

Summary
Oncogenic KIT or PDGFRA receptor tyrosine kinase mutations are compelling therapeutic targets in gastrointestinal stromal tumors (GISTs), and the KIT/PDGFRA kinase inhibitor, imatinib, is standard of care for patients with metastatic GIST. However, most of these patients eventually develop clinical resistance to imatinib and other KIT/PDGFRA kinase inhibitors and there is an urgent need to identify novel therapeutic strategies. We reported previously that protein kinase C theta (PKCθ) is activated in GIST, irrespective of KIT or PDGFRA mutational status, and is expressed at levels unprecedented in other mesenchymal tumors, therefore serving as a diagnostic marker of GIST. Herein, we characterize PKCθ biological functions in imatinib-sensitive and imatinib-resistant GISTs, showing that lentivirus-mediated PKCθ knockdown is accompanied by inhibition of KIT expression in three KIT+/ PKCθ+ GIST cell lines, but not in a comparator KIT+/PKCθ- Ewing sarcoma cell line. PKCθ knockdown in the KIT+ GISTs was associated with inhibition of the PI3-K/AKT signaling pathway, upregulation of the cyclin-dependent kinase inhibitors p21 and p27, anti-proliferative effects due to G1 arrest, and induction of apoptosis, comparable to the effects seen after direct knockdown of KIT expression by KIT shRNA. These novel findings highlight that PKCθ warrants clinical evaluation as a potential therapeutic target in GISTs, including those cases containing mutations which confer resistance to KIT/PDGFRA kinase inhibitors.
Keywords: PRKCQ, KIT, shRNA, gastrointestinal neoplasm, sarcoma, imatinib
Introduction
Tumorigenesis is a complex, multi-step process, and oncogenic tyrosine kinase proteins play key roles in development of many human cancers (Gschwind et al. 2004). Ligand-mediated receptor tyrosine kinase (RTK) activation can regulate proliferation, survival, migration, and invasiveness in non-neoplastic cells, whereas oncogenic RTK mutations can induce constitutive kinase activation, and thereby enhance proliferation and survival in cancer cells (Drevs et al. 2003; Medinger and Drevs 2005). Mutant RTKs are useful therapeutic targets, as shown by the clinical successes of kinase inhibitor therapies in chronic myeloproliferative disorders, metastatic breast cancer, gastrointestinal stromal tumor (GIST), and non-small-cell lung cancer (Gschwind et al. 2004; Paez et al. 2004). GISTs arise from transformed progenitor cells committed to differentiation along the interstitial cell of Cajal lineage and are the most common mesenchymal tumors of the gastrointestinal tract (Fletcher et al. 2002). GISTs are heterogeneous histologically, including spindle cell (70%), epithelioid cell (20%) and mixed types (10%) (Corless et al. 2004). Most GISTs contain oncogenic gain-of-function RTK mutations, involving KIT (∼85%) or PDGFRA (∼5%), which are accompanied by strong expression of the protein products of these oncogenes (Heinrich et al. 2003; Hirota et al. 1998; Corless et al. 2004). KIT oncoproteins remain crucial to the proliferation and survival of GIST cells in patients with metastatic GIST, as evidenced by the clinical successes of KIT kinase inhibition by imatinib (Gleevec) and sunitinib (Sutent), in vitro and in the clinic (Demetri et al. 2002; Tuveson et al. 2001). Indeed, imatinib therapy has rapidly become the standard of care in patients with metastatic GIST (Blay et al. 2005).
PKCθ is a member of the novel family of protein kinase C proteins, and – unlike the conventional protein kinase C proteins – shows a narrow range of expression in human cells. PKCθ expression was described initially in T cells and myogenic cells (Baier et al. 1993; Chang et al. 1993), but can be expressed at even higher levels in GIST (Allander et al. 2001; Medeiros et al. 2004; Nielsen et al. 2002; Duensing et al. 2004a). PKCθ is a calcium-independent but phospholipid-dependent dual-specificity kinase, which is activated by the second messenger diacylglycerol (Teixeira et al. 2003). PKCθ biologic roles have been evaluated extensively in T cells, where PKCθ regulates interleukin-2 production and CD25 expression by activating NFκB and activator protein-1 (AP-1) (Sun et al. 2000; Bi et al. 2001). PKCθ: NFκB interactions can be bidirectional, as in the case of T cell receptor (TCR)-induced NFκB activation, regulated by 3-phosphoinositide-dependent kinase 1 (PDK1), which can activate PKCθ, resulting in PKCθ recruitment to lipid rafts (Lee et al. 2005). TCR pathways also regulate PKCθ tyrosine phosphorylation, which is mediated by SRC family protein tyrosine kinases such as LCK (Liu et al. 2000). In addition, PKCθ can regulate gene expression, functioning as a positive modulator of retinoid X receptor responsive element-dependent transcription during T cell activation (Ishaq et al. 2002). PKCθ also has biologic roles outside of T cell activation, including phosphorylation-mediated regulation of cytoskeletal assembly and barrier permeability in intestinal monolayers (Banan et al. 2004; Banan et al. 2005). Equally compelling roles for PKCθ are likely in GIST, given the strong expression and activation of PKCθ in these tumors (Blay et al. 2004; Duensing et al. 2004a; Motegi et al. 2005). Although preliminary studies were consistent with a PKCθ pro-oncogenic role in GIST (Duensing et al. 2004a), conventional PKCs can inhibit ligand-mediated KIT activation, in a negative feedback loop, by phosphorylating KIT on S741 and S746 (Blume-Jensen et al. 1993; Blume-Jensen et al. 1994; Blume-Jensen et al. 1995). Therefore, it is unclear whether PKCθ has positive or negative regulatory roles, or both, in KIT-mutant GISTs.
Although KIT inhibition by imatinib represents a major therapeutic advance for patients with inoperable GIST, most patients eventually experience clinical progression due to the emergence and outgrowth of multiple imatinib-resistant GIST clones (Heinrich et al. 2006). These imatinib-resistant clones often contain secondary mutations in the KIT kinase domain (Heinrich et al. 2006). Novel multi-targeted kinase inhibitors such as sunitinib show activity in some patients with imatinib-resistant GIST, but are not uniformly effective, and it is therefore important to identify alternative signaling targets for therapies in GIST. PKCθ is an attractive therapeutic target in GIST because it is expressed in few normal cell types. Therefore, highly selective PKCθ inhibitor therapies might be accompanied by minimal toxicities in GIST patients.
In the present study, PKCθ and KIT protein expression were stably silenced by lentiviral PKCθ and KIT short hairpin RNA (shRNA) constructs in a KIT+/PKCθ+ imatinib-sensitive GIST cell line (GIST882), two KIT+/PKCθ+ imatinib-resistant GIST cell lines (GIST48 and GIST430), and a control KIT+/PKCθ- Ewing sarcoma cell line (EWS502). Our findings show that PKCθ regulates KIT expression, cell proliferation, and cell survival in GISTs, suggesting that PKCθ warrants evaluation as a therapeutic target in GISTs, including those with resistance to KIT kinase inhibitors such as imatinib.
Results
PKCθ is expressed and strongly phosphorylated in KIT+ GIST cell lines
PKCθ expression and phosphorylation were evaluated in three GIST cell lines (GIST882, GIST48 and GIST430) that express KIT oncoproteins strongly, and are therefore referred to as “KIT-positive”, and in two GIST cell lines (GIST62 and GIST522) that lack KIT expression, both of which were established from KIT-positive GISTs, and contain genomic KIT mutations. (Figure 1). PKCθ expression – and phosphorylation at T538 and S676, which are required for PKCθ activation – were comparable, or greater, in the KIT-positive GIST lines than in Jurkat T-cell leukemia, which has previously provided a benchmark for high levels of PKCθ expression (Figure 1). By contrast, the two KIT-negative GIST lines showed little or no demonstrable PKCθ expression, and PKCθ S676 phosphorylation was nearly undetectable (Figure 1). In general, PKCθ phosphorylation in the various GIST cell lines paralleled the expression of total PKCθ (Figure 1).
Figure 1.
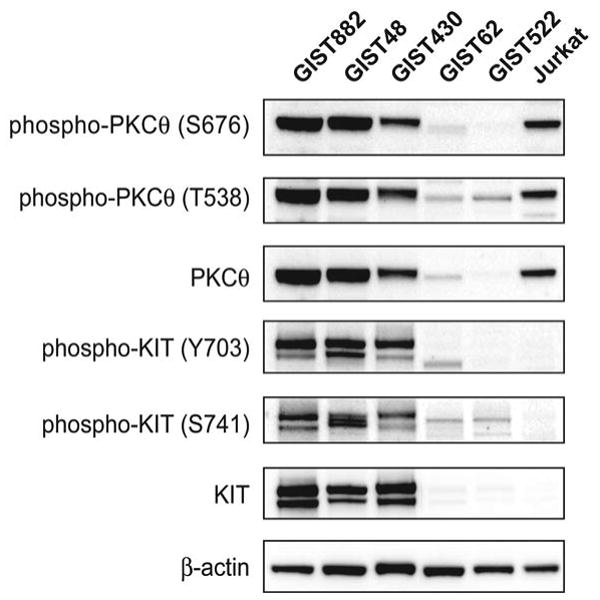
Immunoblotting evaluations of GIST cell lines show strong PKCθ activation and expression in KIT-positive lines (GIST882, GIST430 and GIST48) but not in KIT-negative lines (GIST62 and GIST522). The Jurkat T-cell acute lymphoblastic leukemia provides a positive control for PKCθ activation and expression.
PKCθ regulates KIT oncoprotein expression
To evaluate PKCθ roles in imatinib-sensitive and imatinib-resistant GISTs, PKCθ gene expression was stably silenced by lentivirus-mediated shRNA in GIST882, GIST48, and GIST430, and immunoblotting studies were then performed at 96 h post-infection. KIT gene silencing, also using lentiviral shRNA constructs, was evaluated in parallel studies (Figure 2A; expression quantitations provided in Supplemental Figure 1). The shRNA transductions resulted in greater than 60% inhibition of their intended targets. PKCθ and KIT phosphorylation were also inhibited, commensurate with the reductions in total protein expression, after the shRNA interventions (Figure 2A). KIT oncoprotein expression and phosphorylation, evaluated 96 hours after PKCθ shRNA infection, were substantially inhibited in the GIST48 and GIST430 imatinib-resistant GIST lines, but not in GIST882 (Figure 2A). However, KIT shRNA knockdown did not inhibit PKCθ expression in any of the GIST lines. All lentiviral experimental results were corroborated by at least two separate infections of the various GIST cell lines, and using at least one additional KIT and PKCθ shRNA sequence (data not shown).
Figure 2.
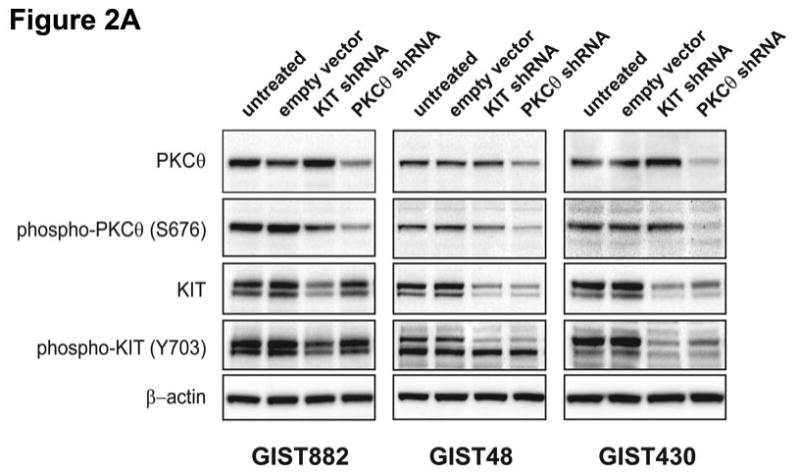
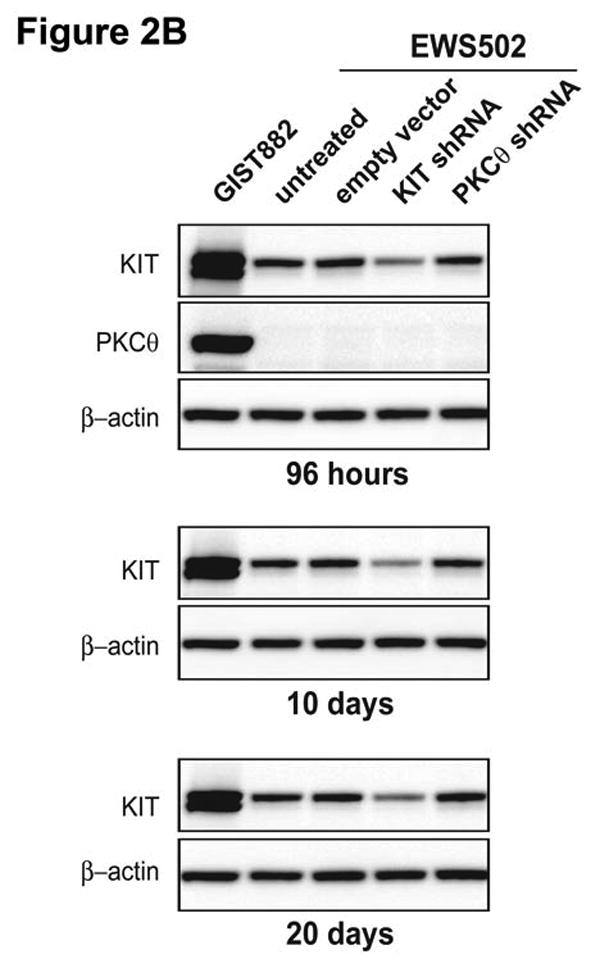
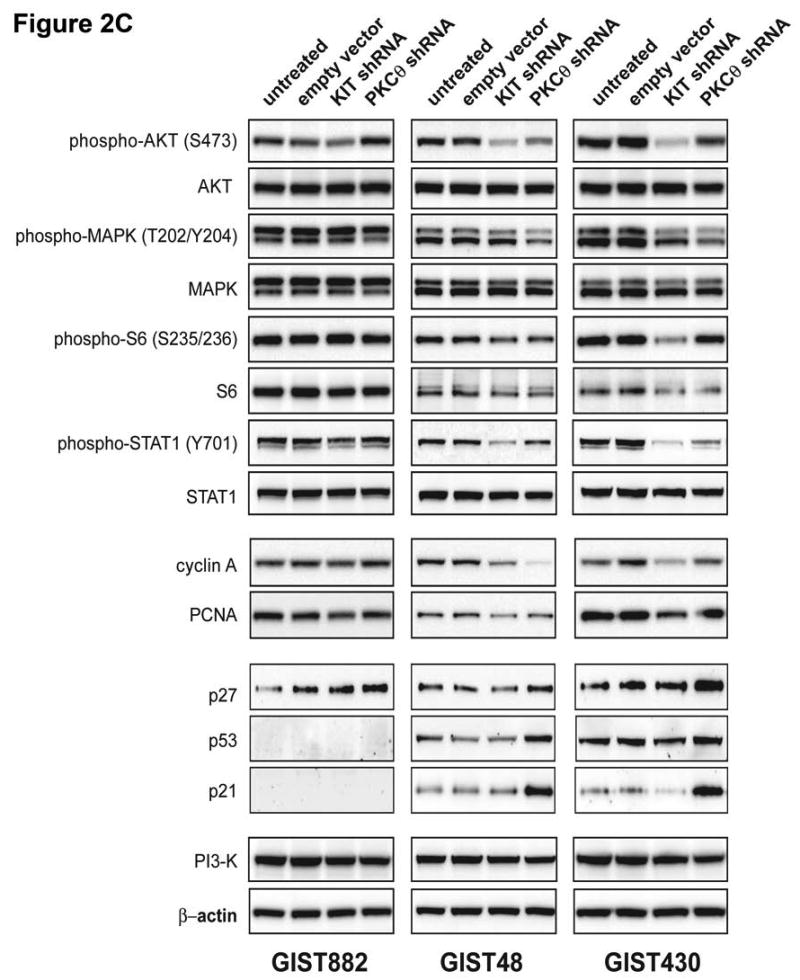
A) Immunoblotting evaluations of KIT and PKCθ expression and activation, in KIT-positive GIST cell lines (GIST882, GIST430 and GIST48) at 96 hours after infection by lentiviral KIT or PKCθ shRNA constructs. KIT shRNA infection inhibited KIT expression and activation in each cell line. PKCθ shRNA inhibition inhibited PKCθ expression and activation in each cell line, and also inhibited KIT expression and activation in the imatinib-resistant GIST48 and GIST430 cell lines. Control lanes, for each cell line, include uninfected cells (untreated lane) and cells infected with empty lentiviral vector. B) Comparison immunoblotting evaluations of KIT and PKCθ expression, after infection of KIT-positive Ewing's sarcoma (EWS502) cells with lentiviral KIT or PKCθ shRNA constructs. These studies show that PKCθ shRNA treatment does not inhibit KIT expression, in the absence of PKCθ expression, indicating that PKCθ shRNA-mediated KIT inhibition in GIST (2A) does not result from nonspecific interactions between the PKCθ shRNA and KIT mRNA. The GIST882 cells provide a positive control for KIT and PKCθ expression. EWS502 no-treatment controls include uninfected cells (untreated lane) and EWS502 infected with empty lentiviral vector. C) Immunoblotting evaluations of KIT-positive GIST cell lines (GIST882, GIST430 and GIST48) at 96 hours after infection by lentiviral KIT or PKCθ shRNA constructs. The immunoblotting assays evaluated affects of KIT and PKCθ knockdown on signaling intermediates (AKT, MAPK p42/44, S6, STAT1), proliferation markers (Cyclin A and PCNA), and cell cycle checkpoint proteins (p27, p21 and p53). The empty vector lane is a parallel control. The PI3-K and Actin immunostains show equivalence of lane loading. Control lanes, for each cell line, include uninfected cells (untreated lane) and cells infected with empty lentiviral vector.
To confirm that the PKCθ shRNA construct was specific for its intended target, and not binding directly to KIT mRNA, control transductions were performed in the KIT+/PKCθ- Ewing sarcoma cell line, EWS502. EWS502 cells express wild-type KIT protein, but do not express PKCθ (Figure 2B). EWS502 KIT expression was inhibited at 96 h after infection with KIT lentiviral shRNA, but was unaffected after infection with lentiviral PKCθ shRNA (Figure 2B). Similarly, reduction in KIT expression was seen in EWS502 cells stably expressing KIT shRNA, but not in EWS502 cells stably expressing PKCθ shRNA, after 10 and 20 days of selection with 2.5 μg/mL puromycin (Figure 2B).
Effects of PKCθ and KIT shRNA knockdown on GIST signaling pathways
Alterations in proliferation and survival signaling pathways were determined by immunoblotting in the GIST882, GIST48, and GIST430 cell lines after shRNA-mediated inhhibition of KIT and PKCθ expression (Figure 2C; expression quantitations provided in Supplemental Figure 2). These studies evaluated phosphorylation of AKT and MAPK, amongst other signaling intermediates which have been shown to be KIT-dependent in GISTs (Duensing et al. 2004b; Corless et al. 2004; Bauer et al. 2007). Evaluations at 4 days after lentiviral shRNA infection showed that KIT silencing resulted in dramatic inactivation of AKT and S6 in the imatinib-resistant GIST48 and GIST430 cells, whereas MAPK was inactivated partially only in GIST430 (Figure 2C). PKCθ knockdown resulted in at least 50% MAPK inactivation in both GIST48 and GIST430, but not in GIST882 (Figure 2C). Likewise, PKCθ knockdown partially inactivated AKT, S6 and STAT1 in GIST48 and GIST430, but not in GIST882 (Figure 2C). Evaluations, performed in puromycin selected GIST882 cells with stable KIT and PKCθ shRNA expression, at 10 and 20 days after lentiviral infection, confirmed that AKT, but not MAPK, was inactivated after KIT silencing (Figure 3A; expression quantitations provided in Supplemental Figure 3). PKCθ knockdown, after 10 days and particularly after 20 days of antibiotic selection, resulted in substantial inactivation of AKT, and marked reduction in expression of total and phosphorylated KIT (Figure 3A). Notably, these studies also reveal that substantial PDGFRA expression is substantially repressed after PKCθ knockdown, but is unaffected after KIT knockdown (Supplemental Figure 3).
Figure 3.
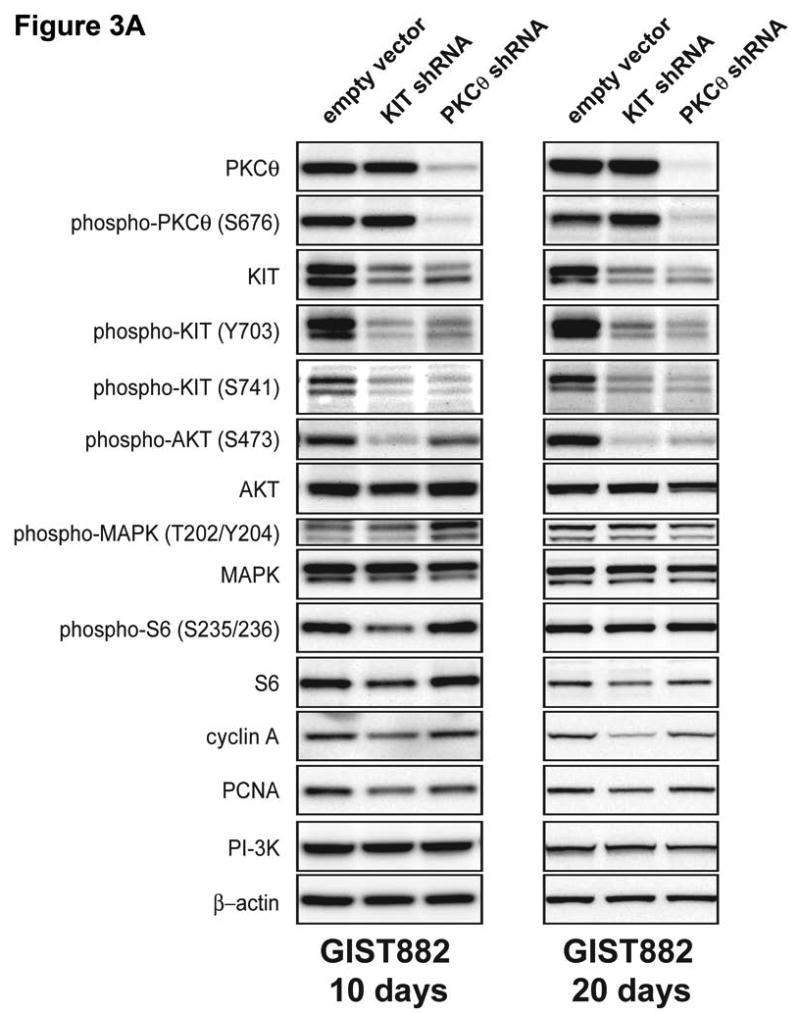
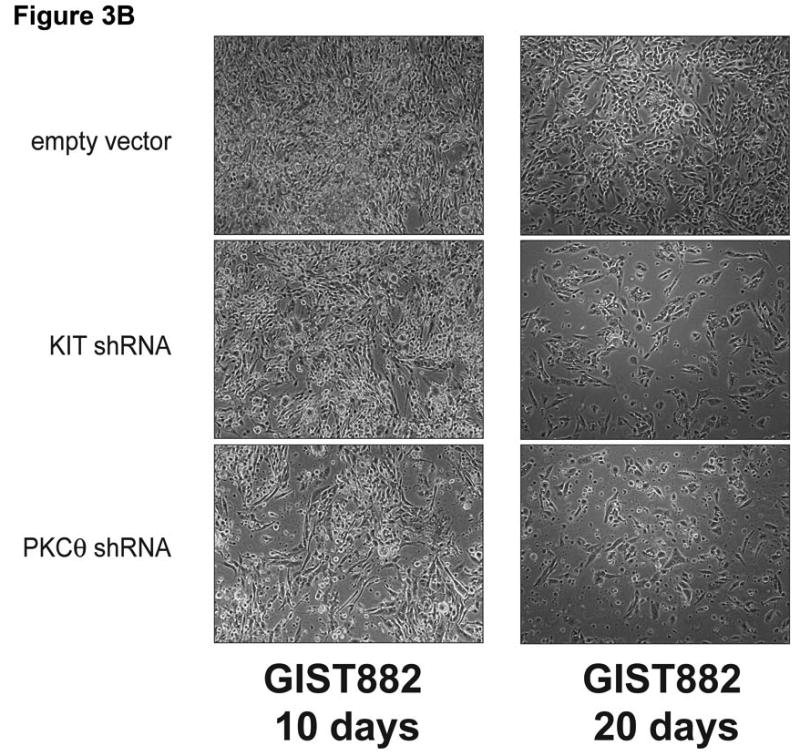
A) Immunoblotting evaluations of GIST882 cells, at 10 and 20 days after infection by lentiviral KIT or PKCθ shRNA constructs. KIT shRNA resulted in decreased expression of KIT, phospho-AKT, phospho-S6, and the proliferation markers Cyclin A and PCNA. PKCθ shRNA resulted in decreased expression of PKCθ, KIT, phospho-AKT, and Cyclin A. The PI3-K and β-actin immunostains show equivalence of lane loading. B) GIST882 cell culture appearance, when evaluated at 10 and 20 days after infection by lentiviral KIT or PKCθ shRNA constructs, showing growth inhibition compared to control GIST882 cultures infected with lentiviral empty constructs.
Effects of PKCθ and KIT shRNA knockdown on GIST proliferation
KIT shRNA knockdown inhibited expression of the cyclin A and PCNA proliferation markers in the imatinib-resistant GIST48 and GIST430 lines (Figures 2C). PKCθ shRNA knockdown also inhibited cyclin A expression in GIST48 and GIST430, and inhibited PCNA expression in GIST48 (Figure 2C). Cyclin A and PCNA expression were unaffected by PKCθ shRNA knockdown in GIST882 at 96 hours (Figure 2C), and were not substantially downregulated in these cells after 10 and 20 day puromycin selections for stable expression of KIT and PKCθ shRNAs (Figure 3A). However, GIST882 cell growth was reduced dramatically, when evaluated after 10 and 20 days of KIT and PKCθ shRNA expression (Figure 3B).
Cellular proliferation was evaluated by an ATP-based cell viability assay (CellTiter-Glo) in GIST882, GIST48, and GIST430 after transduction with KIT or PKCθ shRNAs. Anti-proliferative effects were greater in GIST882 and GIST48 cells after treatment with PKCθ shRNA than after treatment with KIT shRNA. Anti-proliferative effects in GIST430 cells were comparable after treatment with PKCθ and KIT shRNAs (Figures 2C and 4A). All proliferation studies were corroborated by at least two independent shRNA transductions.
Figure 4.
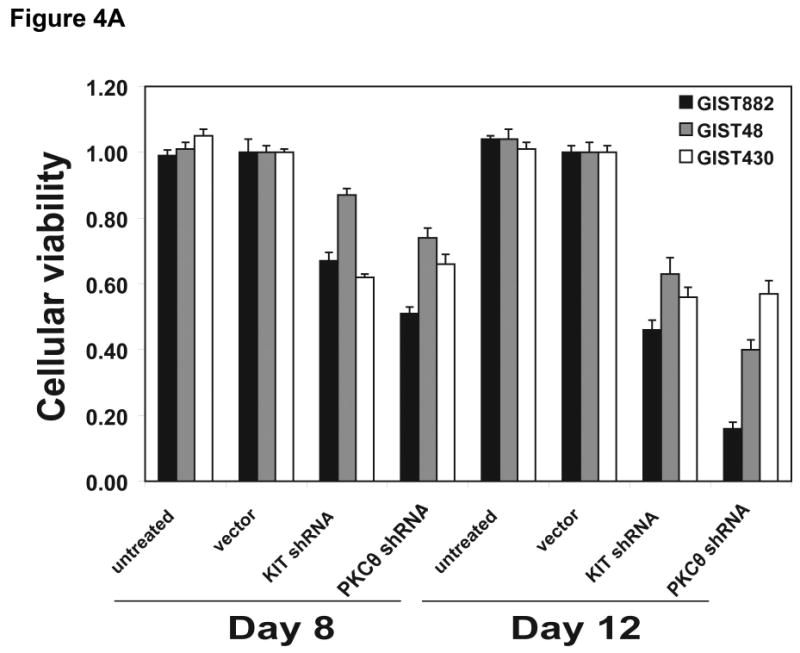

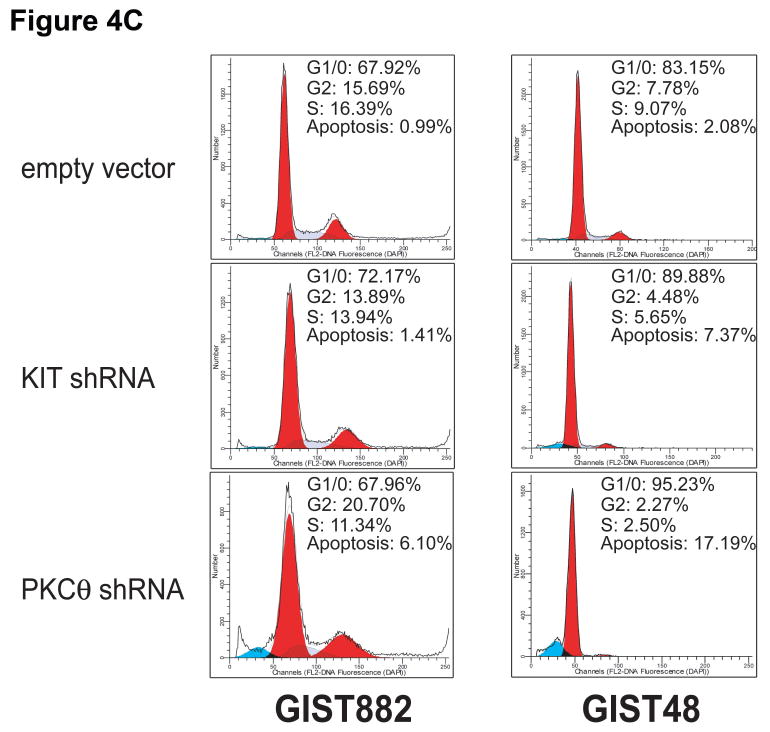
A) Cell viability was evaluated in KIT-positive GIST882 (black bars), GIST48 (gray bars), and GIST430 (white bars) cell lines, at day 8 and day 12 after infection with lentiviral KIT and PKCθ shRNAs. Viability was analyzed using Cell-titer Glo® ATP-based luminescence assay. The data were normalized to the empty lentivirus control infections, and represent the mean values (± s.d.) from quadruplicate cultures. B) Apoptosis was evaluated in GIST882 (black bars) and GIST48 (gray bars) cell lines, at day 4 after infection with lentiviral KIT and PKCθ shRNAs. Caspase 3/7 activity was measured using a Caspase-Glo® luminescence assay. The data were normalized to the empty lentivirus control infections, and represent the mean values (± s.d.) from quadruplicate cultures. C) Cell cycle analyses were performed in GIST882 and GIST48 cells at 8 days and 4 days after infection by lentiviral KIT or PKCθ shRNA constructs, respectively. Apoptotic response was greater, in both cell lines, after treatment with PKCθ shRNA than after treatment with KIT shRNA.
Proliferation-related consequences of KIT and PKCθ knockdown were also determined by assessing immunoblot expression of p53 and the p21 and p27 cyclin dependent kinase (CDK) inhibitors (Figure 2C). GIST48 and GIST430 cells demonstrated upregulation of p53, p21, and p27 expression after PKCθ knockdown, but not after KIT knockdown. Likewise, p27 expression was upregulated in GIST882 cells after PKCθ knockdown, whereas p21 and p53 were not demonstrably expressed in these cells (Figure 2C).
Apoptosis and Cell cycle analysis after PKCθ and KIT gene knockdown
Apoptosis was evaluated by assaying functional caspase 3/7 activation in GIST882 and GIST48 after transduction with KIT or PKCθ shRNAs. Apoptosis was more strongly induced in these GIST cells after treatment with PKCθ shRNA, compared to treatment with KIT shRNA (Figure 4B). Cell cycle analysis in GIST48 showed a G1-block after KIT and PKCθ silencing with an increase in the G1/0 peak from 83.2% after empty vector transduction to 89.9% and 95.2% after transduction with KIT and PKCθ shRNAs, respectively (Figure 4C). This was accompanied by a decrease in the S-phase population from 9% with the empty vector control to 5.6% and 2.5% with KIT and PKCθ shRNAs, respectively (Figure 4C). In addition, the pre-G1 nuclear fragmentation peak increased from 2% with empty vector control to 7% and 17% with KIT and PKCθ shRNAs, respectively (Figure 4C). Cell cycle analysis in GIST882 showed a G1-block after KIT silencing with an increase in the G1/0 peak from 67.9% after empty vector transduction to 72.2% after transduction with KIT shRNA (Figure 4C). The pre-G1 nuclear fragmentation peak in GIST882 increased from 1% with empty vector control to 1.4% and 6.1% with KIT and PKCθ shRNAs, respectively (Figure 4C)
Discussion
Most GISTs contain oncogenic gain-of-function mutations in the KIT or PDGFRA receptor tyrosine kinase proteins. These mutations can be early – and even initiating – transforming events (Corless et al. 2004). Therefore, KIT/PDGFRA inhibition by small molecule kinase inhibitors such as imatinib mesylate or sunitinib malate has become the mainstay of treatment in patients with inoperable GIST. At the same time, studies of human GIST cell lines and transgenic mouse models have enabled substantial advances in understanding the central roles of KIT/PDGFRA signaling pathways in GIST cell proliferation and survival (Demetri et al. 2002; Heinrich et al. 2003; Corless et al. 2005; Rossi et al. 2006; Duensing et al. 2004b; Bauer et al. 2007; Zhu et al. 2007).
Various studies have shown that PKCθ is expressed strongly in GISTs, but not in other sarcoma histotypes (Duensing et al. 2004a; Blay et al. 2004; Motegi et al. 2005). These studies established PKCθ as a diagnostic marker of GIST, but did not illuminate PKCθ biological functions. In the present work, we find strong PKCθ expression in each of three KIT-positive GIST cell lines, whereas PKCθ expression was weak-to-undetectable in two KIT-negative GIST lines (Figure 1). These observations suggest that loss of PKCθ expression could be responsible for inhibition of KIT expression in the KIT-negative GIST lines, both of which were established from KIT-positive GISTs, and contain genomic KIT mutations. By contrast, we have reported that PKCθ is expressed in untreated KIT-negative GISTs that lack KIT mutations but often contain oncogenic PDGFRA mutations as an alternate receptor tyrosine kinase activation mechanism. Although PKCθ is therefore not the sole determinant of KIT expression in GISTs, our data support a key PKCθ role in enabling KIT oncogenic function in GISTs. It is also possible that KIT oncogenic signaling contributes to PKCθ activation in GISTs. KIT activation is known to result in binding and activation of PI3-K and phospholipase Cγ, which regulate, respectively, membrane translocation of PKCθ and synthesis of the PKCθ cofactor diacylglycerol (Altman and Villalba 2003; Villalba et al. 2002). Further, we have shown that KIT oncoproteins complex with and tyrosine phosphorylate PKCθ in GISTs, and might thereby directly activate PKCθ (Zhu et al. 2007). These observations suggest that KIT and PKCθ participate in a positive feedback loop in GIST.
PKCθ-mediated regulation of KIT oncoprotein expression was demonstrated in each of three KIT-positive GIST cell lines, including the imatinib-sensitive line, GIST882, and the imatinib-resistant lines, GIST48 and GIST430. However, KIT oncoprotein expression was inhibited within four days of PKCθ shRNA transduction in GIST48 and GIST430, whereas similar effects were seen only at 10-20 days after PKCθ shRNA transduction in GIST882 (Figure 2A and 3A). One explanation for these observations is that the KIT oncoprotein in GIST882 (K642E mutant) might be more stable after PKCθ knockdown compared to the hyperactivated (Bauer et al. 2007) double-mutant KIT oncoproteins in GIST48 and GIST430. Alternately, other biological variables in these unique GIST models might account for the different times of onset for KIT oncoprotein inhibition, after PKCθ knockdown. The specificity of these findings was substantiated in an Ewing's sarcoma cell line (EWS502) which expresses KIT but not PKCθ, and in which PKCθ shRNA infection had no impact on KIT expression (Figure 2B). These studies confirm that perturbations of KIT expression after PKCθ shRNA infection in GIST lines were indeed mediated by the observed PKCθ silencing in those lines. Our preliminary studies (data not shown) suggest that PKCθ modulation of KIT expression in GIST involves regulation of KIT transcription. However, further studies are needed to determine whether KIT expression is regulated by PKCθ biochemical activation or by PKCθ scaffolding functions, but – either way – our findings show that KIT and PKCθ have highly integrated biologic functions in GIST. In addition, preliminary studies suggest that PDGFRA – an alternate oncoprotein in GIST – is also repressed after PKCθ knockdown (Supplemental Figure 3). These findings highlight that PKCθ pathway therapeutic inhibition warrants clinical evaluation as a novel strategy to downregulate KIT and PDGFRA oncoproteins, including those with imatinib-resistance mutations. PKCθ targeting might also be useful therapeutically in pediatric GISTs which co-express KIT and PKCθ (Janeway et al. 2007), and respond poorly to imatinib (Katie Janeway, personal communication). Further studies are needed to determine whether PKCθ inactivation by small molecule kinase inhibitiors can reproduce the spectrum of findings, demonstrated here, that result from downregulation of PKCθ expression in GIST.
It is particularly intriguing that PKCθ regulates KIT expression in GIST, given that PKCθ and KIT have restricted ranges of expression in human cells, and are both coexpressed at unusually high levels in interstitial cells of Cajal, which are nonneoplastic counterparts of GIST (Southwell 2003). Although the mechanisms by which PKCθ regulates KIT gene expression remain to be determined, there is precedent – in T cells – for PKCθ modulation of transcriptional regulators. Namely, PKCθ regulates retinoid X receptors (RXRs) in T cells, and the RXRs are key nuclear transcription factors that homodimerize or heterodimerize with other steroid and retinoid receptor family transcription factors (Rastinejad 2001). T cell activation signals regulate expression and transactivation of RXRα, and PKCθ participates in these signaling pathways through interactions with calcineurin, resulting in increased RXR responsive element (RXRE) dependent transcription (Ishaq et al. 2002). Further, catalytically inactive PKCθ does not attenuate RXRE-dependent transcription in the T cell models, and PKCθ cooperates with calcineurin to induce Fas ligand expression during activation-induced T cell death (Villalba et al. 1999).
Previous studies of the GIST882 line showed parallel inactivation of KIT, AKT and MAPK, after treatment with imatinib (Duensing et al. 2004b), suggesting that AKT and MAPK were KIT-dependent in GISTs. However, our present studies, based on KIT knockdown by shRNA, while confirming that AKT is KIT-dependent, show that MAPK can be KIT-independent, in both imatinib-sensitive and imatinib-resistant GIST lines (Figures 2C and 3A). These findings suggest that imatinib-dependent MAPK inhibition, in GIST, might depend in part on inactivation of an alternative imatinib target, other than KIT. And these findings are consistent with those reported in EGFR-mutant lung carcinomas and in GISTs, which emphasize that the PI3-K/AKT survival pathway has a crucial role in oncogenic signaling from mutant tyrosine kinase proteins (Sordella et al. 2004; Tarn et al. 2006). However, substantial MAPK inactivation resulted from PKCθ knockdown in the imatinib-resistant GIST48 and GIST430 lines (Figure 2C), and therefore PKCθ inhibition can inactivate both PI3-K/AKT survival pathways and MAPK-dependent proliferation pathways in GISTs. This might explain why PKCθ knockdown had more impact than KIT knockdown on inhibition of cyclin A expression, and on cell cycle arrest and apoptosis in imatinib-resistant GIST48 cells (Figures 2C, 4B and 4C). In addition, the cellular responses seen after PKCθ knockdown in GIST likely depend on dysregulation of pathways beyond those evaluated in the present study. For example, PKCθ mediates activation of the NFκB transcription factor during T cell activation (Sun et al. 2000; Wang et al. 2004), and this PKCθ function is performed through interactions with AKT (Bauer et al. 2001). Therefore, it will be worthwhile in future studies to determine whether NFκB is a PKCθ effector in GIST.
The protein kinase C family has crucial regulatory roles in cell growth and cell cycle progression (Frey et al. 1997; Ashton et al. 1999; Jiang et al. 2002; Cerda et al. 2006; Deeds et al. 2003). Various studies have shown that PKC suppression generally inhibits cell cycle progression, whereas PKC activation stimulates cell cycle progression (Frey et al. 1997; Cerda et al. 2006; Deeds et al. 2003). Therefore, we evaluated cell cycle checkpoints after PKCθ silencing in our GIST models (Figure 2C). PKCθ shRNA knockdown resulted in over-expression of the CDK inhibitors p27 and p21, and the p53 cell cycle checkpoint protein, all of which have tumor suppressor functions in human cancer. These observations suggest that PKCθ regulates cell cycle checkpoint pathways in GIST, and there is precedent for such cell cycle control mechanisms in T-cells (Deeds et al. 2003).
Lentivirus-mediated KIT shRNA knockdown resulted in profound anti-proliferative and pro-apoptotic effects in both imatinib-sensitive and imatinib-resistant GIST cell lines, and was associated with PI3-K/AKT signaling pathway inhibition. These findings show that KIT activation continues to serve a crucial oncogenic role in some GISTs which develop clinical resistance to imatinib (Heinrich et al. 2006). Notably, individual patients can demonstrate heterogeneous imatinib resistance mechanisms, for example having different KIT secondary imatinib resistance mutations in separate clinically-progressing GIST metastases (Debiec-Rychter et al. 2005; Heinrich et al. 2006). This resistance heterogeneity, with multiple different kinase resistance mutations in the same patient, poses a challenge to salvage therapy with alternate KIT kinase inhibitors. Specifically, it is unlikely that a given KIT kinase inhibitor will effectively inhibit the myriad different KIT structural oncoprotein variants that can be encountered in a given patient, at time of clinical progression on imatinib therapy. Therefore, the mechanism revealed in this report, whereby PKCθ knockdown inhibits KIT expression, is of substantial clinical relevance, suggesting that therapeutic PKCθ inhibition might repress expression of imatinib-resistant KIT oncoproteins, irrespective of whatever KIT kinase domain mutations are responsible for the imatinib-resistance. The concept of PKCθ therapeutic inhibition is all the more appealing in that PKCθ RNAi knockdown was associated with more apoptosis than seen after KIT RNAi knockdown. Most patients with metastatic GIST benefit from imatinib therapy, having major responses, but nonetheless do not have complete responses. The residual GIST in these treated patients is generally quiescent metabolically, as evidenced by persistent negative PET scans, and cell proliferation arrest. Nonetheless, a subset of the cells survive, and can eventually progress due to imatinib-resistance mutations, suggesting that GIST survival pathways are not fully inhibited by imatinib. The observations reported here suggest that a PKCθ inhibitor might lead to increased GIST apoptosis and thereby maximize the clinical response. In summary, our findings indicate that PKCθ can serve not only as a diagnostic marker in GIST, but perhaps also as a novel therapeutic target of relevance in patients with both imatinib-sensitive and imatinib-resistant GIST.
Materials and Methods
Antibodies and reagents
Polyclonal antibodies to KIT were from Dako (Carpinteria, CA). Polyclonal antibodies to PKCθ and S6 were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies to AKT, and all phospho-antibodies except phospho-KIT and phospho-STAT1 (Zymed, So. San Francisco, CA), were from Cell Signaling Technologies (Danvers MA). Polyclonal antibodies to MAPK were from Zymed. Polyclonal antibodies to phosphatidylinositol 3′-kinase p85 were from Upstate Biotechnology (Lake Placid, NY). Monoclonal mouse antibodies were to cyclin A (Novocastra, Newcastle upon Tyne, UK), PCNA and p53 (Santa Cruz Biotechnology), STAT1 and p21Cip1/WAF1 (Zymed Laboratories), p27Kip1 (BD Transduction Laboratories, San Jose, CA), and actin (Sigma-Aldrich, St, Louis, MO). Lipofectamine and plus reagent were from Invitrogen. Puromycin and polybrene were from Sigma.
Cell lines
GIST882 is a human cell line established from an untreated GIST with a primary imatinib-sensitive mutation in KIT exon 13 (K642E) (Lux et al. 2000). GIST48 and GIST430 are human cell lines established from GISTs progressing on imatinib therapy. GIST48 has a homozygous KIT exon11 mutation (V560D) and a heterozygous KIT exon17 mutation (D820A). GIST430 has heterozygous mutations in KIT exon11 (in-frame deletion) and KIT exon13 (V654A). GIST62 was established from an untreated GIST. This cell line, like the primary tumor from which it was established, has a heterozygous KIT exon 11 in-frame deletion mutation (resulting in MYEVQWK552-558T), but has essentially undetectable KIT and PKCθ expression. GIST522 was established from a GIST progressing on imatinib therapy, and has a heterozygous KIT exon 11 in-frame deletion mutation (resulting in delEVQWK554-558) but, like GIST62, has essentially undetectable KIT and PKCθ expression. GIST62 and GIST522 are highly imatinib-resistant (data not shown), and these cell lines serve as negative controls for interventions directed to KIT or PKCθ, in a GIST cell context. EWS502 is a Ewing tumor cell line expressing wild-type KIT. 293T cells were used to prepare lentiviral constructs.
PKCθ and KIT shRNA Lentiviral Constructs and Preparations
The pLKO.1puro (7 kb) lentivirus construct contains a U6 promoter and HIV-1 RNA packaging signal with puromycin- and ampicillin-resistance elements cloned 3′ of the human phosphoglycerate kinase (hPGK) promoter. A cpptCTE was inserted 5′ of hPGK promoter. Human PKCθ and KIT shRNA constructs were generated by ligating the following annealed oligomers into the unique AgeI and EcoRI sites of pLKO.1puro: PKCθ forward 5′-CCGGCATCCAAAGCTGCCACAAGTTCTCGAGAACTTGTGGCAGCTTTGGATGTTTTTG-3′ and PKCθ reverse 5′-AATTCAAAAACATCCAAAGCTGCCACAAGTTGTGGCAGCTTTGGATG-3′ and KIT forward 5′-CCGGCCATAAGGTTTCGTTTCTGTACTCGAGTACAGAAACGAAACCTTATGGTTTTTG-3′ and KIT reverse 5′-AATTCAAAAACATAAGGTTTCGTTTCTGTACTCGAGTACAGAAACGAAACCTTATGG-3′.
Lentivirus preparations were produced by cotransfecting pLKO.1puro empty vector with PKCθ or KIT shRNA, and helper virus packaging plasmids pCMVΔR8.91 and pMD.G (at a 10:10:1 ratio) into 293T cells. Transfections were carried out using lipofectamine and PLUS reagent. Lentiviruses were harvested at 24, 36, 48, and 60 h post-transfection. Viral titers were determined according to a protocol from Invitrogen, in GIST882 cells. Viral preparations were then stored at -80°C. Two well-validated shRNAs were used each for KIT and PKCθ knockdown, and these shRNAs were those – screened from a panel of X KIT shRNAs and Y PKCθ shRNAs – which accomplished the most efficient knockdowns of their intended targets.
Cell culture and virus infection
GIST882 and EWS502 were maintained in RPMI 1640 with 15% fetal bovine serum (FBS) containing penicillin/streptomycin and L-glutamine. GIST48 and GIST430 were maintained in F10 containing 15% FBS, penicillin/streptomycin, L-glutamine, amphotericin, Mitotracker+, and bovine pituitary extract. GIST cells and EWS502 cells were seeded in 6-well plates. Infections were carried out in the presence of 8 μg/mL of polybrene. Cells were lysed for western blot analysis at 96 h post-infection or harvested for cell cycle analysis. Following transduction, GIST882 cells were selected for stable expression of the shRNAs using 2.5 μg/mL puromycin. Cell culture images were obtained using Spot software (Version 3.5.9 for MacOS™) and a Nikon Eclipse TE2000-5 microscope.
Western blotting
Whole cell lysates were prepared in lysis buffer (1% NP-40, 50 mM Tris-HCl pH 8.0, 100 mM sodium fluoride, 30 mM sodium pyrophosphate, 2 mM sodium molybdate, 5 mM EDTA, 2 mM sodium orthovanadate) containing protease inhibitors (10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride). Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad Laboratories Hercules, CA, USA). Electrophoresis and Western blotting were performed as described previously (Rubin et al. 2001). Detection was by chemiluminescence (ECL, Amersham Pharmacia Biotechnology), captured using a FUJI LAS1000-plus chemiluminescence imaging system.
Cell viability and apoptosis analyses
GIST882, GIST48, and GIST430 Cells were plated at 5,000 cells/well in 96-well flat-bottom plates from Falcon (Lincoln NJ) and cultured in RPMI1640 or F10 for 2 days before transduction with lentiviral empty vector, PKCθ shRNA or KIT shRNA. Proliferation and apoptosis studies were performed after 4, 8, and 12 days using the CellTiter-Glo luminescent assay and the Caspase-Glo 3/7 assay Kit from Promega (Madison, WI), and were quantitated using a Veritas™ Microplate Luminometer from Turner Biosystems (Sunnyvale, CA). All assays were performed in quadruplicate wells, and were averaged from two independent transductions in each cell line.
Cell cycle analysis
GIST882 and GIST48 cells in 6-well plates were trypsinized, centrifuged, and washed once with Hanks Balanced Salt Solution at room temperature after infection with lentivirus for 8 days and 4 days, respectively. For nuclear staining, a DAPI-containing solution (Nuclear isolation and staining solution, NPE systems, Pembrooke Pines, FL) was added to the cells and the cell suspension was immediately analyzed in a flow cytometer (NPE Quanta, NPE Systems). Data analysis was performed using Modfit LT software 3.1 (Verity Software House, Topsham, ME).
Acknowledgments
This work was supported by grants from an anonymous donor, GI SPORE 1P50CA127003-01, the Life Raft Group, Cesarini Team for the Pan-Massachusetts Challenge, the Virginia and Daniel K. Ludwig Trust for Cancer Research, the Ronald O. Perelman Fund for Cancer Research, the Stutman GIST Cancer Research Fund, the Rubenstein Foundation, and Leslie's Links. We thank Sarah E. Bulmer and William Hahn for providing shRNA constructs.
Reference List
- Allander SV, Nupponen NN, Ringner M, Hostetter G, Maher GW, Goldberger N, Chen Y, Carpten J, Elkahloun AG, Meltzer PS. Cancer Res. 2001;61:8624–8628. [PubMed] [Google Scholar]
- Altman A, Villalba M. Immunol Rev. 2003;192:53–63. doi: 10.1034/j.1600-065x.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- Ashton AW, Watanabe G, Albanese C, Harrington EO, Ware JA, Pestell RG. J Biol Chem. 1999;274:20805–20811. doi: 10.1074/jbc.274.30.20805. [DOI] [PubMed] [Google Scholar]
- Baier G, Telford D, Giampa L, Coggeshall KM, Baier-Bitterlich G, Isakov N, Altman A. J Biol Chem. 1993;268:4997–5004. [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Choudhary S, Forsyth CB, Farhadi A, Keshavarzian A. J Pharmacol Exp Ther. 2005;313:962–982. doi: 10.1124/jpet.104.083428. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Am J Physiol Cell Physiol. 2004;287:C218–C234. doi: 10.1152/ajpcell.00575.2003. [DOI] [PubMed] [Google Scholar]
- Bauer B, Krumbock N, Fresser F, Hochholdinger F, Spitaler M, Simm A, Uberall F, Schraven B, Baier G. J Biol Chem. 2001;276:31627–31634. doi: 10.1074/jbc.M103098200. [DOI] [PubMed] [Google Scholar]
- Bauer S, Duensing A, Demetri GD, Fletcher JA. Oncogene. 2007;26:7560–7568. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]
- Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJ, Altman A. Nat Immunol. 2001;2:556–563. doi: 10.1038/88765. [DOI] [PubMed] [Google Scholar]
- Blay JY, Bonvalot S, Casali P, Choi H, biec-Richter M, Dei Tos AP, Emile JF, Gronchi A, Hogendoorn PC, Joensuu H, Le CA, McClure J, Maurel J, Nupponen N, Ray-Coquard I, Reichardt P, Sciot R, Stroobants S, van GM, van OA, Demetri GD. Ann Oncol. 2005;16:566–578. doi: 10.1093/annonc/mdi127. [DOI] [PubMed] [Google Scholar]
- Blay P, Astudillo A, Buesa JM, Campo E, Abad M, Garcia-Garcia J, Miquel R, Marco V, Sierra M, Losa R, Lacave A, Brana A, Balbin M, Freije JM. Clin Cancer Res. 2004;10:4089–4095. doi: 10.1158/1078-0432.CCR-04-0630. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Ronnstrand L, Gout I, Waterfield MD, Heldin CH. J Biol Chem. 1994;269:21793–21802. [PubMed] [Google Scholar]
- Blume-Jensen P, Siegbahn A, Stabel S, Heldin CH, Ronnstrand L. EMBO J. 1993;12:4199–4209. doi: 10.1002/j.1460-2075.1993.tb06104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Wernstedt C, Heldin CH, Ronnstrand L. J Biol Chem. 1995;270:14192–14200. doi: 10.1074/jbc.270.23.14192. [DOI] [PubMed] [Google Scholar]
- Cerda SR, Mustafi R, Little H, Cohen G, Khare S, Moore C, Majumder P, Bissonnette M. Oncogene. 2006;25:3123–3138. doi: 10.1038/sj.onc.1209360. [DOI] [PubMed] [Google Scholar]
- Chang JD, Xu Y, Raychowdhury MK, Ware JA. J Biol Chem. 1993;268:14208–14214. [PubMed] [Google Scholar]
- Corless CL, Fletcher JA, Heinrich MC. J Clin Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. J Clin Oncol. 2005;23:5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, van Oosterom A, Marynen P. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Deeds L, Teodorescu S, Chu M, Yu Q, Chen CY. J Biol Chem. 2003;278:39782–39793. doi: 10.1074/jbc.M306854200. [DOI] [PubMed] [Google Scholar]
- Demetri GD, von MM, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Drevs J, Medinger M, Schmidt-Gersbach C, Weber R, Unger C. Curr Drug Targets. 2003;4:113–121. doi: 10.2174/1389450033346885. [DOI] [PubMed] [Google Scholar]
- Duensing A, Joseph NE, Medeiros F, Smith F, Hornick JL, Heinrich MC, Corless CL, Demetri GD, Fletcher CD, Fletcher JA. Cancer Res. 2004a;64:5127–5131. doi: 10.1158/0008-5472.CAN-04-0559. [DOI] [PubMed] [Google Scholar]
- Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Oncogene. 2004b;23:3999–4006. doi: 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti H, Rubin BP, Shmookler B, Sobin LH, Weiss SW. Hum Pathol. 2002;33:459–465. doi: 10.1053/hupa.2002.123545. [DOI] [PubMed] [Google Scholar]
- Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD. J Biol Chem. 1997;272:9424–9435. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- Gschwind A, Fischer OM, Ullrich A. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von MM, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA. J Clin Oncol. 2006 doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- Ishaq M, Fan M, Wigmore K, Gaddam A, Natarajan V. J Immunol. 2002;169:732–738. doi: 10.4049/jimmunol.169.2.732. [DOI] [PubMed] [Google Scholar]
- Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Cancer Res. 2007;67:9084–9088. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- Jiang XH, Lam SK, Lin MC, Jiang SH, Kung HF, Slosberg ED, Soh JW, Weinstein IB, Wong BC. Oncogene. 2002;21:6113–6122. doi: 10.1038/sj.onc.1205778. [DOI] [PubMed] [Google Scholar]
- Lee KY, D'Acquisto F, Hayden MS, Shim JH, Ghosh S. Science. 2005;308:114–118. doi: 10.1126/science.1107107. [DOI] [PubMed] [Google Scholar]
- Liu Y, Witte S, Liu YC, Doyle M, Elly C, Altman A. J Biol Chem. 2000;275:3603–3609. doi: 10.1074/jbc.275.5.3603. [DOI] [PubMed] [Google Scholar]
- Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA. Am J Pathol. 2000;156:791–795. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA, Fletcher CD. Am J Surg Pathol. 2004;28:889–894. doi: 10.1097/00000478-200407000-00007. [DOI] [PubMed] [Google Scholar]
- Medinger M, Drevs J. Curr Pharm Des. 2005;11:1139–1149. doi: 10.2174/1381612053507611. [DOI] [PubMed] [Google Scholar]
- Motegi A, Sakurai S, Nakayama H, Sano T, Oyama T, Nakajima T. Pathol Int. 2005;55:106–112. doi: 10.1111/j.1440-1827.2005.01806.x. [DOI] [PubMed] [Google Scholar]
- Nielsen TO, West RB, Linn SC, Alter O, Knowling MA, O'Connell JX, Zhu S, Fero M, Sherlock G, Pollack JR, Brown PO, Botstein D, van de Rijn M. Lancet. 2002;359:1301–1307. doi: 10.1016/S0140-6736(02)08270-3. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Rastinejad F. Curr Opin Struct Biol. 2001;11:33–38. doi: 10.1016/s0959-440x(00)00165-2. [DOI] [PubMed] [Google Scholar]
- Rossi F, Ehlers I, Agosti V, Socci ND, Viale A, Sommer G, Yozgat Y, Manova K, Antonescu CR, Besmer P. Proc Natl Acad Sci U S A. 2006;103:12843–12848. doi: 10.1073/pnas.0511076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Southwell BR. Neurogastroenterol Motil. 2003;15:139–147. doi: 10.1046/j.1365-2982.2003.00394.x. [DOI] [PubMed] [Google Scholar]
- Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, Littman DR. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- Tarn C, Skorobogatko YV, Taguchi T, Eisenberg B, von MM, Godwin AK. Cancer Res. 2006;66:5477–5486. doi: 10.1158/0008-5472.CAN-05-3906. [DOI] [PubMed] [Google Scholar]
- Teixeira C, Stang SL, Zheng Y, Beswick NS, Stone JC. Blood. 2003;102:1414–1420. doi: 10.1182/blood-2002-11-3621. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD. Oncogene. 2001;20:5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bi K, Hu J, Altman Y, Bushway P, Reits E, Neefjes J, Baier G, Abraham RT, Altman A. J Cell Biol. 2002;157:253–263. doi: 10.1083/jcb.200201097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba M, Kasibhatla S, Genestier L, Mahboubi A, Green DR, Altman A. J Immunol. 1999;163:5813–5819. [PubMed] [Google Scholar]
- Wang D, Matsumoto R, You Y, Che T, Lin XY, Gaffen SL, Lin X. Mol Cell Biol. 2004;24:164–171. doi: 10.1128/MCB.24.1.164-171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MJ, Ou WB, Fletcher CD, Cohen PS, Demetri GD, Fletcher JA. Oncogene. 2007;26:6386–6395. doi: 10.1038/sj.onc.1210464. [DOI] [PubMed] [Google Scholar]