

Abstract
We describe a protocol to rapidly and reliably visualize blood vessels in experimental animals. Blood vessels are directly labeled by cardiac perfusion using a specially formulated aqueous solution containing 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), a lipophilic carbocyanine dye, which incorporates into endothelial cell membranes upon contact. By lateral diffusion, DiI also stains membrane structures, including angiogenic sprouts and pseudopodial processes that are not in direct contact. Tissues can be immediately examined by conventional and confocal fluorescence microscopy. High-quality serial optical sections using confocal microscopy are obtainable from thick tissue sections, especially at low magnification, for three-dimensional reconstruction. It takes less than 1 h to stain the vasculature in a whole animal. Compared with alternative techniques to visualize blood vessels, including space-occupying materials such as India ink or fluorescent dye-conjugated dextran, the corrosion casting technique, endothelial cell-specific markers and lectins, the present method simplifies the visualization of blood vessels and data analysis.
INTRODUCTION
We describe here a direct labeling protocol1,2 to visualize blood vessels in small experimental animals rapidly and reliably using a lipophilic carbocyanine dye, DiI (DiIC18(3)). Lipophilic carbocyanine dyes, including DiI, have been widely used to label cell membranes3–8 and to trace neuronal connections in live and fixed tissues7–14. In the cell membrane, a DiI molecule has its two hydrocarbon chains buried in the lipid bilayer parallel to the phospholipid acyl chains, and the chromophores are on the surface of the bilayer15.
The direct labeling technique takes advantage of the lipophilic property of DiI. A special aqueous DiI solution is formulated to have the maximum sodium chloride concentration without causing precipitation of DiI. Glucose is added to maintain the physiological osmolarity. Labeling is achieved by bringing DiI solution in direct contact with endothelial cells through cardiac perfusion. A simple perfusion device is assembled with two three-way stopcocks, a butterfly needle and three 10-ml disposable syringes for phosphate-buffered saline (PBS), DiI solution and fixative, respectively (Fig. 1).
Figure 1.
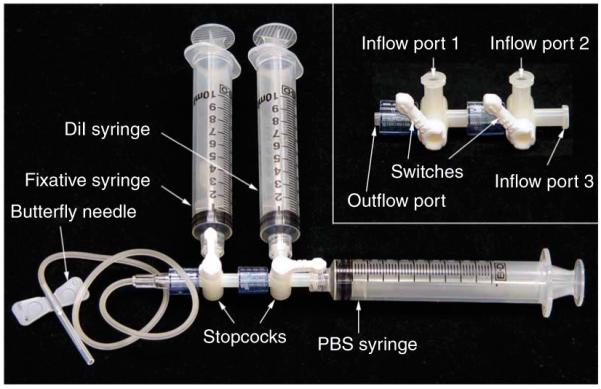
Perfusion device. The main body of the perfusion device is two three-way stopcocks connected in series (inset). The output port of the stopcocks is connected to a butterfly needle. The inflow ports are connected to three 10-ml disposable syringes for PBS, DiI solution and fixative, respectively. The positions of the three syringes depend on the specific stopcock used, as stopcocks of different brands may have different configurations. It is also possible to use a single stopcock for the device. In that case, it will require change of syringes after perfusion with DiI solution. During syringe change, it is important to avoid air from entering the perfusion line.
During perfusion, DiI partitions quickly into the cell membrane (lipid) phase, the endothelial cell membrane lining the blood vessels, resulting in instantaneous labeling of blood vessels (Fig. 2). Once in the membrane, DiI molecules diffuse laterally within the lipid bilayer3. The lateral diffusion helps to stain membrane structures that are not in direct contact with the DiI solution, including angiogenic sprouts and psuedopodial processes of angiogenic endothelial cells (Fig. 3c,d).
Figure 2.

Vasculature visualized by DiI labeling. (a) Retinal whole-mount showing an artery (arrow) on the retinal inner surface along with its branches connecting to the deep capillaries, which are also visible, although out of focus (10 objective lens). (b) Retinal whole-mount-showing another artery (arrow) at a higher magnification (×20 objective lens). The nuclei of endothelial cells are identifiable as oval spots in both a and b. (c) A brain section (100 μm thick, ×10 objective lens) showing three large blood vessels (arrows) located in the midline (dashed line) between the two hemispheres and the vascular network in the tissue. (d) A heart section (left ventricle, 100 μm thick, ×10 objective lens) showing capillaries in the left ventricle wall. In the outer layer (upper part), they are perpendicular to the section plane and appear as bright dots, whereas in the inner layer (lower part), they are parallel to the section plane. Tissues were viewed by fluorescence microscopy (Nikon Eclipse E800 fluorescence microscope with plan fluo ×10/0.3 numerical aperture (NA) or plan fluo ×20/0.5 NA objective lens and Chroma 34,000 rhodamine filter set, with the excitation wavelength of 570 nm and the emission of 590). Scale bars, 200 μm for a, c and d, and 100 μm for b.
Figure 3.
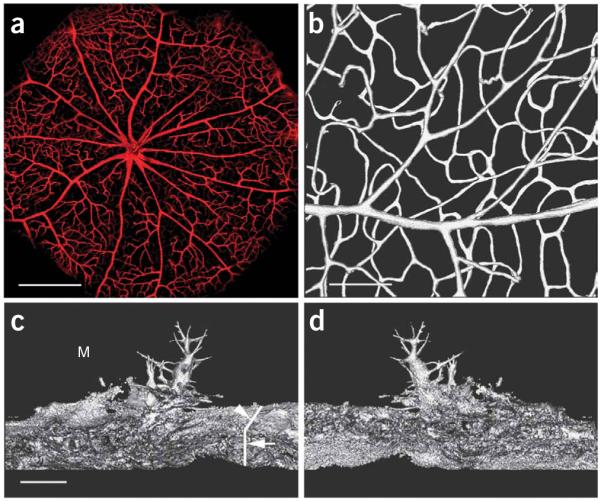
Three-dimensional reconstructed images of retinal vasculature and angiogenic sprouts and pseudopodial processes. (a) A 3-D reconstructed image of the eyecup as a ‘bird’s eye’ view of the vasculature of the posterior retina. The eyecup was placed in a holder and submerged in PBS for direct imaging. A ×5/0.15 NA objective lens was used to take a stack of confocal images, which were used for the reconstruction. DiI signal was detected using Leica DM6000B microscope with the ×5 objective (HC PL Fluotar/0.15) (Leica). Images were acquired by a Leica Application Suite Advanced Fluorescence (LAS AF), 1.7.0, Build 1240 software TCS-SP5 (Leica). The same software was used to create 3-D reconstruction of images and a movie clip, which shows the retinal vasculature rotated around the x axis (Supplementary Movie 1). (b) A 3-D image showing an artery on the retinal surface connected to the deep capillaries. A movie clip shows the artery and its network rotated around the y axis (Supplementary Movie 2). (c) A 3-D image of angiogenic sprouts and pseudopodial processes in a subretinal Matrigel of choroidal neovascularization1 at a viewing angle of 0° (from the coverslip side downward). The vertical line (arrow) indicates the cross section of the choroid. Another line (arrowhead) indicates the surface of the choroicapillaris. Several sprouts from the choriocapillaris invaded the Matrigel layer (M) in the subretinal space. Their pseudopodial processes protruded into the Matrigel deposit (M) in different directions. (d) 3-D image of angiogenic sprouts and pseudopodial processes at a viewing angle of 180°. A movie clip shows the sprouts rotated around the y axis (Supplementary Movie 3). Scale bar, 400 μm for a, 100 μm for b and 20 μm for c and d.
As DiI is directly delivered to the endothelial cell membrane, it is possible to achieve high concentration in cell membrane and thus high signal intensities, which dramatically increases the signal-to-noise ratio. In addition, the dye itself is intensely fluorescent with slow fading when exposed to the excitation light7,8. The strong intensity and slow fading feature are ideal for serial optical sections using laser scanning confocal microscopy, especially at low magnification. Images can be obtained throughout the entire thickness of a tissue sample of 100 μm without noticeable deterioration of image quality. The vasculature in the inner surface of a hollow organ like the eye can be scanned as a single stack of confocal images at very low magnification (×5 objective lens). Digitized data of thick tissue sections can be used to reconstruct 3-dimemsional (3-D) images of vascular network in the samples of interest and/or to perform other analyses with suitable software. The 3-D images have the advantage of viewing the vascular network in its entirety (Fig. 3; Supplementary Movies 1–3 online). In addition, the use of thick tissue sections would save time and effort, as fewer tissue sections need to be prepared to cover the entire tissue sample. This is especially important when many samples are to be examined.
Many alternative techniques are available to visualize blood vessels. Space-occupying high-contrast materials, including India ink16 and fluorescent dye-conjugated dextran (e.g., fluorescein isothiocyanate or FITC)17, are used to fill blood vessels to render them visible. However, the dye molecules are not bound to blood vessels but move freely in the filling solution. When a tissue sample is compressed, especially during sample preparation, the filling solution can be squeezed out of blood vessels, resulting in interrupted or ‘broken’ vascular patterns. This method, however, directly stains the endothelial cell membranes.
The corrosion casting technique using low-viscosity plastics, another type of space-occupying material, to make casts of blood vessel network has been employed to study the three dimensionality of vasculature18,19 in combination with scanning electron microscopy (SEM)20,21. The casts are viewed with SEM to obtain 3-D images of the vasculature. It is technically demanding to make casts and requires access to SEM20,21. It is also difficult to visualize angiogenic sprouts and pseudopodial processes. In comparison, the method described here is technically much simpler in sample preparation and data collection.
Blood vessels can also be visualized by endothelial cell-specific markers, including factor VIII (see ref. 22) or CD34 (see ref. 23). Lectins have been used as surrogate markers for endothelial cells. For example, Ulex europaeus I lectin recognizes human endothelial cells24, and Griffonia simplicifolia isolectin B4 specifically labels endothelial cells in mouse, rat and other laboratory animals25,26. Use of these markers usually requires multistep procedures. In addition, it is difficult for these techniques to achieve the high signal intensities that can be achieved by direct labeling methods described here.
The direct labeling technique with DiI provides an efficient, reliable and low-cost (the amount of DiI used per rat is less than 1 mg that costs less than USD $2) alternative to obtain high intensity, high signal-to-noise ratio labeling of blood vessels in the entire body for both low- and high-power microscopic observation. It can also be used to study angiogenic sprouts and pseudopodial processes. Although described here for use in rodents, this procedure can be adapted easily to the study of vasculature in other vertebrate animals including rabbits, either for the vasculature of the entire body or for ex vivo perfusion of a particular organ, such as rabbit kidneys.
In this method, DiI molecules are directly incorporated into the membrane of endothelial cells. As it is a lipophilic dye, the molecules in the membrane could be washed away by lipophilic solvents. It is therefore not easy to combine DiI staining with other techniques, such as immunohistochemical analysis, if the subsequent procedures involve those lipophilic solvents. This issue could be overcome by using a fixable DiI derivative, CM-DiI (C-7001, Invitrogen/Molecular Probes). CM-DiI is particularly useful in experiments that combine membrane labeling with subsequent immunohistochemical analysis, fluorescence in situ hybridization or electron microscopy. DiI staining is, however, compatible with the nuclear dye DAPI (4′-6-diamidino-2-phenylindole).
MATERIALS
REAGENTS
DiI (D-282, Invitrogen/Molecular Probes; 42364, Sigma-Aldrich)
Ethanol (100% vol/vol)
NaCl (S7653, Sigma-Aldrich)
KCl (P9333, Sigma-Aldrich)
Na2HPO4 (S8282, Sigma-Aldrich)
NaH2PO4 (S7907, Sigma-Aldrich)
Glucose (G7568, Sigma-Aldrich)
HCl (1 M) (13-1700, Sigma-Aldrich)
NaOH (5 M) (S8263, Sigma-Aldrich)
Glycerol (G6279, Sigma-Aldrich)
Paraformaldehyde (158127, Sigma-Aldrich)
Agarose (A5939, Sigma-Aldrich)
Superglue (can be obtained from regular hardware stores)
Experimental animals (rats and mice, Charles River)
 All animal experiments must comply with national laws and institutional regulations.
All animal experiments must comply with national laws and institutional regulations.
EQUIPMENT
25 gauge butterfly needles
Microscope slides
Standard BK7 glass coverslip (no. 1.5, 170 μm thickness)
Three-way stopcocks
10-ml disposable syringes
Aluminum boats
Vibratome (VT1000S, Leica Microsystems)
REAGENT SETUP
DiI stock solution
Dissolve 100 mg of DiI crystal in 16.7 ml of 100% ethanol on a rocker overnight (covered with aluminum foil when rocking) and store in the dark at room temperature (RT, 20–25 °C) for up to 1 year.  DiI solution stains easily on skin and other surfaces. If accidental staining occurs, it can be wiped off easily by 70% ethanol.
DiI solution stains easily on skin and other surfaces. If accidental staining occurs, it can be wiped off easily by 70% ethanol.
PBS (pH 7.4)
Dissolve 8 g of NaCl, 0.2 g of KCl, 1.44 g of Na2HPO4 and 0.23 g of NaH2PO4 in 800 ml of distilled water. Adjust pH to 7.4 with HCl. Add water to 1,000 ml, filter through a 0.22-μm bottle-top filter. Store at RT for up to 1 year.
5% (wt/vol) glucose
Dissolve 50 g of glucose in 1,000 ml of distilled water. Filter through a 0.22-μm bottle-top filter. Store at RT for up to 1 year.
Diluent
Mix PBS and 5% glucose at a ratio of 1:4. Store at RT for up to 1 year.
DiI work solution
Immediately before use, add 200 μl of DiI stock solution into 10 ml of diluent, mix by quick shaking. DiI work solution should be freshly made under room lighting.
Phosphate buffer (pH 7.4)
Prepare separately a solution of 0.2 M NaH2PO4 and another one of 0.2 M Na2HPO4. Mix the two solutions at a ratio of 19:81. Check the pH with a pH meter. Adjust pH with either HCl or NaOH. Store at RT for up to 1 year.
8% (wt/vol) paraformaldehyde
Under a chemical hood, add 8 g of paraformaldehyde to 90 ml of distilled water in a beaker with a magnetic stirrer. Heat to 65 °C and adjust the pH to 7.4 (using pH paper) with 5 M NaOH. Add distilled water to adjust the final volume to 100 ml. Cool the solution and filter. Store at 4 °C for up to 1 month.
4% (wt/vol) paraformaldehyde solution
Mix 8% paraformaldehyde with 0.2 M phosphate buffer at a ratio of 1:1 before use. Store at 4 °C for up to 1 month.
80% (vol/vol) glycerol
Add 1 ml of distilled water into 4 ml of glycerol and mix well. Store at RT for up to 1 year.
5% (wt/vol) agarose
Add 20 ml of distilled water to a beaker, and add 1 g of agarose. Heat on a heat place until agarose melts. Keep 5% agarose to 60 °C. Agarose (5%) can be stored at 4 °C for up to 1 month. To use the agarose again, heat until it melts and keep it at 60 °C.
EQUIPMENT SETUP
Perfusion device
The perfusion device is assembled with two three-way stopcocks, a 25 gauge butterfly needle and three 10-ml syringes (Fig. 1). First, connect the two three-way stopcocks in series (Fig. 1, inset) and then the 25-gauge butterfly needle to the outflow port of the stopcocks (Fig. 1). Load the three syringes with 2 ml of PBS, DiI solution (5 ml for a mouse, 10 ml for a rat) and 10 ml of fixative (4% paraformaldehyde), respectively. Connect the syringe with fixative to the first inflow port and inject solution until there is no air in the line; turn the stopcock to block the port. Then, connect the DiI syringe to the second inflow port, inject solution until there is no air in the line, and turn the stopcock to block the port. Finally, connect the PBS syringe to the last inflow port and inject solution until there is no air in the line. Assembly is under room lighting.  Make sure that no air is trapped in the line or the syringes before proceeding to perfusion. Once in the circulation system, air will block blood vessels, resulting in poor perfusion.
Make sure that no air is trapped in the line or the syringes before proceeding to perfusion. Once in the circulation system, air will block blood vessels, resulting in poor perfusion.
Aluminum boats
Cut a piece of Styrofoam into the shape and size of the samples of interest. Wrap the Styrofoam with a piece of aluminum foil, leaving one side open. Remove the Styrofoam and store the aluminum boats appropriately at RT.
PROCEDURE
Perfusion  ~45 min
~45 min
 All steps are carried out under ambient lighting unless specified.
All steps are carried out under ambient lighting unless specified.
1| Assemble the perfusion device as described in EQUIPMENT SETUP.
![]()
2| Kill a mouse or rat by CO2 overdose in a CO2 chamber, or by sodium phenobarbital overdose (120 mg kg−1, i.p).
 All animal experiments must comply with national laws and institutional regulations.
All animal experiments must comply with national laws and institutional regulations.
3| In a glass tray with an absorbing blue pat or several paper towels, position the animal on its back. Make a transverse incision to open the abdominal cavity. Expose and cut the diaphragm. Cut the chest wall on both sides up to the first or the second rib.
4| Take a small hemostat to hold the lower tip of the sternum and the turn the anterior chest wall toward the head of the animal to expose the chest cavity.
5| (Optional) If the tissues of interest are in the upper body, such as the brain or the eye, there is no need to perfuse the lower body. If this is the case, load half amount of the regular perfusion solutions in Step 1 and clamp the descending aorta to block the flow to the lower body before perfusion. This is not necessary for mice.
6| Insert the butterfly needle of the perfusion device into the left ventricle and then puncture the right atrium with a 16-gauge needle.
 Make sure that the needle is in the left ventricle, which appears lighter than the right ventricle. Do not penetrate the opposite side of the ventricular wall. Such penetration will result in a leak in the left ventricle, making perfusion difficult.
Make sure that the needle is in the left ventricle, which appears lighter than the right ventricle. Do not penetrate the opposite side of the ventricular wall. Such penetration will result in a leak in the left ventricle, making perfusion difficult.
![]()
7| Inject 2 ml of PBS from the perfusion device at the rate of 1–2 ml min−1 (total 1–2 min). A sign of good perfusion is bleeding from the right atrium. After injection, turn the switch to block the port of PBS syringe and open the port to the DiI syringe.
![]()
8| Inject 5–10 ml of the DiI solution at the rate of 1–2 ml min−1 (total 5–10 min). Monitor color change in the ears, nose and palms. DiI appears purple in visible light. During perfusion with DiI solution, the ears, nose and palms will turn slightly purple. After injection, turn the switch to block the port of DiI syringe and open the port to the fixative syringe.
9| Inject 5–10 ml of the fixative at the rate of 1–2 ml min−1 (total 5–10 min).
10| Harvest tissues of interest. To make eyecup preparation, eyes are enucleated and the anterior segments are removed. The retinas are further dissected from eyecups1. Other tissues of interest can be harvested by directly removing them from the body.
 Samples can be left in the fixative or PBS in the dark for up to 1 month at 4 °C.
Samples can be left in the fixative or PBS in the dark for up to 1 month at 4 °C.
 Be sure to place the used needles into a sharp container and to dispose of the animal and waste appropriately.
Be sure to place the used needles into a sharp container and to dispose of the animal and waste appropriately.
![]()
Tissue sectioning  ~1–2 h, depending on the size of the tissue to be sectioned
~1–2 h, depending on the size of the tissue to be sectioned
11| Rinse samples in PBS 2× 15 min.
12| Embed tissue sample in 5% agarose: heat agarose in a 100-ml beaker to 60 °C and pour it (about one-third of the volume required to submerge the sample) into an aluminum boat that fits the sample. Submerge the tissue sample into the agarose and adjust the orientation. Add more agarose into the boat to keep the sample totally submerged. Samples can be kept at 4 °C in the dark up to 1 month when wrapped in plastic wrap to prevent dehydration. Remove the sample block from the aluminum boat before sectioning. Embedding is not necessary for solid tissue such as the brain and the liver.
![]()
13| Trim off excess agarose. Glue the sample to the section stage of the Vibratome using Superglue. Secure the section stage in the section chamber and adjust the sample to the appropriate orientation. Fill the sectioning chamber with PBS to submerge the sample.
![]()
14| Set the start and the end point of sectioning (for additional information regarding operating the Leica VT1000S Vibratome, please read the manufacturer’s manual).
![]()
15| Set the thickness of the section to 100 μm (for additional information regarding operating the Leica VT1000S Vibratome, please read the manufacturer’s manual).
![]()
16| Section the tissue and collect a section with a small (no. 6 or 8) round watercolor brush; put it on a glass slide. One slide may hold several sections.
![]()
17| Use a larger watercolor brush to remove excess PBS, add a small amount of 80% glycerol to the sections and cover with no. 1.5 cover slip. Seal it with nail polish.
 Sections are better viewed as soon as possible but can be kept for several days at 4 °C in the dark.
Sections are better viewed as soon as possible but can be kept for several days at 4 °C in the dark.
![]()
View sections  Depending largely on the number of the sections and the area of interest to be examined, ~1 h to 1 d
Depending largely on the number of the sections and the area of interest to be examined, ~1 h to 1 d
18| View tissue sections under a fluorescence microscope or a confocal microscope using the filter set for rhodamine.
19| Collect a stack of confocal images. Reconstruction of 3-D images can be achieved using any image analysis software that has such a function. We used AutoVisualize 3-D (version 5.5, AutoQuant Image Inc.) and Leica Application Suite Advanced Fluorescence (LAS AF, TCS-SP5) (Leica).
![]()
Steps 1–10: ~45 min
Steps 11–17: ~1–2 h, depending on the size of the tissue to be sectioned
Steps 18 and 19: depending largely on the number of the sections and the area of interest to be examined, ~1 h to 1 d
![]()
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table
| Problem | Possible reason | Possible solution |
|---|---|---|
| Weak signal | Poor perfusion due to air trapped in the system (Steps 1 and 6) | Make sure no air is trapped in the perfusion line before perfusion. When inserting the needle into the left ventricle, slightly press the PBS syringe to allow a fluid drop to appear at the needle tip |
| Poor perfusion because of a leak in the left ventricle (Step 6) | Do not penetrate the opposite side of the ventricle when inserting the butterfly needle. Using a small hemostat to clamp the leaking point sometimes works |
|
| Poor staining because of too much PBS perfusion (Step 7) | Make sure not more than 2 ml of PBS is used before DiI perfusion |
|
| ’Leak’ staining | The inner lining of blood vessels are broken because of rough handling of tissue preparation and sectioning (Steps 10, 12–17) |
DiI will not ‘leak’ to the surrounding tissues if blood vessels are intact. Be gentle during tissue harvesting. View tissue sections as soon as possible |
ANTICIPATED RESULTS
Perfusion with DiI solution delivers dye molecules to endothelial cells lining blood vessels in the entire circulation system, thus blood vessels in the whole body are stained. DiI appears purple in color under visible light, so tissues that are well stained should appear slightly purple. Large blood vessels on the surface should be visible. Blood vessels appear intensely red under a fluorescence microscope using a rhodamine filter set (Fig. 2). Stacks of confocal images could be obtained and used to reconstruct 3-D images in hollow organs like the eye (Fig. 3a) and thick tissue samples (Fig. 3b). Because of lateral diffusion, DiI also stains membrane structures that are not in direct contact with the dye solution, including angiogenic sprouts and pseudopodial processes. Images of angiogenic sprouts with their extruding processes (3-D reconstruction) are shown in Figure 3c,d. Thick tissue sections allow efficient screening of the entire tissue of interest at low magnifications for relatively rare events, which can then be studied in detail at high magnifications and/or using an electronic zoom. For example, to study choroidal neovascularization, it is important to find the site where new blood vessels from the choriocapillaris penetrate the Bruch’s membrane. In our study on choroidal neovascularization in a subretinal Matrigel model of choroidal neovascularization in the rat1, we routinely screen the entire Matrigel-injected area (20–25 sections of 100 μm thick) to identify the penetration sites and then to carefully study them. One such site is shown in Figure 4. The combined DiI staining and DIC (differential interference contrast) image shows the site at which a new blood vessel originated in the choriocapillaris penetrated the Bruch’s membrane and entered the space between the retinal pigment epithelium and the Bruch’s membrane.
Figure 4.

Penetration of Bruch’s membrane by choroidal neovascularization. Choroidal neovascularization was induced by subretinal injection of Matrigel1. To locate the penetration sites of choroidal neovascularization, serial tissue sections (100 μm thick each) were cut through the entire Matrigel injected area (about 2 mm in diameter). Tissue sections were screened at low power (×10 objective) under a fluorescence microscope. Potential penetration sites were further examined by confocal microscopy. (a) A confocal image of a new blood vessel that had penetrated the Bruch’s membrane (square). (b) A superimposed image of DIC (differential interference contrast) and fluorescence (red) shows the penetrating blood vessel (square) relative to Bruch’s membrane (between two arrowheads). (c) The penetration site (arrow) at higher magnification to show Bruch’s membrane (between two arrowheads). Ch, choroid. Scale bar, 50 μm for a and b; 7 μm for c .
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr George McNamara for critical reading of the manuscript. This work was supported by NIH grant EY12727, EY-015289, P30 EY14801, the Karl Kirchgessner Foundation and the Foundation Fighting Blindness.
Footnotes
Note: Supplementary information is available via the HTML version of this article.
References
- 1.Zhao L, et al. Translocation of the retinal pigment epithelium and formation of sub-retinal pigment epithelium deposit induced by subretinal deposit. Mol. Vis. 2007;13:873–880. [PMC free article] [PubMed] [Google Scholar]
- 2.Hadziahmetovic M, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest. Ophthalmol. Vis. Sci. 2008;49:2728–2736. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlessinger J, Axelrod D, Koppel DE, Webb WW, Elson EL. Lateral transport of a lipid probe and labeled proteins on a cell membrane. Science. 1977;195:307–309. doi: 10.1126/science.556653. [DOI] [PubMed] [Google Scholar]
- 4.de Laat SW, van der Saag PT, Elson EL, Schlessinger J. Lateral diffusion of membrane lipids and proteins is increased specifically in neurites of differentiating neuroblastoma cells. Biochim. Biophys. Acta. 1979;558:247–250. doi: 10.1016/0005-2736(79)90064-6. [DOI] [PubMed] [Google Scholar]
- 5.Struck DK, Pagano RE. Insertion of fluorescent phospholipids into the plasma membrane of a mammalian cell. J. Biol. Chem. 1980;255:5404–5410. [PubMed] [Google Scholar]
- 6.Jacobson K, Hou Y, Derzko Z, Wojcieszyn J, Organisciak D. Lipid lateral diffusion in the surface membrane of cells and in multibilayers formed from plasma membrane lipids. Biochemistry. 1981;20:5268–5275. doi: 10.1021/bi00521a027. [DOI] [PubMed] [Google Scholar]
- 7.Honig MG, Hume RI. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. J. Cell. Biol. 1986;103:171–187. doi: 10.1083/jcb.103.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honig MG, Hume RI. DiI and DiO: versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci. 1989;12:333–341. [PubMed] [Google Scholar]
- 9.Godement P, Vanselow J, Thanos S, Bonhoeffer F. A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development. 1987;101:697–713. doi: 10.1242/dev.101.4.697. [DOI] [PubMed] [Google Scholar]
- 10.Molnar Z, Blakemore C. Lack of regional specificity for connections formed between thalamus and cortex in coculture. Nature. 1991;351:475–477. doi: 10.1038/351475a0. [DOI] [PubMed] [Google Scholar]
- 11.Collazo A, Bronner-Fraser M, Fraser SE. Vital dye labelling of Xenopus laevis trunk neural crest reveals multipotency and novel pathways of migration. Development. 1993;118:363–376. doi: 10.1242/dev.118.2.363. [DOI] [PubMed] [Google Scholar]
- 12.Onifer SM, White LA, Whittemore SR, Holets VR. In vitro labeling strategies for identifying primary neural tissue and a neuronal cell line after transplantation in the CNS. Cell Transplant. 1993;2:131–149. doi: 10.1177/096368979300200207. [DOI] [PubMed] [Google Scholar]
- 13.Stark MR, Sechrist J, Bronner-Fraser M, Marcelle C. Neural tube-ectoderm interactions are required for trigeminal placode formation. Development. 1997;124:4287–4295. doi: 10.1242/dev.124.21.4287. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto N, Higashi S, Toyama K. Stop and branch behaviors of geniculocortical axons: a time-lapse study in organotypic cocultures. J. Neurosci. 1997;17:3653–3663. doi: 10.1523/JNEUROSCI.17-10-03653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Axelrod D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 1979;26:557–573. doi: 10.1016/S0006-3495(79)85271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlesinger B. The angioarchitecture of the thalamus in the rabbit. J. Anat. 1941;75:176–183. [PMC free article] [PubMed] [Google Scholar]
- 17.Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 18.Murakami T. Application of the scanning electron microscope to the study of the fine distribution of the blood vessels. Arch. Histol. Jap. 1971;32:445–454. doi: 10.1679/aohc1950.32.445. [DOI] [PubMed] [Google Scholar]
- 19.Ohtani O, Murakami T. Routine methods for vascular casting and SEM. In: Motta PM, Murakami T, Fujita H, editors. Scanning electron microscopy of vascular casting: methods and applications. Kluwer Academic Publishers; Boston: 1992. pp. 13–25. [Google Scholar]
- 20.Lametschwandtner A, Lametschwandtner U, Weiger T. Scanning electron microscopy of vascular corrosion casts—technique and applications. Scan. Electron. Microsc. 1984;2:663–695. [PubMed] [Google Scholar]
- 21.Lametschwandtner A, Lametschwandtner U, Weiger T. Scanning electron microscopy of vascular corrosion casts—technique and applications: updated review. Scanning Microsc. 1990;4:889–940. discussion 941. [PubMed] [Google Scholar]
- 22.Bloom AL, Giddings JC, Wilks CJ. Factor 8 on the vascular intima: possible importance in haemostasis and thrombosis. Nat. New Biol. 1973;241:217–219. doi: 10.1038/newbio241217a0. [DOI] [PubMed] [Google Scholar]
- 23.Fina L, et al. Expression of the CD34 gene in vascular endothelial cells. Blood. 1990;75:2417–2426. [PubMed] [Google Scholar]
- 24.Holthofer H, et al. Ulex europaeus I lectin as a marker for vascular endothelium in human tissues. Lab. Invest. 1982;47:60–66. [PubMed] [Google Scholar]
- 25.Laitinen L. Griffonia simplicifolia lectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem. J. 1987;19:225–234. doi: 10.1007/BF01680633. [DOI] [PubMed] [Google Scholar]
- 26.Hansen-Smith FM, Watson L, Lu DY, Goldstein I. Griffonia simplicifolia I: fluorescent tracer for microcirculatory vessels in nonperfused thin muscles and sectioned muscle. Microvasc. Res. 1988;36:199–215. doi: 10.1016/0026-2862(88)90022-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.