconds at 94°C; annealing for 30 seconds at 60°C; and extension for 2 minutes at 72°C. A final extension was performed for 2 minutes at 72°C. A 2% agarose gel electrophoresis with EtBr detection was used to visualize the PCR reaction products.
Histology and Immunohistochemistry Staining
Whole eyes were dissected from 2- to 3-month-old mice of each genotype and fixed in 10% neutral formalin buffer for 24 hours, then dehydrated, processed, and embedded in paraffin. Paraffin sections of 5-μm thickness were used for hematoxylin and eosin (H&E), Mason’s Trichrome (collagen deposition), or immunohistochemistry staining (α-SMA, fibronectin, and β-crystallin). Fresh eyes from each genotype were also embedded in OCT and frozen sections (5-μm thickness) were used to stain for collagen type I and IV. Staining was visualized with a microscope (Leica, Deerfield, IL) equipped with an immunofluorescence attachment, and images were captured using a high-resolution camera and associated software (OpenLab; Quorum Technologies). Images were reproduced for publication (Photoshop 7.0; Adobe Systems Inc., Mountain View, CA).
For α-SMA, fibronectin, and β-crystallin immunofluorescence localization, paraffin sections were deparaffinized, rehydrated, and incubated with 5% normal goat serum for 20 minutes at room temperature. The sections were then incubated, with mouse anti-α-SMA monoclonal antibody (1:100; Sigma-Aldrich, Inc., St. Louis, MO), rabbit anti-mouse fibronectin (1:500; Cedarlane Laboratories Ltd., Hornby, ON, Canada) or rabbit anti-β-crystallin (1:200; donation from J. Samuel Zigler, National Eye Institute, Bethesda, MD) at room temperature for 1 hour. The bound primary antibodies were visualized with either a fluorescein-isothiocyanate (FITC)-conjugated goat anti-mouse secondary antibody, goat anti-rabbit FITC secondary antibody or tetramethyl rhodamine isothiocyanate (TRITC) conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). All sections were mounted in medium containing 4′, 6′-diamino-2-phenylindole (DAPI; Vector Laboratories Inc., Burlingame, CA), to visualize the nuclei.
Frozen tissue sections were thawed at room temperature, fixed with acetone for 20 minutes at -20°C and used for immunofluorescence localization of collagen type I or IV. The sections were incubated with 5% normal goat serum for 20 minutes at room temperature followed by rabbit anti-collagen type I or rabbit anti-collagen type IV primary antibody (1:100; Cortex Biochem, San Leandro, CA) at room temperature for 1 hour. A goat anti-rabbit FITC secondary antibody was used to visualize the bound primary antibodies. All sections were mounted in medium with DAPI and photographed as for histology.
TUNEL Assay
TUNEL (terminal deoxynucleotidyl transferase mediated dUTP nick end labeling) was used to examine cell death in the lens epithelium of each genotype. Paraffin sections were deparaffinized and a fluorescein in situ apoptosis detection kit (ApopTag plus; Chemicon International Inc., Temecula, CA) was used to detect apoptotic nuclei. The TUNEL procedure was performed in accordance with the manufacturer’s instructions. A positive control was prepared by treating a sample with DNaseI before TUNEL staining. All sections were mounted in medium with DAPI and photographed as for the staining procedures. For quantitative analysis, the percentage of TUNEL-positive cells among 150 lens epithelial cells in three fields per slide was determined at 400-fold magnification in three different samples from each genotype.
Western Blot Analysis
Four lenses from each genotype including TGF-β1/Smad3-/-, TGF-β1/Smad3+/-, TGF-β1/Smad3+/+, Smad3-/-, and wild-type mice were collected and pooled for Western blot analysis of phosphorylated Smad3 (pSmad3) protein expression. In addition two lenses from each genotype were collected and pooled for Western blot analysis of α-SMA protein expression. The lenses were homogenized in Triton X-100 lysis buffer containing protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). The total protein concentration was determined by the Bradford protein assay.34 Equal amounts of total protein from each group of lenses were electrophoresed on 10% Tris-tricine polyacrylamide gel (pSmad3) or 10% SDS polyacrylamide gel (α-SMA). The proteins were electrotransferred onto a nitrocellulose membrane (Pall Corporation, East Hills NY). Membranes were blocked with 5% skimmed milk powder in Tris-buffered saline (50 mM Tris base, NaCl [pH 8.5]) +0.1% Tween-20 and then incubated overnight at 4°C with either a rabbit anti-phospho-Smad3/Smad1 antibody (1:1000; Cell Signaling Inc., Beverly, MA) or mouse anti-α-SMA monoclonal antibody (1:1000). After this incubation, membranes were probed with the appropriate horseradish (HRP)-conjugated anti-rabbit or anti-mouse secondary antibodies (1:5000; GE Healthare, Piscataway, NJ) and ECL detection reagents (GE Healthcare). The Western blot analyses were visualized by x-ray film exposure. The α-SMA membranes were striped and reprobed with mouse anti-β-actin antibody (1:1000; Cedarlane Laboratories Ltd.) as a loading control. The secondary detection for β-actin followed the same procedures as mentioned earlier.
Optical Analysis
Lenses were dissected from 2- to 3-month-old mice after euthanatization with CO2. The lenses were cultured in specialized serum-free medium M199 with Earl’s salts and l-glutamine (product no. 11150-059, Invitrogen Inc., Burlington, ON, Canada) containing penicillin-streptomycin. Lenses were incubated at 37°C with 4.0% CO2 for 24 hours before optical analysis, to ensure that no damage occurred during the dissection. A total of 33 lenses were used for optical measurements: TGF-β1/Smad3-/- (n = 8), TGF-β1/Smad3+/- (n = 8), TGF-β1/Smad3+/+ (n = 11), and wild-type (n = 6) lenses. A laser scanning system (ScanTox) developed at the University of Waterloo (ON, Canada) was used to analyze the quality of the lenses.35-37 The laser scanner consists of a collimated helium neon (HeNe) laser source (on an X-Y table) that projects a beam onto a mirror mounted at 45 ° on a carriage assembly. The reflected beam then goes up through the scanner table surface and through the lens under examination. There are two digital cameras that capture images of the light beam through the lens. The image information collected by the digital cameras is transferred to a computer, and the refracted direction for each of the beam positions is recorded with respect to the optical center, the point at which the slope of the laser beam approaches zero.35-37 Each scan involved 20 laser positions across a 2 mm diameter with a step size of 0.10 mm. The measurements made included average back vertex distance (BVD, a measure of focal length), and back vertex distance variability (BVD error or sharpness of focus) and relative transparency (or scatter). The BVD error is the most sensitive measurement in detecting optical changes in subcapsular cataract development in the mouse lens. Thus, the results will focus on the changes in BVD error as an indication of decreased optical quality in the lens.
Statistical Analysis
The analysis of variance (ANOVA; SPSS, ver. 11.0 statistical software; SPSS, Chicago, IL) was used to assess the optical effects (back vertex variability) of cataract formation between each group of mouse lenses. The unpaired Student’s t-test was used to analyze the apoptotic cell counts for the TUNEL assay. P ≤ 0.05 was considered to be significant with a 95% confidence interval.
Results
Generation of Transgenic TGF-β1/Smad3-Knockout Mice
Figure 1 represents the PCR results for 2- to 3-month-old offspring generated from crosses between the Smad3+/- and TGF-β1 transgenic mice. The genotype of the mice was determined based on detection of the TGF-β1 transgene (300 bp; top gel) and the Smad3 wild-type (400 bp) and Smad3-knockout (250 bp) alleles (bottom gel). TGF-β1/Smad3+/- mice showed all three bands (lane 3) whereas TGF-β1/Smad3-/- and TGF-β1/Smad3+/+ mice exhibited two bands, the transgene and either the wild-type allele or the knockout allele (lanes 2 and 4, respectively). Nontransgenic Smad3+/+ and Smad3-/- mice exhibited a single band at 400 and 250 bp, respectively (lanes 1 and 5).
Figure 1.

Genotyping for TGF-β1/Smad3 knockout mice. Top gel: PCR results for the TGF-β1 (300 bp) transgene; bottom gel: PCR results for the Smad3 wild-type (Wt, 400 bp) and Smad3 knockout (KO, 250 bp) alleles. The mice had the following genotypes: wild type (lane 1), TGF-β1/Smad3-/- (lane 2), TGF-β1/Smad3+/- (lane 3), TGF-β1/Smad3+/+ (lane 4) and Smad3-/- (lane 5).
Western blot analysis for phosphorylated Smad3 provides verification that Smad3 signaling is absent in the lenses of Smad3-knockout mice (Fig. 2). The antibody used for pSmad3 (Ser433/435) detection cross reacts with pSmad1 (Ser463/465; 65 kDa) which was detected in all lenses examined and thus acted as a positive internal control. Both the TGF-β1/Smad3+/+ (lane 1) and TGF-β1/Smad3+/- (lane 5) lenses exhibited pSmad3 (58 kDa) in contrast to the nontransgenic wild-type and Smad3-/- (lanes 2-4) lenses, which did not. p-Smad1 expression was also increased in the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses. Smad1 is a mediator activated mainly by BMP (bone morphogenetic protein) receptors.38,39 Extracellular signal-regulated kinases (ERK) has shown to phosphorylate Smad1 at the linker region, preventing the transcriptional activation by BMP in R-1B/L17 cells.39 Thus, the increase in pSmad1 observed in our Western analysis maybe due to TGF-β induced mitogen-activated protein kinase (MAPK) signaling. Even in the presence of the TGF-β1 transgene, Smad3-/- mice did not show any detectable pSmad3. Therefore, the deletion of exon 8 of the Smad3 gene completely inhibited TGF-β1-mediated Smad3 phosphorylation in the lens.
FIGURE 2.
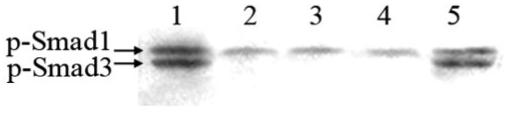
Phosphorylated Smad3. Western blot analysis using an anti-pSmad3/Smad1 antibody for the detection of pSmad3 (58 kDa) protein expression in lens extracts. The antibody cross-reacts with phosphorylated Smad1 (65 kDa) protein, which served as an internal positive control. The lanes show the expression of phospho-Smad3 protein in TGF-β1/Smad3+/+ (lane 1), wild-type (lane 2), TGFβ1/Smad3-/- (lane 3), Smad3-/- (lane 4), and TGF-β1/Smad3+/- (lane 5) mouse lenses. Both TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses showed the presence of pSmad3, whereas wild-type and Smad3-null (TGF-β1/Smad3-/- and Smad3-/-) lenses did not.
Histologic and Immunohistochemical Analyses of the TGF-β1/Smad3 Lenses
Histologic examination of eyes from 3-month-old TGF-β1 transgenic mice on all three Smad3 backgrounds (TGF-β1/Smad3+/+, TGF-β1/Smad3-/-, and TGF-β1/Smad3+/- [not shown]) revealed distinct evidence of ASC formation including extensive multilayering of cells forming plaques beneath the anterior lens capsule (Fig. 3). In contrast to the cataracts of TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- mice, which typically spanned nearly the full width of the anterior region of the lens, the subcapsular plaques in the TGF-β1/Smad3-/- lenses were substantially smaller. In addition, cells in the Smad3-deficient plaques exhibited a more rounded appearance than their wild-type and heterozygous littermates (compare Figs. 3J and 3K). In addition to lens plaque formation, TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- (not shown) mice exhibited overt defects in the cornea including increased thickness and cellularity of the corneal stroma that often resulted in the corneal endothelium’s adhering to the lens capsule (Figs. 3B, 3F). The corneal phenotype in the TGF-β1/Smad3-/- mice was much less severe, and the cornea did not come into contact with the lens. Finally, vacuole formation and nucleation in the posterior cortex of the mature TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses was observed, whereas these features were substantially reduced or absent in the TGF-β1/Smad3-/- lenses (Fig. 4).
FIGURE 3.
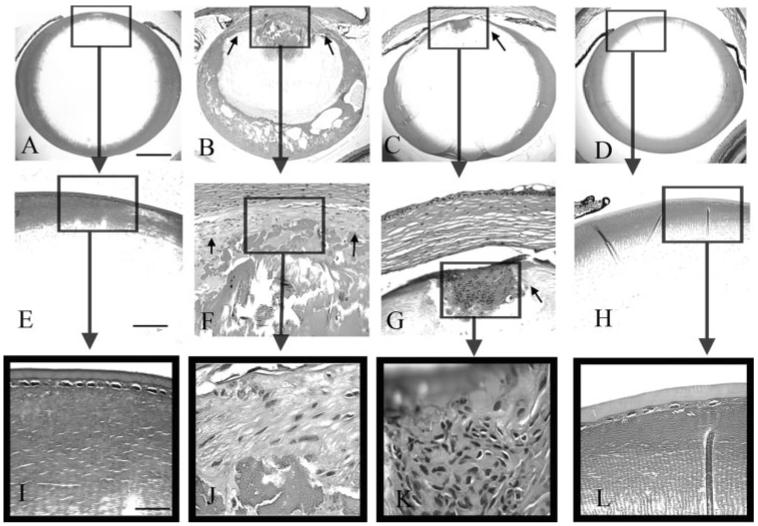
Histologic analysis of the TGF-β1/Smad3 lenses. Wild-type (A, E, I), TGF-β1/Smad3+/+ (B, F, J), TGF-β1/Smad3-/- (C, G, K), and a Smad3-/- (D, H, L) lenses are shown. Boxes: areas in higher-magnification images. The expression of the TGF-β1 transgene induced subcapsular plaque formation in the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses (F, G, small arrows). The subcapsular plaques in TGF-β1/Smad3-/- lenses were smaller than those in the TGF-β1/Smad3+/+ lenses. Both the wild-type and Smad3-/- lenses showed normal morphology of the lens epithelium. The scale bars: (A-D) 400 μm; (E-H) 100 μm; (I-L) 50 μm.
FIGURE 4.
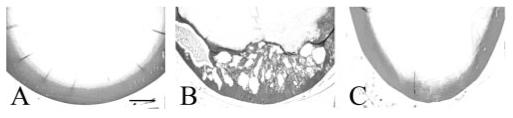
Vacuole formation in the TGF-β1/Smad3 lenses. The posterior lens cortex of wild-type (A), TGF-β1/Smad3+/+ (B), and TGF-β1/Smad3-/- (C) lenses are shown. The expression of TGF-β1 induced nucleation and vacuole formation in the TGF-β1/Smad3+/+ lenses. Both the wild-type and TGF-β/Smad3-/- lenses showed normal morphology of the posterior lens cortex. Scale bar, 200 μm.
The expression of α-SMA was examined because it is a commonly used marker for the EMT that occurs in TGF-β-induced ASC formation (Fig. 5). Expression of α-SMA was observed in the subcapsular plaques of the TGF-β1/Smad3+/+, and TGF-β1/Smad3+/- (not shown) lenses whereas no α-SMA expression was detected in the nontransgenic wild-type and Smad3-/- lenses (Fig. 5). α-SMA expression was also detected in the plaques of lenses from the TGF-β1/Smad3-/- mice, although immunoreactivity appeared reduced compared with the mice with Smad3 wild-type or heterozygote backgrounds. Staining in the iris of the nontransgenic wild-type and Smad3-/- served as a positive control for α-SMA expression. Results from Western blot analysis of α-SMA (42 kDa) expression in lens extracts (Fig. 6) confirmed the immunostaining results. Although all TGF-β1 transgenic mice exhibit distinct expression of α-SMA, lenses from TGFβ1/Smad3-/- mice show reduced levels compared with the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- mice. β-Actin (42 kDa) protein was used as the loading control and showed equal levels of expression in all samples examined.
FIGURE 5.

Immunohistochemical analysis of α-SMA expression (FITC). Wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E), and TGF-β1/Smad3-/- (D, F) lenses are shown. Red arrows: subcapsular plaques. Expression of α- SMA was detected in the subcapsular plaques of the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses. However, there was less α-SMA immunoreactivity detected in TGF-β1/Smad3-/- lenses. Both the wild-type and Smad3-/- lenses showed no expression of α-SMA in the lens and normal expression in the iris (positive control). Scale bars: (A-D) 100 μm; (E, F) 50 μm.
FIGURE 6.
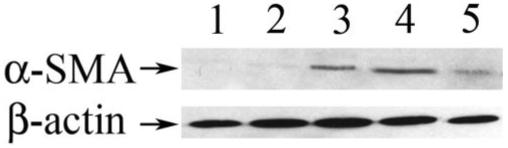
α-Smooth muscle actin protein expression. Western blot analysis using an anti-α-SMA antibody for the detection of α-SMA (42 kDa) protein expression in lens extracts. The membrane was stripped and reprobed for β-actin, which served as a loading control. Lanes 1 to 5: α-SMA and β-actin protein expression in wild-type, Smad3-/-, TGF-β1/Smad3+/-, TGF-β1/Smad3+/+, and TGFβ1/Smad3-/- lenses, respectively. The β-actin signal shows that equal amounts of protein were loaded in all lanes. TGF-β1/Smad3+/+, TGF-β1/Smad3+/-, and TGFβ1/Smad3-/- lenses showed expression of α-SMA, whereas wild-type and Smad3-/- lenses did not. The TGFβ1/Smad3-/- lenses showed reduced levels of α-SMA compared with both the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses.
Fibronectin is a fibrotic marker known to be expressed in ASC plaques.40-43 We therefore examined the expression of fibronectin in the TGF-β1/Smad3 lenses. Unlike non-TGF-β1 transgenic mice in which fibronectin expression was found to be confined to the lens capsule, the cells within the subcapsular plaques of the TGF-β1/Smad3+/+, TGF-β1/Smad3-/-, and TGF-β1/Smad3+/- (not shown) lenses exhibited immunoreactivity to fibronectin (Fig. 7). However, as was shown earlier for α-SMA, the intensity of the immunoreactivity to fibronectin in the cells of the subcapsular plaques of TGF-β1/Smad3-/- lenses was reduced when compared with the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses.
FIGURE 7.

Immunohistochemical analysis of fibronectin expression (TRITC). Wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E) and TGF-β1/Smad3-/- (D, F) lenses are shown. Yellow arrows: location of subcapsular plaques. There was detectable expression of fibronectin in the subcapsular plaques of the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses. The intensity of fibronectin immunoreactivity in the subcapsular plaques of TGF-β1/Smad3-/- lenses was reduced when compared to the TGF-β1/Smad3+/+ lenses. Both the wild-type and Smad3-/- lenses showed normal expression of fibronectin in the lens capsule. Scale bars: (A-D) 100 μm; (E, F) 50 μm.
Mason’s Trichrome stain was next used to observe total collagen deposition (collagen stains blue or aqua) in the mouse lenses (Fig. 8). In the normal lens, collagen expression is confined to the lens capsule, whereas aberrant collagen deposition has been observed in the ASC plaques.43 The expression of the TGF-β1 transgene resulted in substantial collagen deposition in the plaques of TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- (not shown) mice, as previously reported.1,7 In contrast, much less collagen deposition was observed in the subcapsular plaques of the TGF-β1/Smad3-/- mice. Further analysis of collagen type I and IV expression was performed using specific antibodies (Figs. 9, 10), since these isoforms are known to be expressed in ASC.43 Expression of collagen type I was detected in the subcapsular plaques of all the transgenics including TGF-β1/Smad3+/+, TGF-β1/Smad3-/-, and TGF-β1/Smad3+/- mice (data not shown; Fig. 9), whereas non-TGF-β1 transgenic lenses showed no expression of collagen type I. The intensity of immunoreactivity for type I collagen was similar for all the TGF-β1/Smad3-/-, TGF-β1/Smad3+/+, and TGF-β1/Smad3+/- (not shown) lenses. Normal expression of type IV collagen was observed in the lens capsule of all mice examined44 (Fig. 10). A substantial level of collagen type IV immunoreactivity in the subcapsular plaques of TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- (not shown) lenses was observed. In contrast, the plaques of TGF-β1/Smad3-/- mice exhibited little to no type IV collagen staining.
FIGURE 8.

Mason’s trichrome staining for collagen. Wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E), and TGF-β1/Smad3-/- (D, F) lenses are shown. Arrows: subcapsular plaques; (*) collagen expression in the lens capsules. The expression of TGF-β1 increased collagen deposition in the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- plaques. However, the collagen deposition in the subcapsular plaques of TGF-β1/Smad3-/- lenses was substantially reduced compared with the TGF-β1/Smad3+/+ lenses. Both the wild-type and Smad3-/- lenses showed normal collagen expression in the lens capsule. Scale bars: (A-D) 100 μm; (E, F) 50 μm.
FIGURE 9.

Immunohistochemical analysis of collagen type I expression (FITC). Wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E), and TGF-β1/Smad3-/- (D, F) lenses are shown. Arrows: subcapsular plaques. Expression of collagen type I was detected in the subcapsular plaques of the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses. There was no observable difference in collagen I expression between the TGF-β1/Smad3-/- and TGF-β1/Smad3+/+ lenses. Both the wild-type and Smad3-/- lenses show no expression of collagen type I in the lens. Scale bars: (A-D) 100 μm; (E, F) 50 μm.
FIGURE 10.
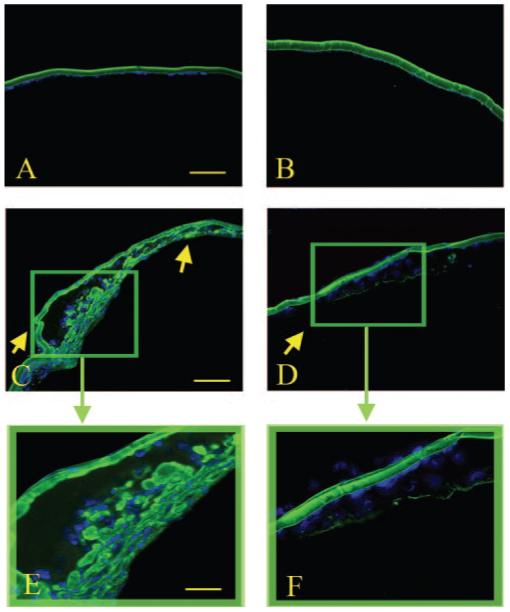
Immunohistochemical analysis of collagen type IV expression (FITC). Wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E), and TGF-β1/Smad3-/- (D, F) lenses are shown. Arrows: subcapsular plaques. Expression of collagen type IV was detected in the subcapsular plaques of the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses. There was considerable reduction in collagen IV immunoreactivity in the TGF-β1/Smad3-/- lenses when compared with the TGF-β1/Smad3+/+ lenses. Both the wild-type and Smad3-/- lenses showed collagen type IV in the lens capsule. Scale bars: (A-D) 100 μm; (E, F) 50 μm.
Previous studies have shown that TGFβ-induced subcapsular plaques consist of a heterogeneous population of cells that are immunoreactive to either αSMA or to β-crystallin, a lens fiber specific marker.45 Expression of β-crystallin was therefore examined to further characterize the cellular makeup of the plaques in the TGF-β1 transgenic lenses (Fig. 11). Expression of β-crystallin was detected in the posterior part of subcapsular plaques (away from the lens capsule) in the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- (not shown) lenses. These results correspond to the β-crystallin immunoreactivity that has been detected in larger human ASC plaques.45 The TGF-β1/Smad3-/- lenses also exhibited immunoreactivity to β-crystallin, demonstrating that although these plaques are reduced in size they still contain a heterogeneous population of cells similar to their wild-type and heterozygote littermates.
FIGURE 11.

Immunohistochemical analysis of β-crystallin expression (FITC). Shown are wild-type (A), Smad3-/- (B), TGF-β1/Smad3+/+ (C, E), and TGF-β1/Smad3-/- (D, F) mouse lenses. Arrows: subcapsular plaques. Expression of β-crystallin was detected in the posterior aspect (abutting the lens fiber cell mass) of the subcapsular plaques in both the TGF-β1/Smad3+/+ and TGF-β1/Smad3-/- lenses. Both the wild-type and Smad3-/- lenses showed no expression of β-crystallin in the lens epithelium. Scale bars: (A-D) 100 μm; (E, F) 50 μm.
Apoptotic Cell Death Analyses of TGF-β1/Smad3 Lenses
Because the ASC plaques observed in the TGF-β1/Smad3-/- mice are smaller than their wild-type littermates and apoptosis has been reported to occur in TGF-β-induced ASC,8,46 we used TUNEL labeling to examine the level of apoptosis in the different experimental groups. Both the nontransgenic wild-type and Smad3-null lenses did not exhibit TUNEL-positive nuclei (not shown). TUNEL-positive nuclei were found in the subcapsular plaques of all the TGF-β transgenic mice, including the TGF-β1/Smad3+/+, TGF-β1/Smad3+/- (data not shown), and TGF-β1/Smad3-/- mice (Fig. 12). TGF-β1/Smad3-/- lenses demonstrated a significantly higher percentage of TUNEL-positive cells (20.3% ± 5.8%) than did the TGF-β1/Smad3+/+ (6.1% ± 1.1%) lenses (unpaired Student’s t-test: P ≤ 0.05). The TUNEL-positive cells of TGF-β1/Smad3-/- lenses were mainly situated at the posterior aspect of the plaque, abutting the lens fiber cell mass.
FIGURE 12.
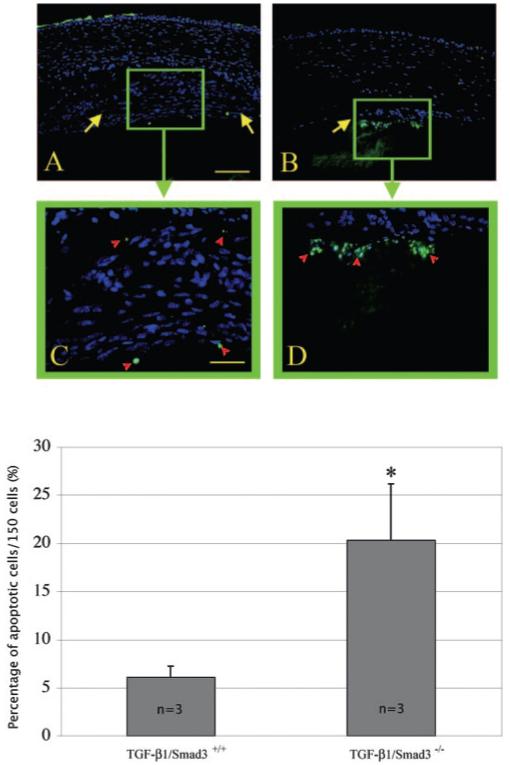
TUNEL staining of apoptotic nuclei (FITC) in TGF-β1/Smad3 lenses. Shown are TGF-β1/Smad3+/+ (A, C) and TGF-β1/Smad3-/- (B, D) mouse lenses. Arrows: subcapsular plaques. The TGF-β1/Smad3+/+ lenses showed a few TUNEL-positive nuclei located in the anterior and posterior regions of the plaques (A, C). The TGF-β1/Smad3-/- lenses exhibited more TUNEL-positive nuclei. The bar graph represents the percentage of apoptotic nuclei per 150 cells. *The TGF-β1/Smad3-/- lenses had significantly more apoptotic nuclei (20.3% ± 5.8%) than did the TGF-β1/Smad3+/+ (6.1% ± 1.1%) lenses (unpaired Student’s t-test: P ≤ 0.05). Scale bars: 100 μm(A, B); 50 μm (C, D).
Effect of the TGF-β1 and Smad3 on the Optical Quality of the Mouse Lens
A laser scanning system was next used to determine quantitative differences in the optical quality of the lenses of TGF-β1 transgenic mice on the different Smad3 backgrounds. As described in detail in the Methods section, the automated laser scanning system consists of a scanning helium-neon laser source that is projected through the lens to measure lens optical quality (the average back vertex distance, BVD) and sharpness of focus (BVD error).35-37 The bar graph in Figure 13 represents the back vertex distance variability (BVD error, mean millimeters ± SEM) for wild-type, TGF-β1/Smad3+/+, TGF-β1/Smad3+/-, and TGF-β1/Smad3-/- mouse lenses. An increase in BVD error signifies a decrease in sharpness of light focus through the lens. The TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses showed the most significant (ANOVA: P ≤ 0.05) BVD errors (0.531 ± 0.071 and 0.486 ± 0.040 mm, respectively) when compared with wild-type lenses (0.067 ± 0.002 mm), indicating that lens optical quality was reduced in the presence of subcapsular plaques in the TGF-β1 transgenic mice. The TGF-β1/Smad3-/- (0.099 ± 0.005 mm) lenses also showed a significantly greater BVD error than the wild-type lenses; however, the error was significantly lower than that in the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses. Thus, the optical results corresponded with the morphologic findings.
FIGURE 13.

Optical effects of subcapsular cataract formation in mouse lenses. Data are the BVD error variability (mm ± SEM) in wild-type, TGF-β1/Smad3+/+, TGF-β1/Smad3+/-, and TGF-β1/Smad3-/- mouse lenses. An increased in BVD error signifies a decrease in sharpness of light focus through the lens. +Statistical analysis (ANOVA: P ≤ 0.05) shows that the TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses had the greatest BVD errors (0.531 ± 0.071 and 0.486 ± 0.040 mm, respectively) when compared to the wild-type (0.067 ± 0.002 mm) lenses. *TGF-β1/Smad3-/- (0.099 ± 0.005 mm) lenses also show a significantly greater BVD error when compared to the wild-type lenses. However, the BVD error of TGF-β1/Smad3-/- lenses is significantly lower than both TGF-β1/Smad3+/+ and TGF-β1/Smad3+/- lenses.
DISCUSSION
TGF-β plays crucial roles during development, homeostasis, and pathologic conditions, including ocular fibrotic diseases, such as ASC formation and glaucoma.1,22,24,47 Typically, TGF-β has inhibitory effects on cells of epithelial origin, whereas it is mitogenic for smooth muscle cells and fibroblasts.24,48 In addition, TGF-β can cause an EMT of various types of epithelial cells in response to injury. This occurs in the lens where active TGF-β has been shown to promote the conversion of lens epithelial cells into myofibroblasts, which form ASC plaques.1,16,20 Smad3 is a major mediator of TGF-β-induced fibrosis in the kidney and lung.27,30,49 However, the requirement of Smad3 in the EMT of epithelial cells remains controversial.50 In the present study, we have shown that transgenic, expression of TGF-β1 in the lens promotes the formation of ASCs in the absence of Smad3, demonstrating that Smad3-independent signaling pathways are activated.
Smad2 is another receptor-mediated Smad that complexes with the co-Smad, Smad4. Thus, one could surmise that Smad2 activation may have compensated for the loss of Smad3 and contributed to the TGF-β-induced EMT in the TGF-β1/Smad3-/- lens epithelium. However, although both Smad2 and -3 can mediate TGF-β and activin signaling, they clearly have nonredundant functions. For example, homozygous mice with targeted disruptions in the Smad2 gene exhibit embryonic lethality during gastrulation,51,52 whereas homozygous Smad3-null mice are viable and survive to adulthood.32,53 Smad3 and -2 have also been shown to have different gene-regulatory functions and Smad3, but not Smad2, has been linked to fibrotic events.25,53,54 Finally, pSmad2 has been shown to be constitutively expressed in the lens epithelium and to be unaffected by TGF-β signaling.55
Although the predominant signaling pathways activated by TGF-β involve the Smads, additional Smad-independent TGF-β signaling pathways exist that could regulate the EMT of lens epithelial cells. These include the MAPKs, such as c-Jun-N-terminal protein kinases (JNK), extracellular signal-regulated kinases (ERK), and p38.50,56-58 In particular, the p38 pathway has been shown to play a key role in TGF-β stimulated EMT and cell migration in many systems, including mouse mammary and human breast epithelial cells.56,59 p38 activation occurs through the TGF-β-activated kinase-1 (TAK1) upstream mediator.57,60 Of note, p38 has also been shown to converge with the Smad pathway to produce full effects of EMT in some cell lines,61 which may explain why we observe a less severe corneal and lens phenotype, including smaller sized plaques, on the Smad3-null background. Phosphadtidyl-inositol 3-kinase (PI3K) and RhoA are also alternative parallel pathways shown to be involved in TGF-β-mediated EMT of epithelial cells62-64 and could therefore be candidate signaling molecules for the EMT of lens epithelial cells in ASC formation (Fig. 14). More recently, it has been shown that other growth factors may be able to mediate EMT independent of TGF-β signaling pathways (Garcia CM, et al. IOVS 2005;46:ARVO E-Abstract 4641).
FIGURE 14.
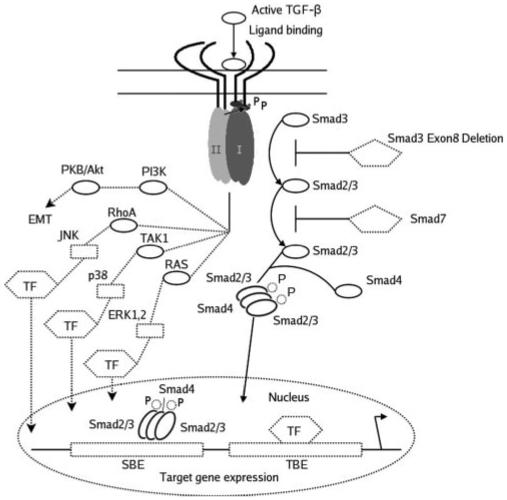
TGF-β induced Smad-dependent and -independent signaling pathways. On ligand binding, type I and type II receptors are activated, and phosphorylation of R-Smads (Smad2/3) occurs. Phosphorylated R-Smads form heterotrimeric complexes with co-Smad (Smad 4) and translocate to the nucleus. The Smad complexes interact with other transcription factors (TF) at DNA sequence-specific binding sites (transcription factor binding element [TBE] Smad binding element [SBE]) to regulate gene expression. Smad7 is an inhibitory Smad that prevents receptor activation of R-Smads. Smad3 phosphorylation is prevented in Smad3-null mice, and so TGF-β-induced responses are presumed to occur through activation of other TGF-β-induced signaling pathways. TGF-β also induces activation of the MAPK pathways (JNK, p38 and ERK1, 2) through the upstream mediators RhoA, Ras, and TAK1. Additional pathways involving PI3K have been shown to mediate EMT. Modified with permission from Roberts AB, Derynck R. Meeting Report: Signaling Schemes for TGF-β. Sci. STKE 2001;pe43. http://stke.sciencemag.org/cgi/content/full/OC sigtrans;2001/113/pe43. © 2001 AAAS.
The EMT of lens epithelial cells into myofibroblasts has also been shown to occur in a lens injury model in which the lens capsule is disrupted with a hypodermic needle. Furthermore, lens injury-induced EMT in the lens was shown to be completely inhibited on the Smad3-null background.31 This is in contrast to our current findings, which show evidence of EMT and ASC formation in the Smad3-null lens in the presence of the TGF-β1 transgene. Reasons for the difference in these findings may relate to the difference in the mode of delivery of TGF-β between the two models that could affect the levels and exposure time of TGF-β. For example, the TGF-β1 transgenic model used in the present study involves the expression of active TGF-β1 in lens fiber cells, under the control of the αA-crystallin promoter, which is initiated during early embryogenesis and continues throughout the life of the animal. In contrast, in the lens injury model, postnatal mice are used, and latent TGF-β in lens cells or the aqueous humor is thought to be activated after a puncture wound to the capsule.31 Crystallin promoters are typically strong promoters and, when used to drive active TGF-β1 expression in the lens, produce dramatic lens phenotypes and corneal defects, including a thickened, hypercellular corneal stroma and lack of a stratified corneal epithelium.1,24 In comparison, in the lens injury model, corneal phenotypes have not been reported, perhaps because the cornea is fully differentiated in the postnatal mice used for the injury studies. However, it may also reflect that the levels of TGF-β, and/or length of exposure are greater in transgenic models than in lens injury models. As discussed earlier, it is becoming increasingly clear that additional Smad-independent pathways can regulate EMT and fibrosis. Thus, it is possible that Smad-independent signaling pathways are differentially induced in the two models and that this results in EMT and subsequent ASC development in the absence of Smad3 in the transgenic model.
Aberrant ECM deposition is a typical feature of ASC plaques.1,7,45 We observed ECM deposition in the subcapsular plaques of all the TGF-β1 transgenic lenses examined. However, the plaques of TGF-β1/Smad3-/- lenses showed only trace amounts of collagen type IV and substantially less fibronectin staining than did the other TGF-β1 transgenic lenses. This reduced ECM deposition correlated with smaller sized plaques in the TGF-β1/Smad3-/- mice that contained fewer myofibroblasts which secrete aberrant ECM. However, the fact that collagen type IV and fibronectin appeared to be selectively reduced, whereas collagen I expression did not seemed to change dramatically, suggests that Smad3 controls specific aspects of the ASC phenotype. Selective changes in expression of ECM have been reported for the Smad3-null mice. For example, Smad3-null mice when challenged with streptozotocin to induce diabetes and glomerular fibrosis show attenuated induction of fibronectin and α3Col4 expression when compared to their wild-type littermates.66 A selective induction of fibrotic markers, such as αSMA, has also been shown to occur after corneal injury in the Smad3-deficient mice.67 Thus, although it is clear that TGF-β-induced EMT in the lens can occur in the absence of Smad3, other fibrotic gene programs induced during ASC formation may be Smad3-dependent.
Apoptosis has been shown to be an additional feature of both TGF-β-induced ASC and posterior capsule opacification (PCO).7,8,46 In this study, we observed apoptotic cells in the plaques of the TGF-β1 transgenic mice, with more significant detection in the TGF-β1/Smad3-/- mice than in the transgenic wild-type littermates. This observation may explain why the plaques in the TGF-β1/Smad3-/- mice remain small relative to their wild-type and heterozygote littermates. The mechanism by which TGF-β induces apoptosis in the lens is not known. However, epithelial cells when separated from their basement membrane have been shown to undergo apoptosis and specifically, type IV collagen, a component of the lens capsule (basement membrane), has been shown to protect lens epithelial cells from undergoing Fas-stimulated apoptosis.44 Thus, the deposition of type IV collagen within the plaques of the TGF-β transgenic mice, which has also been reported in human ASC samples,43 may permit survival of these cells. The fact that we observed an increased number of apoptotic cells in the TGF-β1/Smad3-/- plaques, in which there was little or no type IV collagen deposition, further supports a role for type IV collagen in survival of the plaque cells.
We also used a laser-scanning instrument (ScanTox) to obtain quantitative measures of the optical quality of the TGF-β transgenic versus nontransgenic lenses. The optical measurements obtained were very consistent with the morphologic and molecular data. For example, all the TGF-β1 transgenic lenses, which exhibited ASC plaques, showed significantly larger BVD errors (decrease sharpness of focus) relative to the BVD errors of the wild-type lenses, which are devoid of plaques. The scanning laser instrument was able to discern the effects of size of the subcapsular plaques among the different genotypes on optical quality. This instrument is likely to be a useful tool in future transgenic/knockout lens studies involving quantitative measures of lens optical quality.
In conclusion, findings in the present study show that Smad3 deficiency does not prevent ASC formation in an in vivo transgenic TGF-β1 mouse model. The fact that the subcapsular plaques were reduced in size in the TGF-β1 transgenic mice on the Smad3-null background with less type IV collagen and fibronectin deposition compared with their wild-type and heterozygote littermates suggests that the Smad3 pathway may selectively regulate aspects of the ASC phenotype. Uncovering additional TGF-β signaling mechanisms may provide potential therapeutic targets to help prevent ASC and closely related ocular fibrotic diseases, such as PCO.
Acknowledgments
The authors thank Sam Zigler (National Eye Institute) for his generous donation of the β-crystallin antibody.
Supported by National Eye Institute Grants EY015006 and EY017146 (JAW-M) and the Natural Sciences and Engineering Research Council of Canada (JGS).
Footnotes
Disclosure: A. Banh, None; P.A. Deschamps, None; J. Gauldie, None; P.A. Overbeek, None; J.G. Sivak, None; J.A. West-Mays, None
References
- 1.Srinivasan Y, Lovicu FJ, Overbeek PA. Lens-specific expression of transforming growth factor beta1 in transgenic mice causes anterior subcapsular cataracts. J Clin Invest. 1998;101:625–634. doi: 10.1172/JCI1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurisaki A, Kose S, Yoneda Y, Heldin CH, Moustakas A. Transforming growth factor-beta induces nuclear import of Smad3 in an importin-beta1 and Ran-dependent manner. Mol Biol Cell. 2001;12:1079–1091. doi: 10.1091/mbc.12.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qing J, Zhang Y, Derynck R. Structural and functional characterization of the transforming growth factor-beta-induced Smad3/c-Jun transcriptional cooperativity. J Biol Chem. 2000;275:38802–38812. doi: 10.1074/jbc.M004731200. [DOI] [PubMed] [Google Scholar]
- 5.Dunker N, Krieglstein K. Reduced programmed cell death in the retina and defects in lens and cornea of Tgfbeta2(-/-) Tgf-beta3(-/-) double-deficient mice. Cell Tissue Res. 2003;313:1–10. doi: 10.1007/s00441-003-0761-x. [DOI] [PubMed] [Google Scholar]
- 6.Lee EH, Seomun Y, Hwang KH, et al. Overexpression of the transforming growth factor-β-inducible gene βIg-h3 in anterior polar cataracts. Invest Ophthalmol Vis Sci. 2000;41:1840–1845. [PubMed] [Google Scholar]
- 7.Lovicu FJ, Schulz MW, Hales AM, et al. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br J Ophthalmol. 2002;86:220–226. doi: 10.1136/bjo.86.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maruno KA, Lovicu FJ, Chamberlain CG, McAvoy JW. Apoptosis is a feature of TGF beta-induced cataract. Clin Exp Optom. 2002;85:76–82. doi: 10.1111/j.1444-0938.2002.tb03012.x. [DOI] [PubMed] [Google Scholar]
- 9.de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs. 2005;179:43–55. doi: 10.1159/000084508. [DOI] [PubMed] [Google Scholar]
- 10.Dwivedi DJ, Pino G, Banh A, et al. Matrix metalloproteinase inhibitors suppress transforming growth factor-β-induced subcapsular cataract formation. Am J Pathol. 2006;168:69–79. doi: 10.2353/ajpath.2006.041089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang RA, McAvoy JW. Growth factors in lens development. In: Lovicu FJ, Robinson ML, editors. Development of the Ocular Lens. Cambridge University Press; New York: 2004. pp. 261–289. [Google Scholar]
- 12.Sasaki K, Kojima M, Nakaizumi H, Kitagawa K, Yamada Y, Ishizaki H. Early lens changes seen in patients with atopic dermatitis applying image analysis processing of Scheimpflug and specular microscopic images. Ophthalmologica. 1998;212:88–94. doi: 10.1159/000027285. [DOI] [PubMed] [Google Scholar]
- 13.Font R, Brownstein S. A light and electron microscopic study of anterior subcapsular cataracts. Am J Ophthalmol. 1974;78:972–984. doi: 10.1016/0002-9394(74)90811-3. [DOI] [PubMed] [Google Scholar]
- 14.Novotny GE, Pau H. Myofibroblast-like cells in human anterior capsular cataract. Virchows Arch A Pathol Anat Histopathol. 1984;404:393–401. doi: 10.1007/BF00695223. [DOI] [PubMed] [Google Scholar]
- 15.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel). 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 16.Hales AM, Schulz MW, Chamberlain CG, McAvoy JW. TGF-beta 1 induces lens cells to accumulate alpha-smooth muscle actin, a marker for subcapsular cataracts. Curr Eye Res. 1994;13:885–890. doi: 10.3109/02713689409015091. [DOI] [PubMed] [Google Scholar]
- 17.Hales AM, Chamberlain CG, McAvoy JW. Cataract induction in lenses cultured with transforming growth factor-β. Invest Ophthalmol Vis Sci. 1995;36:1709–713. [PubMed] [Google Scholar]
- 18.Kappelhof JP, Vrensen GF. The pathology of after-cataract: a minireview. Acta Ophthalmol Suppl. 1992:13–24. doi: 10.1111/j.1755-3768.1992.tb02176.x. [DOI] [PubMed] [Google Scholar]
- 19.Marcantonio JM, Syam PP, Liu CS, Duncan G. Epithelial transdifferentiation and cataract in the human lens. Exp Eye Res. 2003;77:339–346. doi: 10.1016/s0014-4835(03)00125-8. [DOI] [PubMed] [Google Scholar]
- 20.Cousins SW, McCabe MM, Danielpour D, Streilein JW. Identification of transforming growth factor-beta as an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 1991;32:2201–2211. [PubMed] [Google Scholar]
- 21.Wallentin N, Wickstrom K, Lundberg C. Effect of cataract surgery on aqueous TGF-beta and lens epithelial cell proliferation. Invest Ophthalmol Vis Sci. 1998;39:1410–1418. [PubMed] [Google Scholar]
- 22.Rooke HM, Crosier KE. The smad proteins and TGFbeta signalling: uncovering a pathway critical in cancer. Pathology. 2001;33:73–84. [PubMed] [Google Scholar]
- 23.Gordon-Thomson C, de Iongh RU, Hales AM, Chamberlain CG, McAvoy JW. Differential cataractogenic potency of TGF-beta1, -beta2, and -beta3 and their expression in the postnatal rat eye. Invest Ophthalmol Vis Sci. 1998;39:1399–1409. [PubMed] [Google Scholar]
- 24.Flugel-Koch C, Ohlmann A, Piatigorsky J, Tamm ER. Disruption of anterior segment development by TGF-beta1 overexpression in the eyes of transgenic mice. Dev Dyn. 2002;225:111–125. doi: 10.1002/dvdy.10144. [DOI] [PubMed] [Google Scholar]
- 25.Greene RM, Nugent P, Mukhopadhyay P, Warner DR, Pisano MM. Intracellular dynamics of Smad-mediated TGFbeta signaling. J Cell Physiol. 2003;197:261–271. doi: 10.1002/jcp.10355. [DOI] [PubMed] [Google Scholar]
- 26.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 27.Inazaki K, Kanamaru Y, Kojima Y, et al. Smad3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]
- 28.Ramirez AM, Takagawa S, Sekosan M, Jaffe HA, Varga J, Roman J. Smad3 deficiency ameliorates experimental obliterative bronchiolitis in a heterotopic tracheal transplantation model. Am J Pathol. 2004;165:1223–1232. doi: 10.1016/S0002-9440(10)63382-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao J, Shi W, Wang YL, et al. Smad3 deficiency attenuates bleo-mycin-induced pulmonary fibrosis in mice. Am J Physiol. 2002;282:L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 31.Saika S, Kono-Saika S, Ohnishi Y, et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651–663. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashcroft GS, Yang X, Glick AB, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 33.de Iongh RU, Lovicu FJ, Overbeek PA, et al. Requirement for TGFbeta receptor signaling during terminal lens fiber differentiation. Development. 2001;128:3995–4010. doi: 10.1242/dev.128.20.3995. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.Priolo S, Sivak JG, Kuszak JR, Irving EL. Effects of experimentally induced ametropia on the morphology and optical quality of the avian crystalline lens. Invest Ophthalmol Vis Sci. 2000;41:3516–3522. [PubMed] [Google Scholar]
- 36.Oriowo OM, Cullen AP, Chou BR, Sivak JG. Action spectrum and recovery for in vitro UV-induced cataract using whole lenses. Invest Ophthalmol Vis Sci. 2001;42:2596–2602. [PubMed] [Google Scholar]
- 37.Bantseev V, McCanna D, Banh A, et al. Mechanisms of ocular toxicity using the in vitro bovine lens and sodium dodecyl sulfate as a chemical model. Toxicol Sci. 2003;73:98–107. doi: 10.1093/toxsci/kfg060. [DOI] [PubMed] [Google Scholar]
- 38.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 39.Kretzschmar M, Doody J, Massague J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 40.Symonds JG, Lovicu FJ, Chamberlain CG. Posterior capsule opacification-like changes in rat lens explants cultured with TGFbeta and FGF: effects of cell coverage and regional differences. Exp Eye Res. 2006;82:693–699. doi: 10.1016/j.exer.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 41.de Jong-Hesse Y, Kampmeier J, Lang GK, Lang GE. Effect of extracellular matrix on proliferation and differentiation of porcine lens epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2005;243:695–700. doi: 10.1007/s00417-004-1116-3. [DOI] [PubMed] [Google Scholar]
- 42.Oharazawa H, Ibaraki N, Lin LR, Reddy VN. The effects of extracellular matrix on cell attachment, proliferation and migration in a human lens epithelial cell line. Exp Eye Res. 1999;69:603–610. doi: 10.1006/exer.1999.0723. [DOI] [PubMed] [Google Scholar]
- 43.Ishida I, Saika S, Okada Y, Ohnishi Y. Growth factor deposition in anterior subcapsular cataract. J Cataract Refract Surg. 2005;31:1219–1225. doi: 10.1016/j.jcrs.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 44.Futter CE, Crowston JG, Allan BD. Interaction with collagen IV protects lens epithelial cells from Fas-dependent apoptosis by stimulating the production of soluble survival factors. Invest Ophthalmol Vis Sci. 2005;46:3256–3262. doi: 10.1167/iovs.05-0086. [DOI] [PubMed] [Google Scholar]
- 45.Lovicu FJ, Steven P, Saika S, McAvoy JW. Aberrant lens fiber differentiation in anterior subcapsular cataract formation: a process dependent on reduced levels of Pax6. Invest Ophthalmol Vis Sci. 2004;45:1946–1953. doi: 10.1167/iovs.03-1206. [DOI] [PubMed] [Google Scholar]
- 46.Lee JH, Wan XH, Song J, et al. TGF-beta-induced apoptosis and reduction of Bcl-2 in human lens epithelial cells in vitro. Curr Eye Res. 2002;25:147–153. doi: 10.1076/ceyr.25.3.147.13475. [DOI] [PubMed] [Google Scholar]
- 47.Lütjen-Drecoll E. Morphological changes in glaucomatous eyes and the role of TGFbeta(2) for the pathogenesis of the disease. Exp Eye Res. 2005;81:1–4. doi: 10.1016/j.exer.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 48.Horowitz JC, Lee DY, Waghray M, et al. Activation of the prosurvival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonniaud P, Kolb M, Galt T, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 50.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 51.Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- 52.Weinstein M, Yang X, Li C, et al. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci USA. 1998;95:9378–9383. doi: 10.1073/pnas.95.16.9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Massague J. Wounding Smad. Nat Cell Biol. 1999;1:E117–E119. doi: 10.1038/12944. [DOI] [PubMed] [Google Scholar]
- 55.Beebe D, Garcia C, Wang X, et al. Contributions by members of the TGFbeta superfamily to lens development. Int J Dev Biol. 2004;48:845–856. doi: 10.1387/ijdb.041869db. [DOI] [PubMed] [Google Scholar]
- 56.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibro-blastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 57.Hanafusa H, Ninomiya-Tsuji J, Masuyama N, et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem. 1999;274:27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- 58.Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW. TGF-beta1 activates MAP kinase in human mesangial cells: a possible role in collagen expression. Kidney Int. 1999;56:1710–1720. doi: 10.1046/j.1523-1755.1999.00733.x. [DOI] [PubMed] [Google Scholar]
- 59.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- 60.Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 61.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 63.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001;276:46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 64.Masszi A, Di Ciano C, Sirokmany G, et al. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 65.Roberts AB, Derynck R. Meeting report: signaling schemes for TGF-beta. Sci STKE. 2001;2001:PE43. doi: 10.1126/stke.2001.113.pe43. [DOI] [PubMed] [Google Scholar]
- 66.Fujimoto M, Maezawa Y, Yokote K, et al. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun. 2003;305:1002–1007. doi: 10.1016/s0006-291x(03)00885-4. [DOI] [PubMed] [Google Scholar]
- 67.Stramer BM, Austin JS, Roberts AB, Fini ME. Selective reduction of fibrotic markers in repairing corneas of mice deficient in Smad3. J Cell Physiol. 2005;203:226–232. doi: 10.1002/jcp.20215. [DOI] [PubMed] [Google Scholar]