

Abstract
Although several lines of evidence support a role for accumulating somatic mitochondrial DNA (mtDNA) mutations in the etiology of aging, it remains unclear if they are a major cause of age-related deterioration and death. Mouse models that harbor elevated mtDNA mutation frequencies age prematurely; these findings were thought to provide conclusive evidence for a causal role of such mutations in aging. Yet, the presence of several conflicting reports has sparked controversy in the field and this is further aggravated by discrepancies in the estimates of mtDNA mutant fractions, which disagree by orders of magnitude. Here, we briefly review the evidence and some of the unresolved questions surrounding a causative role for accumulating mtDNA mutations in aging.
Mitochondria: molecular clocks of aging?
Mitochondria are crucially important cellular organelles: they provide the cell with most of its energy and regulate several signaling cascades, including apoptosis. A central role for mitochondria in aging was first proposed >30 years ago by Denham Harman, based on his original theory that aging is caused by the accumulation of damage resulting from reactive oxygen species (ROS) [1]. ROS are the inevitable by-products of normal cellular processes, most notably, oxidative phosphorylation (which occurs in mitochondria) (Box 1). Harman postulated that as one of the most important sources of ROS, mitochondria should also be one of its most important targets. Hence, as the parts of the cell most vulnerable to ROS, mitochondria could function as an ‘aging clock’. Later, it was realized that errors made during the repair or replication of damaged DNA lead to mutations, which are irreversible changes in the DNA sequence that can vary from single base pair changes to large deletions [2] (Box 1). Mutations in the mitochondrial genome were specifically implicated as the potential causal factor in aging by Linnane and colleagues [3]. A possible role for mutations in aging was corroborated by an observation that inherited pathogenic mitochondrial DNA (mtDNA) mutations cause a variety of deteriorative age-progressive diseases somewhat reminiscent of aging [4]. Since then, progress has been made in at least two major areas. First, a few tissues (i.e. brain, muscle and colon) have been identified in which high local levels of mtDNA mutations make their causal relationship to the aging process more plausible. Second, in an attempt to directly test cause and effect, various mouse models have been generated that harbor increased levels of mtDNA mutations; some models display multiple symptoms of premature aging. Intriguingly, the data from these models have been interpreted in nearly opposite ways, both supporting and rejecting a role for mtDNA mutations in aging. Here, we critically review the current highly dynamic state of the mitochondrial DNA mutation theory of aging.
Box 1. Mitochondria, mtDNA and mtDNA mutations.
Mitochondria are distinct cellular organelles that generate ATP, the energy carrier of the cell, by oxidizing glucose and fatty acids. Several pathways involved in this process converge upon oxidative phosphorylation, a chain of reactions performed by a set of five multi-subunit enzyme complexes called the electron transport chain (ETC) [5].
Mitochondria are unique because they are the only organelles in mammalian cells with their own genome. They harbor a small (~16 000 base pairs in mammals) circular genome (mtDNA) encoding 13 polypeptide subunits scattered among the ETC complexes; all other ETC subunits (~80) are encoded in the nuclear DNA [5], as are the rest of several thousand mitochondrial proteins. The 13 mitochondrially encoded polypeptides are transcribed and translated within the mitochondrion by its own gene expression system [57,58]. For this purpose, mtDNA additionally encodes the full set of 22 tRNAs and two rRNAs for the translation machinery.
Mitochondria are dynamic structures subject to fusion and fission and the rates of these processes can differ between cell types. Thus, a given mtDNA copy does not ‘belong’ to a given mitochondrion, but rather is part of the fluid mitochondrial landscape of a cell [59]. During cell division, mtDNAs distribute between daughter cells and then replicate at random (although only a subpopulation of mtDNAs might be replicating at any given time) to replenish the mtDNA content. In non-dividing cells, mtDNA also replicates to replace those that are lost when old mitochondria are degraded by lysosomes [60].
The mitochondrial genome principally suffers from two types of mutations: point mutations, which are changes of one or a few nucleotides; and large deletions, which involve the removal of large portions of the genome (from a few hundred base pairs to almost the entire genome). The exact sources of mtDNA mutations are a matter of debate, but might be similar to those causing nuclear DNA mutations, that is, errors during DNA damage processing or spontaneous polymerase errors [2].
Do mtDNA mutations cause aging by impeding oxidative phosphorylation?
Because the genes encoded in the mitochondrial genome are required for respiratory function, any pathogenic mutations in mtDNA could induce respiration defects leading to various disease phenotypes. Indeed, maternally inherited pathogenic mutations in human mtDNA can cause disease through respiration defects [5]. Therefore, defects in the electron transport chain (ETC) that impede oxidative phosphorylation are the likely consequence of randomly accumulating detrimental mtDNA mutations in somatic cells (Box 1). Yet, owing to functional complementation among different copies of mtDNA (and between mitochondria [6,7]), a loss-of-function mutation must first reach a threshold level by clonally expanding within a cell (Box 2) before it can cause adverse effects. This idea is in accordance with the observation that ETC defects in aging tissues are typically highly focal, that is, limited to individual cells (in which a clonal expansion presumably occurred), or groups of closely related cells, such as those originating from a stem cell with clonal expansion (Figure 1). Initially, results obtained with aged muscle, brain or heart from both mice and humans indicated very low frequencies of ETC-deficient cells (i.e. too low to substantially affect tissue function) [8]. However, recent work has shown that, in at least some human tissues such as brain, muscle and colon, the cells harboring clonal expansions of mutant mtDNA molecules and resultant respiratory deficient cells might be abundant enough to impact the whole organism functionally.
Box 2. Physiological effects of mtDNA mutations.
The most obvious expected adverse effect of mtDNA mutations is the impediment of ETC function. Depending on the nature of the disrupted gene (structural, tRNA, rRNA), point mutations could directly inactivate a particular ETC polypeptide subunit or cause a translation defect that affects all mtDNA-encoded polypeptides. Deletions typically remove several structural genes and a few tRNA and/or rRNA genes, thus exerting both effects. mtDNA mutations vary from being severely pathogenic mutations to neutral polymorphisms (http://www.mitomap.org). Despite such diversity, a total fraction of somatic mutations in a tissue is a useful measure because a higher total mutant fraction signifies a proportionally higher fraction of all mutations, including the most severe.
Phenotypic threshold
The effects of loss-of-function mtDNA mutations depend on the percent of mutated mtDNA (heteroplasmy level) in a given cell. Typically, almost no effect is observed until heteroplasmy reaches a certain value (i.e. the phenotypic threshold), above which the defect presents in full strength. The steepness of the transition and the value of the threshold vary from mutation to mutation with values typically reported from 60 to 95%. Apparently, the threshold exists because mitochondria share their components via fusion and fission [7] and because most components are present in some excess. Thus, small amounts of a defective component encoded by a low frequency mutant mtDNA will be compensated by the shared access of the normal component. As the fraction of mutant DNA increases via clonal expansion, the shared excess of the normal component is exhausted in all mitochondria at once, causing an abrupt ETC shutdown [6,61].
Clonal expansions of mtDNA mutations
Any somatic mutation initially occurs in a single DNA molecule and thus exists below the physiological threshold. Somewhat paradoxically, it is common for somatic mitochondrial mutations (including non-detrimental silent mutations) to clonally expand within cells, (the progeny of a single mutant molecule is favored over the non-mutants and eventually comprises a large portion or even the entire population of cellular mtDNA molecules). Clonal mtDNA mutation expansions can extend beyond the single cell level, for example, in colonic crypts fed by a stem cell with a clonal expansion [22], or in tumors originating from progenitor cells with clonal expansions [62]. The mechanisms of expansion remain a matter of debate. Expansion could be driven by random genetic drift within intracellular populations [62,63]. Alternatively, mutations might have a selective advantage; this has been convincingly shown for deletions [15]. Thus, clonally expanding detrimental mutations ultimately might exceed the phenotypic threshold, resulting in focal ETC defects (Figure 1). It should be noted that this review deals primarily with the overall accumulation of mutations, without distinguishing between accumulation of de novo mutations and clonal expansion of pre-existing mutations. The relative importance of these two processes awaits rigorous exploration.
Figure 1.
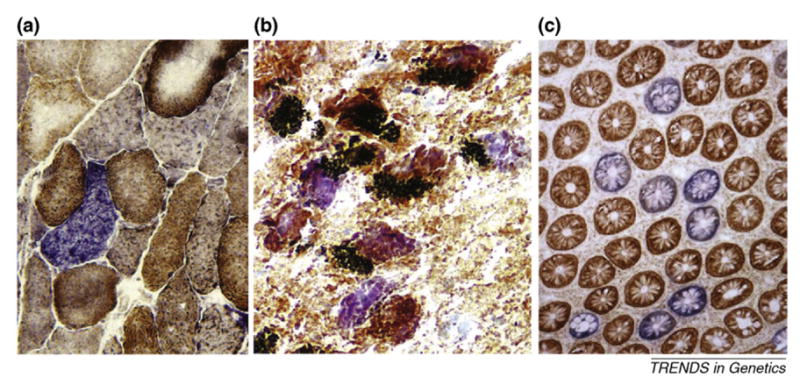
Focal mitochondrial deficiency: a signature of old age. Clonal expansions of mtDNA mutations that cause focal cellular defects in the ETC present similar patterns in various aged human tissues. (a) Muscle cut across the fibers (courtesy of J. Aiken; http://www.biomedexperts.com). (b) Substantia nigra in which black neuromelanin accumulates in pigmented neurons. (c) Colon cut across the crypts. Each round body is a cross-section of a crypt fed by a single stem cell (courtesy of D. Turnbull; http://www.ncl.ac.uk). All samples were stained for the activity of cytochrome c oxidase, a mtDNA-encoded enzyme (brown) and counterstained for succinate dehydrogenase, a nuclear-encoded enzyme (muscle and colon) or a neuron-specific dye, cresyl violet, in substantia nigra. Thus, the blue color indicates diminished cytochrome c oxidase activity (i.e. an ETC-deficient cell). Mutational analysis of individual cells reveals that, in most cases, ETC defects result from the clonal expansion of detrimental mtDNA mutations – deletions in brain and muscle and point mutations in colon.
Muscle
Muscle atrophy, or sarcopenia, is a serious human age-related health problem in which up to 40% of muscle mass disappears by age 80. Muscle was also one of the first tissues in which an accumulation of mtDNA deletions with age was reported [9]. The fraction of such deleted mtDNA molecules in aged muscle is ~0.1%, which at first glance seems too low to be of any importance for the dramatic age-dependent muscle loss observed in aged humans and animals. The deletions, however, are not uniformly distributed; instead, they are clonally expanded in short segments of muscle fibers. The local mutant fraction in these segments approaches 100%, resulting in focal ETC deficiency [10,11] (Figure 1a). Notably, mtDNA deletions are nearly absent outside these ETC-deficient segments. Because each deletion-rich segment occupies only a very short portion (e.g. ~1/300) of the overall fiber length, the average deletion fraction of 0.1% signifies that approximately one-third (0.1% × 300 = 1/3) of all fibers could harbor an ETC-defective segment somewhere along their length [11]. Given that an ETC-defective segment eventually undergoes local fiber degeneration and splitting [12], these seemingly rare deletions seem to be potentially capable of causing substantial muscle fiber loss, assuming that local fiber splitting ultimately leads to whole fiber degeneration [12].
The possibility that mtDNA deletions cause adverse effects in aging muscle is corroborated by the observation that individuals with a mildly increased frequency of mtDNA deletions in muscle typically present a muscle wasting phenotype [13]. By contrast, the idea seems to be refuted by results obtained with mito-mice (i.e. mice that were generated by introducing various amounts of mtDNA with a defined 5-kb deletion into zygotes) in which deleted mtDNA is uniformly distributed among tissues and is transmitted to the offspring [14]. For example, a mito-mouse with 5% deleted mtDNA at birth lived >130 weeks and showed no signs of disease despite the fact that, at the end of life, mtDNA deletion load in its tissue increased to 30–40% [15]. This amount is over an order of magnitude greater than the ~0.1% observed in normal aged mouse or human muscle, which seems to contradict the idea that mtDNA deletions cause sarcopenia. It should be noted, however, that the distribution of deleted mtDNA in mito-mice is very different from that in normally aged mice (or humans, for that matter). In contrast to highly punctuated distribution of deletions observed in normal aging, the deleted mtDNAs in mito-mice are uniformly distributed among and along fibers. Essentially all fibers contain similar fractions of deleted mtDNA and no segments harbor highly concentrated deletions. Moreover, in contrast to a normal mouse, in which ETC defects are present at overall fractions of deletions of 0.1%, mito-mouse muscle remains free of ETC defects until the overall fraction of deleted mtDNA exceeds ~80% [6]. Owing to these differences, evidence derived from mito-mice that argues against a causal role of mtDNA deletions in aging deserves caution.
Substantia nigra
For many years, the substantia nigra of the midbrain, the target site of lesions that cause Parkinson’s disease in humans, was thought to sustain particularly high mtDNA mutation levels compared with other cell types [16]. Single cell analysis of mtDNA (Box 3) demonstrated [17,18] that mtDNA deletions in individual pigmented neurons of substantia nigra are clonal, with the total fraction of deleted mtDNA approaching 80% in cells from very old individuals. Moreover, the deleted mtDNA fraction was found to be significantly higher in cytochrome c oxidase (COX)-deficient neurons [18] than in COX-positive neurons, indicating that mtDNA deletions might be directly responsible for impaired cellular respiration (Figure 1b). It is tempting to speculate that abundant ETC defects in pigmented neurons cause some age-related degenerative process, such as ‘mild parkinsonian signs’, a syndrome characterized by slowness, tremor and gait problems, which is present in many people over 85. However, it remains unknown whether the ETC defects observed in old pigmented neurons are sufficient to disrupt their activity to the extent that would cause parkinsonian signs or any other age-related health problems. Interestingly, a mouse chimera with a mosaic of ETC-positive and -negative neurons (owing to a nuclear DNA mutation) presents neurological symptoms when the amount of ETC-negative neurons in the forebrain approaches 20% [19], indicating that ETC defects in human substantia nigra at old age (with up to 30% ETC-defective cells [20]) could have a functional impact.
Box 3. Measuring mtDNA mutations.
The main challenge in analyzing somatic mutations is the low frequency of each mutation. Unfortunately, mutation frequencies are usually in the same range as the artificial mutations caused by the procedures involved in the measurement process, most notably, by PCR. There are several approaches to avoid, or to control for, such artifacts. To avoid PCR errors altogether, it is possible to clone mtDNA directly, followed by sequencing a large number of clones to establish the mutation frequency [64]. This procedure apparently introduces no artificial mutations but is unpopular for being too laborious. Alternatively, it is possible to avoid PCR errors by analyzing individual cells rather than tissue homogenates [21]. This approach circumvents the problem of the low frequency of somatic mutations by taking advantage of intracellular mutational clonal expansion (Box 2). By definition, in a cell with a clonal expansion (or in a cluster of such cells, e.g. a mutant colonic crypt [22]), a mutation constitutes ~10% of the total mtDNA copies in a cell or higher, that is, frequencies at which mutations can be measured by conventional approaches including direct sequencing. Artificial mutations do not expand clonally; thus, they arise at the low levels typical of these artifacts in tissue homogenates (usually in the order of 10−4) and, therefore, are not detectable (and hence do not interfere with conventional methods). A third possibility to avoid PCR errors involves the removal of all wild-type copies of the target sequence before PCR, for example, by restriction digestion of wild-type template, as in the random mutation capture (RMC) assay [65], or by separation of the wild type from mutant molecules by partially denaturing electrophoresis (PDE) [52]. Because most artificial mutations are created on wild-type molecules, removal of the latter greatly reduces the artificial mutation rate. Most of the currently used protocols do not eliminate PCR errors, but rather limit their occurrence by using high-fidelity polymerases in PCR-cloning [66]. To account for PCR errors in PCR-cloning approaches, the clones (used merely as mutation-free ‘control’ DNA) are PCR amplified in a repeat reaction and when no (or substantially fewer) mutations are found in these secondary clones than in primary clones, it is assumed that the mutations found in the primary PCR amplicons are mostly real.
Dividing tissues: mtDNA mutations in stem cells
Age-related accumulation of mtDNA mutations to high levels have been found mainly in non-dividing tissues. However, mitotically active tissues can also harbor cells with large proportions of clonally expanded somatic point mutations (but apparently not deletions) [21]. In a series of elegant experiments, Taylor et al. [22] showed that a high proportion of human colonic crypts are fed by stem cells with clonally expanded mtDNA point mutations. Furthermore, they demonstrated that ~15% of colonic crypts are ETC defective by the age of 80 (Figure 1c). A majority of ETC-deficient crypts were found to contain expanded point mutations that could reasonably account for the defect [22]. Although it is alarming that 15% of our colon is apparently deficient for a vital cellular function by old age, it is still unknown whether these defects are of any importance in the aging process.
Thus, there is little doubt that naturally accumulating mtDNA mutations in tissues of humans and rodents can cause adverse effects (i.e. focal cellular ETC defects), which makes mtDNA mutation accumulation an attractive candidate for a universal, primary aging mechanism. However, we still do not know if the observed mtDNA-driven defects cause any age-related phenotypes.
Mitochondrial mutator mice: testing the theory?
Ideally, to establish a causal relationship between mtDNA mutations and aging, we would need to decrease the natural mtDNA mutation frequency, which should then slow aging. This has been attempted, albeit indirectly, through a reduction in ROS production, which is widely considered to be an important cause of both aging and mtDNA mutation accumulation. The frequency of mtDNA point mutations is decreased in mCAT mice [23], that is, transgenic mice that overexpress a mitochondrially localized version of the human ROS-scavenging enzyme, catalase [24]. Similarly, the frequency of clonally expanded mtDNA deletions is decreased in rats subjected to calorie restriction [25], which is underfeeding without malnutrition. This promotes increased longevity across many animal species because of metabolic changes associated with reduced ROS production and/or increased antioxidant defense. However, both interventions can have many benefits and the increased lifespan in these animals does not necessarily result from reduced mtDNA mutation rates. Thus far, it has not been possible to specifically decrease mtDNA mutation frequency. Instead, it has been possible to do the reverse, that is, to increase mtDNA mutation rates in various organs and tissues of the mouse through genetic manipulation. Intriguingly, this approach resulted not only in an increase in the fraction of mtDNA mutants and in the frequency of cells with ETC defects, but also in the premature appearance of multiple symptoms reminiscent of aging and a shorter life span. However, the interpretation of these results is not always as straightforward as originally anticipated; indeed, these results have led to as many questions as answers.
Does premature aging in mtDNA mutator mice imply that mtDNA mutations cause normal aging?
If mtDNA mutations cause any of the deteriorative processes of normal aging, then an increased mutation rate should result in premature aging and reduced life span. To test this hypothesis, a series of ‘mutator’ mice, that is, mice with increased mtDNA mutation rates (both point mutations and deletions), were constructed. In these mice, mitochondrial DNA polymerase γ (Polg), which is required for mtDNA replication, was rendered error-prone by introducing deleterious amino acid changes that impede its proofreading 3′–5′ exonuclease activity. We refer to the error-prone (‘mutator’) mitochondrial polymerase variants used in these models collectively as ‘Polgmut’. In the tissue-specific mutator mice, a Polgmut transgene was expressed in the heart [26], neurons [27] or in pancreatic β-cells [28]. By contrast, in the so-called whole body mutator mice, an error-prone Polgmut operates in all tissues. In this knock-in model, either one or both alleles of the native Polg are replaced in the mouse germline by Polgmut [29,30].
Reassuringly, mice with tissue-specific expression of Polgmut displayed increased mtDNA mutation accumulation in the respective tissues, which was accompanied by accelerated age-dependent deterioration, for example, severe cardiomyopathy and β-cell death [26,28]. Excessive accumulation of mtDNA mutations was not associated with increased markers of oxidative stress [31] (as might be expected if ROS were the driver), but rather with markers of apoptosis [32]. This finding challenges the hypothesis that the ETC defects caused by increased mtDNA mutations would lead to increased ROS production, which in turn would cause more mtDNA mutations (i.e. vicious cycle hypothesis [33]). As a note of caution, transgenic experiments are commonly prone to artifacts. It remains possible that the phenotype is non-specifically caused by increased levels of Polg protein rather than mtDNA mutations, as shown for a green fluorescent protein (GFP) heart-specific transgene, which drives cardiomyopathy in a dose-dependent manner [34]. In support of a causative role for mtDNA mutations in tissue deterioration, a similarly constructed wild-type Polg+ transgene did not result in cardiomyopathy; however, Polg protein levels were not compared between the mutant and wild-type transgenic lines [35].
In keeping with expectations, the whole-body mutator mice (i.e. the homozygous Polgmut/mut knock-in mice) showed increased mtDNA mutation frequencies in all tissues accompanied by multiple progressive symptoms of premature aging, including dilated cardiomyopathy, thymic involution, testicular atrophy, sarcopenia, loss of bone mass, loss of intestinal crypts and hearing loss [29,30,36]. Similarly to the heart-specific mutator mice, the whole-body mutators did not show signs of increased oxidative stress, but instead displayed evidence of increased apoptosis.
Taken at face value, these results seem to support a causal role for mtDNA mutations in normal aging, possibly through apoptosis. However, the level of mtDNA mutations in tissues from these mice, ~10–20 × 10−4 mutations per base pair (bp) depending on tissue and method of measurement, was approximately one to two orders of magnitude higher than in normally aged mice or humans (depending on which mutant fraction estimates are used; Box 3). In spite of this high mutation load, which was largely established very early in life, these animals were able to live for >1 year [30].
Heterozygous mutator mice
Interestingly, whereas homozygous Polgmut/mut knock-in mice age prematurely, their heterozygous Polgmut/+ littermates seem to age normally in spite of a substantially higher mtDNA mutation load [23]. Indeed, although initial reports showed no evidence for increased mutation frequency in the heterozygotes, subsequent work using a new method known as random mutation capture (RMC) analysis (Box 3) indicated that the point mutation frequency (1.6–3.3 × 10−4 mutations per bp) was ~200–500-fold that of age-matched young wild-type control animals, and ~30-fold that of old normal mice. However, caution is warranted when considering the data obtained through different methods (Box 4). Because the mutation frequencies in the heterozygotes were ~5–10 times lower than in the homozygous mutator mice, we would call them ‘mild mutator mice’. The difference in mutation frequencies measured by the RMC method compared with the levels given in the original reports resides in the much lower mutation frequencies now observed in the wild-type animals. Indeed, the originally reported levels could possibly represent artifacts owing to the use of PCR (Box 3). This difference would vastly overestimate normal mtDNA mutation frequencies. Of note, this might not necessarily be true for all mtDNA mutation frequencies reported; indeed, some reports are not based on PCR, whereas others included thorough controls (Box 3,4).
Box 4. Different methods – different numbers.
Strikingly, estimates of mtDNA point mutant fractions (Table 1) vary over almost three orders of magnitude. In some cases, differences might be potentially explained by differences between ages, species and tissues but even estimates made in one particular species, age and tissue, such as young mouse brain (Table 1, bold typeface), vary by over two orders of magnitude. Interestingly, methods that are supposedly less prone to artifacts (e.g. RMC, single cell analysis, direct cloning) yield lower estimates, indicating that artifacts might lead to higher estimates. However, all high estimate studies include reasonable controls (Box 3). The caveat exists, however, that some of the artificial mutations might have originated not from spontaneous error of the thermostable DNA polymerase, but from conversion of DNA-damaged nucleotides into mutations by the same polymerase. Because control DNA used in PCR-cloning experiments might not bear the same chemical damage as the original sample DNA, artificial mutations at damaged nucleotides might go unaccounted for. By contrast, the RMC method could potentially underestimate mutations because it uses short target sequences (just 4 bp long) and, thus, might conceivably miss mutational hotspots (i.e. sites highly susceptible to mutation) and clonally expanded mutations if they happen to be located outside the target sequence. Furthermore, RMC estimates, unlike those from the cloning procedures, rely on DNA quantification, which could cause additional uncertainty. The discrepancies in the results call for cross-evaluation of the different methodologies. Unfortunately, the use of different methods to estimate mutant fractions across tissues, ages and species, make it impossible to determine whether discrepancies arise from methodological problems or natural variation in mutation levels. We believe that the systematic use of a combination of diverse methods would help to resolve the issue.
Intriguingly, these new results, using a presumably superior method of mutation analysis, seem to demonstrate that mtDNA point mutations cannot cause normal aging; otherwise a 200–500-fold increase would probably accelerate this process and result in a markedly shorter life span of the mild mutator mouse. Because this is not the case, it was concluded that mtDNA point mutations do not limit the lifespan of wild-type mice [23]. Although this is indeed the simplest explanation, it should be noted that the time course of mtDNA mutation accumulation, and possibly the type of mutations and their tissue distribution, is very different between normal and mutator mice. Whereas the proofreading defect in knock-in mutator mice gives rise to increased mtDNA mutagenesis approximately from day 7 post conception, when mtDNA synthesis begins in the developing embryo, mtDNA mutations in an aging animal would presumably be driven by factors such as ROS production [23], which is likely to be time- and tissue-dependent. Hence, increased mutation levels in Polgmut/+ mice might not induce premature aging because they do not affect the right cells at the right time. Moreover, the heart-specific mutator mouse developed severe cardiomyopathy at ~1.5 × 10−4 mutations per nucleotide [35] (a frequency similar to that found in the healthy whole body Polgmut/+ mice). One possible explanation for this discrepancy is that the time course of age-related mutation accumulation in the heart-specific mouse model differs from that in the Polgmut/+ knock-in and might be more similar to normal aging. Other reports indicate that mtDNA mutation fractions, in aged mice and humans, number up to several point mutations per genome (Box 4; Table 1), which is close to the levels reported for the heart-specific mutator mouse. Hence, it is too early to rule out a causal role of mtDNA point mutations in aging. What about mtDNA deletions?
Table 1.
Levels of mtDNA mutations as estimated by different methods
| Species | Tissue(s) | Agea | Method | Mutations per 104 | Refs |
|---|---|---|---|---|---|
| Mouse | Heart | 2 | RMC | 0.006 | [23] |
| Mouseb | Brain | 6 | RMC | 0.01 | [23] |
| Human | Oocyte | 0 | Single cell | 0.03 | [51] |
| Human | Lung | 1 | PDE | 0.03 | [52] |
| Mouse | Brain | 6 | Direct cloning | 0.04 | [53] |
| Mouse | Heart | 30 | RMC | 0.05 | [23] |
| Mouse | Various | 1 | Direct cloning | 0.06 | [35] |
| Mouse | Brain | 30 | RMC | 0.1 | [23] |
| Mouse | Muscle | 6 | Direct cloning | 0.1 | [53] |
| Human | Lung | 55 | PDE | 0.1 | [52] |
| Human | Colon | 60 | Single cell | 0.2 | [22] |
| Rat | Muscle | 6 | PCR-cloning | 0.3 | [50] |
| Rat | Muscle | 24 | PCR-cloning | 0.4 | [50] |
| Human | Muscle | 55 | PCR-cloning | 0.6 | [54] |
| Human | Brain | 75 | PCR-cloning | 0.9 | [55] |
| Human | Neurons | 70 | PCR-cloning | 2 | [56] |
| Murine | Brain | 6 | PCR-cloning | 2 | [47,50] |
| Mouse | Brain | 10 | PCR-cloning | 5 | [47] |
| Human | Brain | 75 | PCR-cloning (D-loop) | 6 | [55] |
Average age in a study given in months for mice and rats and in years for humans. Note that life expectancy for laboratory mice and rats is ~3 years.
Young mouse brain, which is the type of tissue investigated by all three methods, is given in bold typeface to facilitate comparison.
Is premature aging in mutator mice driven by mtDNA deletions?
In addition to point mutations, mtDNA Polgmut/mut mutator mice bear an excess of mtDNA deletions [35]. Interestingly, a variation of the RMC method that is capable of measuring mtDNA deletions showed that heterozygous Polgmut/+ mild mutator mice harbor normal mtDNA deletion levels in heart and brain. By contrast, the prematurely aging Polgmut/mut mice showed an increase by one or two orders of magnitude, depending on the part of the mitochondrial genome that was analyzed [37]. Is it possible to conclude, following Vermulst et al. [37], that premature aging in mutator mice (or normal aging, for that matter) is driven by mtDNA deletions? Such a conclusion seems premature for at least two reasons. First, the 5–10 fold increase in point mutations, from 2–5 mutations per mitochondrial genome in mild mutator mice to 20–30 in the homozygous mutator mice, probably contributes to the premature aging symptoms observed in the latter. Of course, not all mtDNA mutations are pathogenic, but it is unlikely that none out of 20–30 are detrimental. Point mutations were indeed shown to cause isolated ETC defects in mild mutator duodenum [37], so they are the most likely cause of these defects in homozygous mutator mice because they are present at higher fractions in this model. The second reason for pause in considering mtDNA deletions as a cause of premature aging stems from the aforementioned data obtained from the mito-mouse, which indicate that mtDNA deletions might be benign even at very high fractions [6,15].
Is premature aging in mtDNA mutator mice caused by ETC-deficiency-driven cell death?
Whether point mutations or deletions, it is crucially important to know exactly how an excess of mtDNA mutations results in premature or normal aging. As mentioned earlier, the premature aging phenotype in the Polgmut/mut mice might be driven by accelerated cell death in multiple tissues [30,32]. In this respect, clonal expansion of mtDNA mutations might lead to ETC deficiency, which in turn could promote apoptosis through mitochondrial permeability transition [30]. Indeed, when excessive ETC defects are generated in the absence of mtDNA mutations, for example, by genetically engineered mtDNA depletion, phenotypes similar to those seen in mutator mice are observed, including dilated cardiomyopathy [38,39], loss of pancreatic β-cells [40] and neuronal loss [19,41,42]. Yet, ETC deficiency does not necessarily cause apoptosis. For example, ETC-deficient stem cells that feed ETC-deficient colonic crypts can survive for many generations [43]. Furthermore, ETC defects apparently are not required to induce apoptosis in mutator mice: increased apoptosis in the heart-specific mutator mice is not associated with any substantial ETC deficiency [32]. Thus, although ETC defects or apoptosis caused by ETC defects probably contribute to normal or premature aging, these are not necessarily the only mechanisms that drive premature aging in mtDNA mutator mice.
Dominant lethal mutations and aging
The marked apoptosis observed in the heart of the heart-specific mtDNA mutator mice in the absence of ETC deficiency implies that mtDNA mutations might cause apoptosis without inducing ETC deficiency. Apoptosis in these mice has been proposed to result from rare (presumably occurring once in ~20 000 conventional mutations) but deadly ‘dominant lethal mutations’ [44]. These mutations are postulated to kill a cell even when present at low number, by altering a mtDNA-encoded peptide into an apoptotic signaling molecule [44]. We note that the same effect could be achieved by exerting any other strong ‘toxic’ effect. For example, it is conceivable that a mutation might act by sharply increasing local mitochondrial ROS production. Indeed, ROS-mediated mitochondrial damage can facilitate apoptotic induction via cytochrome c release [45]. Regardless of the mechanism, the concept of dominant lethal mutations is attractive because it additionally explains why heart-specific mutator mice present a severe phenotype while the whole body heterozygous mutators are healthy despite their similar mutational loads. In the whole-body mutators, either homo- or heterozygous, excessive mutational generation [44] should begin around day 7 post conception, when mtDNA synthesis is resumed after a pause [46]; by day 13.5, approximately half the total mutations observed in the adult have accumulated [47]. By contrast, in the cardiac-specific mutator mouse, excessive mutations are generated only after birth (owing to the transgene promoter) [35]. Because both models bear similar mutant fractions at two months of age, the post-natal mutational rate (i.e. change in mutant fraction over time) in the heart-specific mutator should be much higher than in the heterozygous mutator. This prediction is in agreement with the observation that the post-natal mutation rate in the heart-specific mutator mice (i.e. approximately 10−4 per month) [35] is similar to that observed in the homozygous whole body mutator [47], because mtDNA is replicated by a proofreading-deficient polymerase in both models.
The difference in time of onset of mtDNA mutation generation in a mutator mouse could enable mutations to affect the organism differentially. Whereas in the whole body mutator, excess mutations arise mostly during rapid mtDNA replication in the growing embryo, in the cardiac-specific mutator, the promoter of the transgene dictates excess mutagenesis no sooner than after birth, when cell proliferation in the heart has stopped and switched to hypertrophic growth of non-dividing cardiomyocytes. Mature cardiomyocytes contain far more mtDNA copies than small, embryonic cells and, therefore, according to the argument by Zassenhaus and colleagues, have a proportionally higher chance to acquire a lethal mutation (assuming similar mutant frequencies per mtDNA copy) [44]. Therefore, small embryonic cells are expected to be much less prone to killing than large cardiomyocytes; moreover, those killed are probably more easily replaced than the post-mitotic cardiomyocytes. Furthermore, excess mtDNA mutations might induce an anti-apoptotic response [44,48] that protects the heterozygous mutator cardiomyocytes, but comes too late in the heart-specific mutator. However attractive, the dominant lethal mutation model has several drawbacks: the molecular mechanism(s) of how such mutations might kill cells is yet to be elucidated and the observed phenotypes in the heart-specific Polgmut transgenic mice might be artifacts of an overexpressed transgene (see earlier). Yet, this less-known model has enough merit to warrant consideration along with other models in the field.
Could dominant lethal mutations have a role in normal aging? The likelihood of lethal mutations should increase with overall mutation fraction – in a stochastic fashion –and their impact should be higher for those generated later in life. Thus, the answer depends on the actual mutant fractions and the kinetics of mutant accumulation during normal aging. If mtDNA mutation levels in normal adults are ~10−4, as in heart-specific mutator mice, and accumulate late in life, then dominant lethal mutations could have a role in normal aging. Unfortunately, the data for normal mtDNA mutations remain unclear. According to the RMC assay, mutations in normal mice do accumulate late in life, but reach only ~10−5 mutations per bp [23], whereas PCR-cloning approaches report >10−4 mutations per bp (Box 4; Table 1). Yet, the existence of tissue-wide clonal expansions of somatic mtDNA mutations indicates that a substantial fraction of mutations in aged humans might originate early in development [49,50].
Concluding remarks and future perspectives
Despite decades of research and recent advances in generating mouse models with increased mutational loads, the study of mitochondrial DNA mutations in aging still has not reached a stage at which clear, definitive conclusions can be drawn regarding causal relationships. Although multiple elegant and plausible hypotheses have been formulated (Figure 2), the functional relevance of mtDNA mutation accumulation in human and mouse aging, or even in mutator mouse premature aging, remains unclear (Box 5). Testing these hypotheses is limited largely by the lack of sufficiently reliable and detailed (both spatially and temporally) information about mutant fractions in the various species and model systems. New, sensitive methods capable of accurately measuring a broad range of mtDNA mutations in single cells are necessary to eliminate the devils in the details.
Figure 2.

MtDNA mutations and the aging process: a summary of hypotheses. Whereas all of these hypotheses are plausible, it is not clear which pathway(s), if any, are important for normal aging. More research, in particular detailed measurements of mutant fractions in normal and mutator mice and in humans, is necessary to evaluate these diverse hypotheses. The hypotheses do not need to be mutually exclusive; in fact, it is quite possible that different types of mutations have different roles in various cell types.
Box 5. Outstanding questions.
What are the actual fractions of mtDNA mutations?
What are the reasons for current discrepancies in mutant fraction estimates?
What are the kinetics of mutant fraction accumulation with age?
What mechanisms underlie premature aging in mtDNA mutator mice and how relevant are they to normal aging in mice and humans?
Acknowledgments
The authors are grateful to Nils Larsson and three anonymous reviewers for criticisms and suggestions, which improved the manuscript and to Jun-Ichi Hayashi, Doug Turnbull and Judd Aiken for helpful discussions. This work was supported in part by grants from the NIH: AG17242 and AG20438 to J.V., and NS058988 and AG19787 to K.K., and by a grant from the Ellison Medical Foundation to J.V. and from the United Mitochondrial Disease Foundation to K.K.
References
- 1.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Vijg J. Aging of the Genome. Oxford University Press; 2007. [Google Scholar]
- 3.Linnane AW, et al. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 5.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 6.Nakada K, et al. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- 7.Ono T, et al. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs HT. The mitochondrial theory of aging: dead or alive? Aging Cell. 2003;2:11–17. doi: 10.1046/j.1474-9728.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 9.Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18:6927–6933. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wanagat J, et al. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001;15:322–332. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- 11.Bua E, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herbst A, et al. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnston W, et al. Late-onset mitochondrial myopathy. Ann Neurol. 1995;37:16–23. doi: 10.1002/ana.410370106. [DOI] [PubMed] [Google Scholar]
- 14.Inoue K, et al. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- 15.Sato A, et al. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci U S A. 2005;102:16765–16770. doi: 10.1073/pnas.0506197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soong NW, et al. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet. 1992;2:318–323. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- 17.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 18.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 19.Dufour E, et al. Age-associated mosaic respiratory chain deficiency causes trans-neuronal degeneration. Hum Mol Genet. 2008;17:1418–1426. doi: 10.1093/hmg/ddn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh K, et al. Cytochrome c oxidase defects of the human substantia nigra in normal aging. Neurobiol Aging. 1996;17:843–848. doi: 10.1016/s0197-4580(96)00168-6. [DOI] [PubMed] [Google Scholar]
- 21.Nekhaeva E, et al. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. Proc Natl Acad Sci U S A. 2002;99:5521–5526. doi: 10.1073/pnas.072670199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor RW, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112:1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermulst M, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 24.Schriner SE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 25.Bua E, et al. Calorie restriction limits the generation but not the progression of mitochondrial abnormalities in aging skeletal muscle. FASEB J. 2004;18:582–584. doi: 10.1096/fj.03-0668fje. [DOI] [PubMed] [Google Scholar]
- 26.Mott JL, et al. Low frequencies of mitochondrial DNA mutations cause cardiac disease in the mouse. Ann N Y Acad Sci. 1999;893:353–357. doi: 10.1111/j.1749-6632.1999.tb07853.x. [DOI] [PubMed] [Google Scholar]
- 27.Kasahara T, et al. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006;11:577–593. doi: 10.1038/sj.mp.4001824. [DOI] [PubMed] [Google Scholar]
- 28.Bensch KG, et al. A transgenic model to study the pathogenesis of somatic mtDNA mutation accumulation in β-cells. Diabetes Obes Metab. 2007;9 (Suppl 2):74–80. doi: 10.1111/j.1463-1326.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- 29.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 30.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 31.Mott JL, et al. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474:35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, et al. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res. 2003;57:147–157. doi: 10.1016/s0008-6363(02)00695-8. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka M, et al. Accumulation of deletions and point mutations in mitochondrial genome in degenerative diseases. Ann N Y Acad Sci. 1996;786:102–111. doi: 10.1111/j.1749-6632.1996.tb39055.x. [DOI] [PubMed] [Google Scholar]
- 34.Huang WY, et al. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, et al. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- 36.Niu X, et al. Somatic mtDNA mutations cause progressive hearing loss in the mouse. Exp Cell Res. 2007;313:3924–3934. doi: 10.1016/j.yexcr.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 37.Vermulst M, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 38.Park CB, et al. MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell. 2007;130:273–285. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 39.Hansson A, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva JP, et al. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 41.Sorensen L, et al. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci. 2001;21:8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekstrand MI, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greaves LC, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci U S A. 2006;103:714–719. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubec SJ, et al. Mitochondrial DNA mutations may contribute to aging via cell death caused by peptides that induce cytochrome c release. Rejuvenation Res. 2008;11:611–619. doi: 10.1089/rej.2007.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orrenius S, et al. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 46.Cree LM, et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 47.Trifunovic A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D, et al. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol. 2005;288:H2476–H2483. doi: 10.1152/ajpheart.00670.2004. [DOI] [PubMed] [Google Scholar]
- 49.Khrapko K, et al. Clonal expansions of mitochondrial genomes: implications for in vivo mutational spectra. Mutat Res. 2003;522:13–19. doi: 10.1016/s0027-5107(02)00306-8. [DOI] [PubMed] [Google Scholar]
- 50.Khaidakov M, et al. Contribution of de novo point mutations to the overall mutational burden in mitochondrial DNA of adult rats. Exp Gerontol. 2005;40:396–402. doi: 10.1016/j.exger.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs L, et al. mtDNA point mutations are present at various levels of heteroplasmy in human oocytes. Mol Hum Reprod. 2007;13:149–154. doi: 10.1093/molehr/gal112. [DOI] [PubMed] [Google Scholar]
- 52.Coller HA, et al. Clustering of mutant mitochondrial DNA copies suggests stem cells are common in human bronchial epithelium. Mutat Res. 2005;578:256–271. doi: 10.1016/j.mrfmmm.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 53.Sato A, et al. Rare creation of recombinant mtDNA haplotypes in mammalian tissues. Proc Natl Acad Sci U S A. 2005;102:6057–6062. doi: 10.1073/pnas.0408666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wanrooij S, et al. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–3064. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smigrodzki R, et al. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol Aging. 2004;25:1273–1281. doi: 10.1016/j.neurobiolaging.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 56.Cantuti-Castelvetri I, et al. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol Aging. 2005;26:1343–1355. doi: 10.1016/j.neurobiolaging.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Falkenberg M, et al. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 58.Rorbach J, et al. How do mammalian mitochondria synthesize proteins? Biochem Soc Trans. 2007;35:1290–1291. doi: 10.1042/BST0351290. [DOI] [PubMed] [Google Scholar]
- 59.Legros F, et al. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 60.Terman A, et al. Autophagy, organelles and ageing. J Pathol. 2007;211:134–143. doi: 10.1002/path.2094. [DOI] [PubMed] [Google Scholar]
- 61.Yoneda M, et al. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol. 1994;14:2699–2712. doi: 10.1128/mcb.14.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coller HA, et al. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001;28:147–150. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- 63.Elson JL, et al. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Monnat RJ, Jr, Loeb LA. Nucleotide sequence preservation of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1985;82:2895–2899. doi: 10.1073/pnas.82.9.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bielas JH, Loeb LA. Quantification of random genomic mutations. Nat Methods. 2005;2:285–290. doi: 10.1038/nmeth751. [DOI] [PubMed] [Google Scholar]
- 66.Simon DK, et al. Low mutational burden of individual acquired mitochondrial DNA mutations in brain. Genomics. 2001;73:113–116. doi: 10.1006/geno.2001.6515. [DOI] [PubMed] [Google Scholar]