

Abstract
In vertebrates and bacteria, heparosan the precursor of heparin is synthesized by glycosyltransferases via the stepwise addition of UDP-N-acetylglucosamine and UDP-glucuronic acid. As heparin-like molecules represent a great interest in the pharmaceutical area, the cryptic Pasteurella multocida heparosan synthase PmHS2 found to catalyze heparosan synthesis using substrate analogs has been studied. In this paper, we report an efficient way to purify PmHS2 and to maintain its activity stable during 6 months storage at −80 °C using His-tag purification and a desalting step. In the presence of 1 mM of each nucleotide sugar, purified PmHS2 synthesized polymers up to an average molecular weight of 130 kDa. With 5 mM of UDP-GlcUA and 5 mM of UDP-GlcNAc, an optimal specific activity, from 3 to 6 h of incubation, was found to be about 0.145 nmol/μg/min, and polymers up to an average of 102 kDa were synthesized in 24 h. In this study, we show that the chain length distribution of heparosan polymers can be controlled by change of the initial nucleotide sugar concentration. It was observed that low substrate concentration favors the formation of high molecular weight heparosan polymer with a low polydispersity while high substrate concentration did the opposite. Similarities in the polymerization mechanism between PmHS2, PmHS1, and PmHAS are discussed.
Keywords: Heparosan synthase, Pasteurella multocida, Glycosyltransferase, Glycosaminoglycan, Nucleotide sugar, Polymerase
Introduction
Bioactive carbohydrates play an important role in many organisms. In vertebrates, they are implicated in many physiological processes and biological functions at the molecular level from gene expression to protein regulation and interaction (Jackson et al. 1991). Therefore, some of these carbohydrates, especially glycosaminoglycans (GAGs), are of great interest in medicine. GAGs are linear polysaccharides, sulfated or not, made of repeated disaccharides units of hexose or hexuronic acid and N-acetyl-hexosamine. The GAG heparosan (-α4-GlcNAc-β1,4GlcUA-1-) is the unsulfated precursor of heparin. Heparin acts as an anticoagulant and is known for its use in surgery to prevent, e.g., vein thrombosis (reviewed in Rabenstein 2002). Recent studies have evidenced that the molecular weight of heparin greatly influences its biological activity. Low molecular weight heparin (LMWH) ranging from 2.4 to 5.4 kDa (8 to 18 monosaccharides units) may have an antitumor effect by reducing or suppressing tumor growth and metastasis in some cases (Castelli et al. 2004; Lee 2007; Norrby 2006). This finding implies that a tight control of the molecular weight of heparin and heparin-like molecules during production seems to be the clue to enlarge its therapeutic use. However, the current ways to produce heparin and LMWH are far from being optimal with respect to the control of the chain length distribution. Monodisperse heparin fractions with low molecular weight are difficult to obtain from animal tissue, and the chemical synthesis of polymers longer than six units is not economically feasible (Chen et al. 2006).
In vivo GAGs are polymerized by glycosyltransferase (GT) enzymes that catalyze the polymer elongation through stepwise addition of α-linked uridine nucleotide sugars. Depending on the polysaccharide structures, the polymerization involves GT able to perform an inverting (α → β) or a retaining (α → α) mechanism (Coutinho et al. 2003). The polymerization step involving the (α → β) mechanism is well known, however, the mechanism (α → α) is less described and still remains not fully understood. Due to the nature of the heparosan polymer, the heparosan synthase exhibits both of these mechanisms to elongate heparosan chains.
Bacterial capsules composed of heparosan have been reported in Escherichia coli K5 (Vann et al. 1981) and Pasteurella multocida Type D (DeAngelis et al. 2002; Pandit and Smith 1993; Rimler 1994). In E. coli K5, the synthesis of heparosan is regulated by four genes, KfiA, KfiB, KfiC, and KfiD, located on the same operon. However, only two of them, KfiA and KfiC, encoding respectively for the N-acetylglucosaminyltransferase and for the D-glucuronyltransferase seem to be involved in the elongation of heparosan polymers (Chen et al. 2006; Griffiths et al. 1998; Hodson et al. 2000). Different from what is observed in E. coli, in P. multocida Type D, the synthesis of heparosan is performed by only one enzyme with two glycosyltransferase activities; the heparosan synthase PmHS1. The P. multocida Type D heparosan synthase gene pmhssA also known as pmhs1 located on the putative capsule locus was cloned, and PmHS1 active proteins were expressed (DeAngelis and White 2002). Based on homology with pmhssA, a cryptic gene pmhssB (described also as pglA) encoding for an active recombinant heparosan synthase PmHS2 was discovered in P. multocida Type A, D, and F (DeAngelis and White 2002; May et al. 2001). Characterization of recombinant PmHS1 and PmHS2 enzymes showed that despite their high amino acid level homology (73%), the enzymes differ considerably with respect to their kinetic properties and the molecular weight distribution of the synthesized heparosan polymers (DeAngelis and White 2004; Sismey-Ragatz et al. 2007).
Kinetic studies on both P. multocida heparosan synthase enzymes report an initiation rate and an elongation rate of 2.6 and 76 pmol/μg/min, respectively, for PmHS1, while 5.2 and 28 pmol/μg/min were observed for PmHS2 (Sismey-Ragatz et al. 2007). When incubated without acceptor, the polymerization properties of PmHS1 and PmHS2 were largely different with respect to molecular weight of the synthesized polymer. PmHS1 synthesized heparosan polymers with an average molecular weight of 800 kDa, while heparosan of 28 kDa were observed when PmHS2 was used as a catalyst. The addition of heparosan oligosaccharide acceptors lowered the molecular weight of heparosan polymers synthesized by PmHS1 while it had almost no effect on the PmHS2 polymerization process. Sismey-Ragatz et al. (2007) also showed that contrary to PmHS1, PmHS2 is able to synthesize glycosaminoglycan polymers with new biological properties using unnatural donor sugar analogs. These heparin analog molecules with potentially new biological activities represent a great interest to enlarge therapeutic uses. It was indeed observed that depending on the sugar substitution, digestion of the polymer by heparinase III could be avoided (Sismey-Ragatz et al. 2007).
Because of the above-mentioned polymerization properties, PmHS2 was investigated in detail in this study. An efficient way to express, purify, and store the PmHS2 enzyme is reported. The PmHS2 polymerization activity in time as well as the heparosan polymer elongation process has been characterized by use of a coupled enzyme assay, agarose gel electrophoresis, and high-performance size-exclusion chromatographic (HPSEC) analysis. The influence of parameters such as the nucleotide sugar, the cofactor, and the UDP concentrations on the enzyme activity and the product molecular weight are described. In this report, we show that PmHS2 is capable of synthesizing heparosan polymers with a high molecular weight (Mw) and that heparosan polymer average Mw and size distribution can be controlled by the initial nucleotide sugar concentration present in the polymerization reaction.
Materials and methods
Cloning, expression, and purification of recombinant P. multocida heparosan synthase 2
Based on the sequence of the P. multocida Type D heparosan synthase cryptic gene pmhssB (GenBank account number AY292200), the pmhssB forward primer CGCCATGAAGGGAAAAAAAGAGATG and reverse primer GGATCCTTATAAAAAATAAAAAGGTAAAC were designed. Using a high-fidelity polymerase chain reaction (PCR) mix (Roche) on genomic DNA isolated from a P. multocida Type D (strain number: 40456 from the Wageningen-UR Central Veterinary Institute collection), the pmhssB open reading frame was amplified by 30 cycles of PCR (94 °C, 30 s; 60 °C, 30 s; 72 °C, 1 min). The PCR fragment was ligated into pCR-BluntII vector and transformed into E. coli Top10 cells (Invitrogen). The identity of the pmhssB gene was confirmed by sequencing (Base Clear B.V). Using the forward primer CACCATGAAGGGAAAAAAAGAGATG and the reverse primer TAAAAAATAAAAAGGTAAACAGGGGATA, pmhssB fragments were obtained by applying 30 cycles of PCR (94 °C, 30 s; 55 °C, 30 s; 72 °C, 1 min) which were cloned in the pET101-D-TOPO expression vector (Invitrogen).
The pET101-D-TOPO vector allowing the fusion of a V5 epitope and a C-terminal His-tag to the pmhssB fragment was first transferred into the E. coli Top10. Subsequently, positive plasmids were isolated and transformed into the E. coli BL21*(DE3) expression strain (Invitrogen). Recombinant protein was expressed according to the manufacturer's instruction. Briefly, a culture inoculated with an overnight preculture was grown in LB ampicillin (50 μg/ml) media at 37 °C under shaking condition (250 rpm). When the optical density600 (OD) was around 0.6, the protein expression was induced by addition of 0.5 mM isopropyl b-D-1-thiogalactopyranoside, and growth was continued for 2 h at 30 °C/250 rpm. Cells were harvested by centrifugation (Allegra 6R centrifuge, Beckman Coulter, Rotor: GG-3.8A.) at 5 °C/3,000 g/20 min, and the pellet was stored at −80 °C. For enzyme purification, the pellet (from 75 ml culture) was thawed on ice and resuspended in 4 ml phosphate buffer (50 mM NaH2PO4 and 300 mM NaCl, pH 8) before being sonicated 4 × 30 s on ice with a sonication tip (Vibra cell, Sonic and materials, Inc, output control set on 40). Prior to centrifugation at 5 °C/18,000 g/15 min (Eppendorf centrifuge 5417R), MgCl2 was added to the lysate to a final concentration of 6 mM (Kane et al. 2006). After centrifugation, the supernatant was kept on ice for further purification, and the pellet was discarded. In order to perform an optimal His-tag purification, imidazole was added to the supernatant to a final concentration of 10 mM. To 4 ml of supernatant, 1 ml of 50% Ni-NTA Superflow resin slurry (Qiagen) was added and gently mixed for 1 h at 4 °C on a rotating shaker. The resin slurry and supernatant mix was washed with increasing imidazole concentrations, respectively, 10 mM and 20 mM. To allow the elution of PmHS2, 250 mM of imidazole was present in the elution buffer. The first three eluted fractions of 1 ml known to have the highest PmHS2 concentrations were pooled and then desalted by gel filtration using a PD10 column with Sephadex G-25 Medium resin (GEA Healthcare) with 50 mM Tris pH 7.2 as eluent. Ethylene glycol to a final concentration of 1 M was added to the desalted PmHS2 fraction before storage at −80 °C.
Purity of the PmHS2 desalted fraction was monitored by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) stained with SimplyBlue SafeStain (Invitrogen), and the protein content was quantified by the bicinchoninic acid (BCA) method (Pierce) using bovine serum albumin as reference.
Characterization of the PmHS2 polymerization process
Standard polymerization conditions
Unless noted, polymerization assays with a final volume between 30 to 200 μl contained 40 mM Tris pH 7.2, 4 mM MnCl2, 4 mM MgCl2, 5 mM UDP-glucuronic acid, 5 mM UDP-N-acetylglucosamine, and 55–60 µg/ml of 95% pure PmHS2 enzyme. The reaction occurred in the dark at 30–32 °C under mild shaking conditions for time ranging from 1.5 to 48 h depending on the experiment. In order to study the effect of different parameters, the composition of the polymerization mix was changed. Modifications in the standard incubation conditions are stated in the legend to the figures.
Note that in this study, no oligosaccharide acceptor was added to the polymerization reaction.
Determination of the heparosan polymer molecular weight and polydispersity
After synthesis, the reactions were quenched at 99 °C for 10 min and then cooled on ice. The polymer chain length and the molecular weight distribution of the synthesized products were assessed by agarose gel electrophoresis and by HPSEC analysis. For gel analysis ,the samples were mixed with glycerol to a final concentration of 12%, loaded on a 2% agarose gel, and run for 2 h at 50 V in Tris-acetate EDTA buffer (Lee and Cowman 1994). The gels were stained overnight in the dark in 50% ethanol per 0.005% Stains-All (Sigma). The destaining of the gels was carried out in pure water for 30 min in the dark followed by an exposure to the light till background discoloration. Images from the gel were acquired with a BioRad GS-800 calibrated densitometer and analyzed by the Quantity One program. Selected hyaluronan molecular weight marker ranging from 27 to 495 kDa (Select-HALoLadder, Hyalose, OK, USA) was used to estimate the heparosan molecular weight. Since Stains-All tends to overstain the polysaccharides on an agarose gel, it was difficult to estimate the average molecular weight of the polymers in an accurate way by this type of analysis.
Alternatively, the size distribution and the molecular weight of the polymers were analyzed by HPSEC on a SpectraSystem HPLC (Thermo Separation Products, USA) using three TosoHaas TSK-gel columns in series (4,000, 3,000, and 2,500 PWx1, 300 × 7.5 mm; TosoHaas, Japan) preceded by a TSK PWx1 guard column (TosoHass, Japan). Samples (20 μL) were injected and eluted at 30 °C using 0.8 ml/min NaNO2 (0.2 M). Detection was performed using a Shodex RI 71 refractive index detector (Showa Denko K.K., Japan). Hyaluronan molecular weight standards of, respectively, 30, 160, and 262 kDa were used for calibration. From the logarithmic model fitting the elution time of the hyaluronan molecular weight standards, we established the following equation: 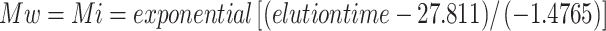 . By assuming that heparosan and hyaluronan elution patterns are comparable, the molecular weight of the eluted samples (Mi) was determined by applying the equation mentioned above. The molecular weight distribution (polydispersity index (PDI)) was determined using the following formula:
. By assuming that heparosan and hyaluronan elution patterns are comparable, the molecular weight of the eluted samples (Mi) was determined by applying the equation mentioned above. The molecular weight distribution (polydispersity index (PDI)) was determined using the following formula: 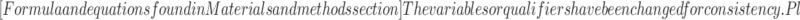
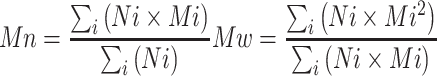 |
The refractive index (RI) signal, known to be proportional to the relative amount of eluted product, was considered as being equivalent to the amount of polymer chains (Ni) of molecular weight Mi.
Determination of PmHS2 specific activity
The polymerization reaction was performed with 95% pure PmHS2 and in presence of nucleotide sugars. Both nucleotide sugars (UDP-glucuronic acid and UDP-N-acetylglucosamine) were added in equimolar ratio and in sufficient amount in order to initiate the polymerization and enable us to follow the elongation process in time by means of the coupled enzyme assay and agarose gel analysis. The activity of PmHS2 was assessed by measuring the UDP formation with a noncontinuous spectrophotometric assay at 340 nm (Gosselin et al. 1994; Krupa et al. 2007). In this assay, the UDP released from the conversion of the nucleotide sugars during the polymerization is coupled via pyruvate kinase (PK) and lactate dehydrogenase (LDH) to the oxidation of NADH into NAD+. With 0.3 mM NADH as initial concentration and in presence of UDP, the NADH conversion into NAD+ was established to be linear. According to the stoichiometric equation, 1 mM UDP will convert 1 mM NADH. This was confirmed with preliminary experiments in which the NADH/UDP conversion ratio was found to be 1.016.
Due to the degradation of nucleotide sugars at high temperature, heat shock was not used to quench the reactions. Polymerization reactions were stopped by immersion in liquid nitrogen and stored at −20 °C. Prior to the UDP measurement, the samples were thawed on ice and diluted to contain less than 0.3 mM of UDP. No residual PmHS2 activity was detected using this procedure. The following compounds were added to the diluted samples: 0.3 mM NADH, 2 mM phospho(enol) pyruvic acid trisodium salt heptahydrate 98% (Across Organic, Belgium), 112.5 mM KCl, and 25 U/ml LDH (l-lactate dehydrogenase from Rabbit muscle, Fluka). The mix with a final volume of 140 μl was preincubated in a Tecan Safire thermostated spectrophotometer during 10 min at 30 °C, and its absorbance was measured at 340 nm. The assay was performed in microtiter plate wells (UV-star plate 96 wells, flat bottom, Greiner Bio-One). After the preincubation, PK (pyruvate kinase from rabbit muscle, Sigma) was added to a final concentration of 10 U/ml. The final reaction volume was 150 μl. The OD was measured at 340 nm for 30 min at 30 °C. A stable absorbance indicated full conversion of the UDP. The preincubation and incubation absorbance data were combined to calculate the amount of UDP released in the polymerization reactions and thus, estimate the enzymatic activity using the conversion ratio of 1.016. All experiments were performed in independent duplicate, and the average is presented in the “Results” section.
Identification of the polymer chains synthesized by PmHS2
The identification of the polymer obtained by enzymatic activity of the recombinant PmHS2 was carried out using Heparinase III from Flavobacterium heparinum (Sigma). Heparinase III is known to specifically cleave at the α1–4 linkage between the hexosamine and glucuronic acid residues of heparosan and heparan sulfate. First, the polymerization assay was quenched at 99 °C for 30 min, then Heparinase III (0.5 U/μl in 0.05 M sodium phosphate buffer pH 7.6) was added to it, and the mixture was incubated for 24 h at 30 °C (DeAngelis et al. 2002). The samples obtained after polymerization and incubated with and without Heparinase III were analyzed on a 2% agarose gel. Full degradation of the polymer confirmed the identity of PmHS2 synthesized product as being heparosan polymers (data not shown).
Results
Cloning, expression, and purification of the recombinant P. multocida heparosan synthase PmHS2
The P. multocida pmhssB gene was cloned, and PmHS2 recombinant proteins were expressed using the vector pET101 in E. coli BL21*(DE3) strain. After sonication of the cell suspension, 15–20% of PmHS2 protein was found in the soluble fraction (Fig. 1). Most of the PmHS2 remained in the membrane fraction. After Ni-NTA purification and PD10 desalting steps, we estimated the purity of PmHS2 enzyme to be about 95% based on SDS-PAGE gel analysis. BCA assay showed that 5% to 10% of pure PmHS2 enzyme was lost during the PD10 desalting step. We estimated that about 7 ± 1 mg of 95% purified PmHS2 was obtained from a 1 L of E. coli culture after 2 h of induction at 30 °C.
Fig. 1.

SDS-PAGE analysis of recombinant PmHS2 protein. E. coli protein fractions were separated on a 10% polyacrylamide gel and stained with SimplyBlue SafeStain (Invitrogen). The expressed PmHS2 is present in the total E. coli cell fraction (dilution × 4; lane1), in the soluble fraction after cold sonication and centrifugation (dilution × 2; lane2), and in the His-tag purified and PD10 desalted fraction (dilution × 1; lane3). The approximate protein molecular weight (around 77 kDa) was estimated by comparison with the SeeBlue Plus2 Prestain (Invitrogen) molecular mass standard (lane 4)
Characterization of the PmHS2 polymerization process
Storage stability of PmHS2
The storage stability of PmHS2 was determined by comparing the catalytic activity of the newly extracted and purified PmHS2 enzyme with the PmHS2 enzyme stored at −80 °C. The catalytic activity of PmHS2 was estimated by gel electrophoresis analysis of the polymerization products obtained under standard conditions. Addition of MnCl2 and MgCl2 to the storage buffer increased enzyme instability at −80 °C. Under these storage conditions, a significant loss of activity was observed after 3 weeks at −80 °C. Addition of ethylene glycol to a final concentration of 1 M in the storage buffer was crucial to ensure the enzyme stability. With ethylene glycol, both the activity of the enzyme expressed as the amount of nucleotide sugars converted by PmHS2 after 24 h of incubation at 30 °C and the chain length of the synthesized heparosan were the same before and after 6 months at −80 °C (Fig. 2). After 1 year of storage, a decrease of about 10% to 20% of the enzymatic activity was observed. SDS-PAGE analysis showed that PmHS2 was not degraded during the storage at −80 °C (data not shown).
Fig. 2.

Influence of the time of storage at −80 °C on PmHS2 polymerization activity. After several months of storage at −80 °C, purified PmHS2 (55–60 μg/ml) was thawed on ice, and its activity was assayed in the standard polymerization buffer described in the Materials and methods section. Enzyme activity expressed as the amount of substrate converted after 24 h of incubation at 30 °C was determined using the coupled enzyme assay. One hundred percent activity corresponds to the amount of substrate converted by freshly extracted enzyme
Process stability during the polymerization
In presence of 55–60 μg/ml of 95% pure PmHS2 and 20 mM of each UDP-sugar, we observed that PmHS2 maintained at least 80% of its initial activity during the incubation time from 24 to 48 h. After 24 h of incubation in the presence of 5 mM or 20 mM of both UDP-sugars, PmHS2 converted comparable amount of nucleotide sugars. Based on these results, we assumed that PmHS2 process stability was constant for 24 h under the assay conditions used. Thus, we considered 24 h of incubation as being the reference time point to study the influence of different parameters such as the divalent metal ion, the nucleotide sugar, and the UDP concentrations on the polymerization activity.
Influence of the divalent metal ions Mn2+ and Mg2+
Divalent metal ions such Mn2+and Mg2+ have been described as being cofactors of some glycosyltransferases (Markovitz et al. 1959; Stoolmiller and Dorfman 1969). DeAngelis and White showed that in the case of PmHS2, Mn2+ is the optimal divalent metal ion to support the polymerization, but Mg2+ also contributes to the substrate conversion (DeAngelis and White 2004). To study the influence of these divalent metal ions on the PmHS2 catalytic activity, an equimolar mixture of MnCl2 and MgCl2 ranging from 0 to 16 mM was investigated (Fig. 3). The polymerization was not initiated in absence of metal cofactor. PmHS2 activity after 24 h of reaction time was optimal with 4 mM of both MgCl2 and MnCl2. With 0.5 mM of MgCl2 and 0.5 mM of MnCl2, a 40% decrease of the activity was found. In the range from 2 to 6 mM of each divalent metal ion, a decrease of activity of not more than 10% was observed. In presence of 8 to 16 mM of each divalent metal ion, a decrease of activity, respectively, about 20% to 50% was observed. Not only the amount of substrate converted in 24 h was affected by the cofactor concentrations but also the average polymer molecular weight. Based on agarose gel electrophoresis analysis, PmHS2 incubated in presence of 4 mM of Mg2+ and 4 mM of Mn2+ synthesized heparosan polymers with an average of 102 kDa while 30–40 kDa heparosan polymers were obtained in presence of 10 mM of each ion.
Fig. 3.

Effect of the MnCl2 and MgCl2 concentration on PmHS2 activity. Enzymatic activity of purified PmHS2 (55–60 μg/ml) was assayed in the standard polymerization buffer described in the Materials and methods section with an equimolar mixture of divalent metal ions (MnCl2 and MgCl2) ranging from 0 to 16 mM. One hundred percent activity corresponds to the amount of substrate converted after 24 h of incubation at 30 °C in presence of 4 mM of MnCl2 and 4 mM of MgCl2. The data represent the average of three independent experiments ± standard deviation (SD)
Influence of the nucleotide sugar concentrations
Since for each mole of nucleotide sugar converted 1 mol of UDP is released, the free UDP generated during the polymerization reaction was used to estimate the substrate conversion by PmHS2. The molecular weight and the size distribution of heparosan polymer were determined by agarose gel electrophoresis and HPSEC using hyaluronan polymers with Mw ranging from 27 to 495 kDa as standards.
Nucleotide sugars, UDP-GlcUA, and UDP-GlcNAc, mixed in an equimolar ratio, were added to the polymerization buffer in a sufficient amount to initiate the polymerization and enable us to follow the elongation process in time by means of the coupled enzyme assay and agarose gel analysis. In order to study the influence of the nucleotide sugar concentration on the polymerization process, substrate concentrations of 1, 5, and 20 mM of both UDP-GlcUA and UDP-GlcNAc were added to the polymerization reaction with purified PmHS2.
It is important to notice that for the three equimolar nucleotide sugar concentrations chosen in this study, the specific activity of PmHS2 remained constant from 180 to 360 min (3 to 6 h) of incubation despite the decrease of available nucleotide sugars and the subsequent increase in UDP, a possible inhibitor of the polymerization reaction (Fig. 4) (Baggenstoss and Weigel 2006; Tlapak-Simmons et al. 2004).
Fig. 4.

Influence of the substrate concentration on PmHS2 activity during 180 to 360 min of incubation. Purified PmHS2 (55–60 μg/ml) was assayed in the standard polymerization buffer described in the Materials and methods section with increasing substrate concentration ranging from 1 up to 20 mM of both nucleotide sugars. The specific activity observed during 180 to 360 min of incubation for the following substrate concentrations 1 (Δ), 5 (○), and 20 mM (•) of both UDP-GlcUA and UDP-GlcNAc was, respectively, about 0.080, 0.145, and 0.128 nmol/µg/min
We observed that the substrate concentration influenced PmHS2 polymerization process in two distinct manners. First, as shown in Fig. 4, the substrate concentration influenced the specific activity of the enzyme. The highest activity of 0.145 nmol/μg/min was observed between 180 to 360 min of incubation when 5 mM of each UDP-sugar were added. Adding 20 mM of each nucleotide sugar lowered PmHS2 specific activity to 0.128 nmol/μg/min. At a lower substrate concentration, 1 mM of UDP-GlcUA and 1 mM of UDP-GlcNAc, the specific activity was about 0.080 nmol/µg/min. In this last case, based on the amount of substrate converted per unit of time, we calculated that the nucleotide sugars were totally converted after about 8 h of incubation.
Secondly, the substrate concentration greatly influenced the molecular weight and the size distribution of the heparosan polymers synthesized by PmHS2. Both agarose gel electrophoresis and HPSEC analysis showed that PmHS2 elongated high molecular weight heparosan polymers when incubated in presence of 1 mM of each nucleotide sugar while lower molecular weight products were obtained when incubated with 5 mM or 20 mM of each UDP-sugar (Fig. 5a, b). Also, the polydispersity of the formed heparosan was influenced by the UDP-sugar concentration, a low substrate concentration resulted in a lower polydispersity. HPSEC analysis showed that in presence of 1, 5, and 20 mM of both UDP-GlcUA and UDP-GlcNAc, the average molecular weight of the heparosan formed was, respectively, 130, 102, and 45 kDa. These data showed an inverse correlation between the nucleotide sugar concentration and the polymer chain length. Depending on the initial conditions, PmHS2 lengthened heparosan chains to a different polymerization degree. In presence of 1 mM of each nucleotide sugar, polymers with an average of 650 monosaccharide units were synthesized in comparison to 225 monosaccharide units per polymer with 20 mM of each UDP-sugar.
Fig. 5.

Influence of the substrate concentration on PmHS2 polymerization process. Purified PmHS2 (55–60 μg/ml) was incubated at 30 °C in the standard polymerization buffer described in the Materials and methods section with increasing substrate concentration (UDP-GlcUA and UDP-GlcNAc). a Heparosan polymer chain length and size distribution depending on the substrate concentration and incubation time was analyzed on 2% agarose gels stained with Stains-All. b HPSEC analysis of the synthesized heparosan after 24 h of incubation in presence of 1, 5, and 20 mM of both UDP-GlcUA and UDP-GlcNAc. The average molecular weight (Mw) and polydispersity (PDI) of heparosan polymers were, respectively, 130 kDa (PDI = 1.04; Δ), 102 kDa (PDI = 1.36; ○), and 45 kDa (PDI = 1.52)
Table 1 shows, for each substrate concentration studied, the relation between the amount of nucleotide sugars converted after 3 h of incubation and the corresponding average polymer molecular weight observed on agarose gel electrophoresis. From these data, we have calculated the amount of heparosan polymer chains present in each sample and their elongation rates.
Table 1.
Influence of the nucleotide sugar concentration on the PmHS2 polymerization process
| UDP-GlcUA UDP-GlcNAc (mM) | UDP-sugar converted per microgram PmHS2 (nmol; 3 h incubation) | Observed average heparosan Mw (kDa; 3 h incubation)a | Heparosan chain initiated per microgram PmHS2 (nmol; 3 h incubation) | Calculated U bMS | Calculated average heparosan Mw (kDa; 6 h incubation) | Observed average heparosan Mw (kDa; 6 h incubation)a |
|---|---|---|---|---|---|---|
| 1 | 8 ± 0.20 | 70 ± 5 | 0.022 | 3.6 | 198 | 120 ± 10 |
| 5 | 11 ± 0.70 | 28 ± 2 | 0.082 | 1.8 | 84 | 60 ± 5 |
| 20 | 10 ± 0.50 | 10 ± 2 | 0.200 | 0.6 | 33 | 25 ± 2 |
The polymerization reaction was performed in 100-μl reaction volume containing 55–60 μg/ml purified PmHS2. The detail of the calculation is presented in the Results section
aThe polymer average molecular weight (± estimated error) was determined using agarose gel electrophoresis analysis in comparison with hyaluronan molecular weight marker as a reference
bRefers to the calculated average number of monosaccharide units transferred per minute and per heparosan polymer chain
The following equation was used to determine the amount of heparosan polymer chains (nmole P Tn) initiated during the first 3 h of incubation in a 100-μl reaction volume per microgram of PmHS2.
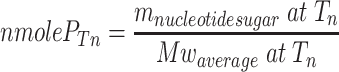 |
During the first 3 h of polymerization, we calculated that about nine times less heparosan polymer chains were initiated and elongated with 1 mM of UDP-GlcUA and 1 mM of UDP-GlcNAc compared to 20 mM of both UDP-sugars.
Assuming that only the oligosaccharides present after 3 h of polymerization reaction will be elongated, in other words by neglecting the initiation of new polymers, we calculated the elongation rate per polymer chain. For this calculation, the PmHS2 specific activity values previously determined for a period of incubation between 3 to 6 h were used (Fig. 4).
At an incubation time T n, we estimated the total amount of nucleotide sugar converted (nmole T Tn).
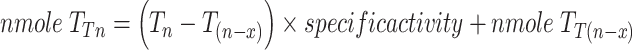 |
For each incubation time T n, the average polymerization degree for each polymer chain could be determined (DPTn).
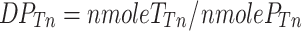 |
From the DPTn, the average Mw of the heparosan polymer chains at T n could be assessed.
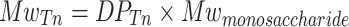 |
The average number of monosaccharide units transferred per minute and per heparosan polymer chain (U MS, equal to elongation rate) has been estimated for the elongation phase using the equation:
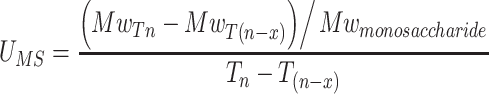 |
The calculated elongation rate was about 3.6 monosaccharide unit per minute per heparosan chain versus 0.6 monosaccharide unit per minute per heparosan chain, respectively, with 1 mM versus 20 mM of each UDP-sugar. Thus, at low substrate concentration, both a decrease in number of polymer chains and an increase in elongation rate per heparosan chain result in longer polysaccharides.
Finally, based on our assumptions and the calculations, we determined the average heparosan polymer Mw (kilodaltons) that should be synthesized after 6 h of polymerization.
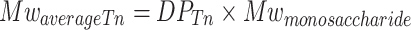 |
We were able to describe PmHS2 polymerization behavior in presence of different substrate concentrations. However, the calculated average heparosan Mw after 6 h of incubation are higher than what is observed on agarose gel electrophoresis (Table 1). For example, in presence of 1 mM of each UDP-sugar, we calculated that after 6 h of incubation heparosan polymers should have an average molecular weight of 198 kDa, while on agarose gel heparosan polymers of about 120 ± 10 kDa were observed. The difference between the calculated and the observed data is even more significant if we consider 24 h of incubation. The increased tailoring of the spots (Fig. 6b) indicated enhanced polydispersity due to initiation of new heparosan chains at the same time as the elongation of heparosan polymers.
Fig. 6.

Analysis of PmHS2 polymerization process in time. The PmHS2 (55–60 μg/ml) enzymatic activity, incubated during 28 h at 30 °C, was analyzed with standard assay conditions as described in the Materials and methods section. At many time points, the amount of substrate converted was determined using the coupled enzyme assay. a The specific activity for the elongation phase (2–12 h) and the level off were, respectively, 0.100 ± 0.010 nmol/μg/min and 0.020 ± 0.005 nmol/μg/min. Five independent experiments were completed for each data set, and their average ± SD is presented. b The molecular weight distribution of heparosan polymer in the samples was analyzed by 2% agarose gels stained with Stains-All
To conclude, the effect of the nucleotide sugar concentration on PmHS2 polymerization process can be described, thanks to equations estimating the amount of polymer chains initiated and their approximate molecular weight. However, the results suggest that the estimation can be further improved when taking into account that, during the elongation, the initiation of new polymer chain occurs.
PmHS2 polymerization process in time
The polymerization process in time was investigated with 55–60 μg/ml of 95% pure PmHS2 using the optimal nucleotide sugar concentrations to get the highest specific activity (5 mM of each UDP-sugar). The quantification of free UDP was used to determine the amount of nucleotide sugar converted in time. We observed that the PmHS2 polymerization process could be divided into phases depending on the specific activity observed in time (Fig. 6a). These polymerization phases have been studied and characterized for the enzyme-specific activity, the product molecular weight synthesized, and the polymer size distribution. The polymerization starts with the synthesis of short oligosaccharide from nucleotide sugars. This phase called initiation is quite short, and in the case of PmHS2, “the level of de novo initiation” is low (Sismey-Ragatz et al. 2007). As shown in Table 1, we observed a correlation between the UDP-sugar concentrations and the amount of heparosan polymer chain initiated.
Following the initiation, the short heparosan chains synthesized will be elongated. This phase is usually referred as the elongation phase and is characterized by a constant ratio of nucleotide sugar conversion over the time (∆P/∆T). Between 2 and 12 h of incubation, the enzyme specific activity was about 0.100 ± 0.01 nmol/μg/min (Fig. 6a.). This corresponds to a transfer of 1.2 monosaccharide unit per minute per heparosan chain. The average molecular weight of representative polymer samples increased significantly in time from 61 kDa (PDI = 1.25) after 4 h to 102 kDa (PDI = 1.36) after 24 h of incubation; meaning that about 500 monosaccharide units in average have been incorporated into each polymer chain. However, as noticed before the increase in polydispersity in time showed that initiation of new chains and elongation proceed simultaneously (Fig. 6b).
The last phase observed is characterized by a sudden level off of the enzyme activity. After about 12 to 13 h of incubation, the specific activity of PmHS2 slowed down to about 0.020 ± 0.005 nmol/μg/min (Fig. 6a). Between 12 and 24 h, the polymer size only slightly increased in contrast with an increase in polydispersity (Fig. 6b).
Inhibition of the PmHS2 polymerization process by UDP
UDP has been reported to inhibit glutathione 5-transferase enzymes activity (Baggenstoss and Weigel 2006; Markovitz et al. 1959; Stoolmiller and Dorfman 1969; Tlapak-Simmons et al. 2004). When added to the polymerization buffer prior to the synthesis, UDP showed severe inhibition of the PmHS2 activity. The inhibition level of 5 mM of UDP on the polymerization activity was around 50% with 5 mM or 20 mM of each nucleotide sugar (Fig. 7a). In the range of 8 to 10 mM of UDP, the level of inhibition was dependent on the nucleotide sugar concentration (Fig. 7b). With 5 mM of each nucleotide sugar, the complete inhibition of the PmHS2 polymerization activity was observed in presence of 8 mM UDP, while with 20 mM of each nucleotide sugar, the full inhibition was obtained with 10 mM of UDP. These results suggest that UDP inhibition is somehow related to the concentration of nucleotide sugars present in the reaction assay. The inhibitory UDP concentration does not seem to be reached during the first hours of incubation. We, indeed, observed that from 2 to 12 h of incubation, the UDP rise, and the decrease of substrate availability did not affect PmHS2 specific activity (Fig. 4 and Fig. 6a). To conclude, the inhibition by UDP does not seem to occur when the UDP concentration is below or equal to 4 mM.
Fig. 7.

Effect of initial UDP concentration on PmHS2 activity. Purified PmHS2 (55–60 μg/ml) was assayed in the standard polymerization buffer described in the Materials and methods section with increasing initial UDP concentrations from 0 to 10 mM. a The nucleotide sugar concentration of both UDP-GlcUA and UDP-GlcNAc was, respectively, 5 mM (○) and 20 mM (•). After 24 h of incubation at 30 °C, the amount of substrate converted by PmHS2 in presence of UDP was determined using the coupled enzyme assay. b Samples from both series were analyzed on a 2% agarose gel stained with Stains-All
Discussion
In this report, we describe the successful cloning of the P. multocida Type D heparosan synthase-2 gene (pmhssb) and the expression of the corresponding PmHS2 protein into E.coli BL21*(DE3) using pET101 as an expression vector. Protein expression and purification allowed the recovery of 7 ± 1 mg of 95% pure and active PmHS2 enzyme per liter of culture. This level of PmHS2 enzyme recovery after purification, which is stable for at least 6 months when stored at –80 °C, has not been described so far.
We found that, in absence of divalent metal ions, PmHS2 did not exhibit activity and that cofactor concentration added in excess inhibit the enzymatic activity. The optimal cofactor concentration for PmHS2 polymerization activity was determined to be 4 mM of both Mn2+ and Mg2+. This finding support the classification reviewed by Coutinho et al. (2003), PmHS2 belongs to the GlycosylTransferase-A (GT-A) superfamily due to the requirement for divalent metal ions in order to be active and its DxD amino acid conserved domains in which D stands for aspartic acid and x for any amino acid.
Polymerization reactions catalyzed by the recombinant PmHS2 showed three distinct polymerization phases. PmHS2 initiates the polymerization process by the synthesis of short oligosaccharides using nucleotide sugars. For PmHS2, the initiation phase has been described to have a slow “de novo” initiation rate (Sismey-Ragatz et al. 2007). We demonstrated that for PmHS2, the nucleotide sugar concentrations during the initiation phase play an important role in the average Mw and the polydispersity of the resulting heparosan polymers. Low substrate concentrations favor the formation of higher molecular weight polymers with a lower polydispersity while high substrate concentrations do the opposite.
On one hand, we assume that, in an early stage of the polymerization, the PmHS2 enzyme has more affinity for oligomers than for nucleotide sugars. On the other hand, we also hypothesize that, in presence of a low substrate concentration, the probability for PmHS2 to encounter a synthesized heparosan oligomers instead of a nucleotide sugar is relatively high. A lower substrate concentration causes less heparosan chains to be initiated. Consequently, lesser heparosan polymers to be elongated, and thus, it will result in the synthesis of longer heparosan polymer chains. This polymerization phenomenon is similar to what has been observed with PmHAS and PmHS1 in presence of oligosaccharide acceptors. For both enzymes, the affinity for the oligosaccharide acceptor was higher than for activated monosaccharides. In addition, a decrease in oligosaccharide acceptor concentration resulted in polymer chains with a higher Mw, while an increase in acceptor lowered the average Mw of the polymer formed (Jing and DeAngelis 2004; Sismey-Ragatz et al. 2007). We demonstrated that for PmHS2, the initiation is a determinant step in the polymer synthesis. During this phase, the amount of heparosan chains initiated is regulated by the nucleotide sugar concentration. Another conclusion from this experiment is that PmHS2 is a nonprocessive enzyme. With a processive enzyme, the dissociation of the polymer and the enzyme implicates the end of the chain elongation. The amount of chains initiated will not differ depending on the nucleotide sugar concentrations, and thus, low substrate concentrations will result in shorter polysaccharides. At the opposite, with a nonprocessive enzyme such as PmHS2, the addition of a single monosaccharide to the growing chain is followed by the dissociation of the polymer from the enzyme. The polymer is recaptured before proceeding to the transfer of new monosaccharide unit. As in presence of low substrate concentration, PmHS2 initiates less chains, and it leads to higher molecular weight products.
The substrate concentration also influenced the specific activity of the PmHS2 enzyme during the elongation phase. The highest activity was observed by adding 5 mM of each UDP-sugars, from 3 to 6 h of incubation, and the specific activity was found to be about 0.145 ± 0.010 nmol/µg/min. In this research, the specific activity observed for PmHS2 is considerably higher than the elongation rate of 28 pmol/μg/min that has been reported so far for PmHS2 (Sismey-Ragatz et al. 2007). When using the optimal nucleotide sugar concentration, PmHS2 showed an elongation phase of about 10 h, and under these conditions, the polymers chains were elongated with average up to 500 to 550 monosaccharides units in 24 h.
Although we have observed that at the first stage of the polymerization that the enzyme has more affinity towards products with a higher molecular weight, both the elongation and the initiation of new heparosan chain occur simultaneously. Weigel stipulated that despite the fact that, in theory, nonprocessive enzyme could elongate the polymer chains till infinite, it has not been observed. Due to the increase of the polymer chain length and the polymer concentration, interaction between the nonreducing end of the polymer chains and the enzyme become more difficult in time. As the system enzyme/polymer is becoming more rigid, it will favor the initiation of new polymer chains from nucleotide sugars or short oligosaccharides that are more mobile than longer chains (Weigel 2002). This phenomenon supports the fact that new heparosan chains are also initiated while the elongation of polymer occurs. As newly elongated heparosan oligomers cannot obtain the same molecular weight as the previously synthesized heparosan polymers during the incubation time, it results in a broader molecular weight distribution and therefore, a higher polydispersity.
In many studies, UDP was found to be an inhibitor of glycosyltransferase activity (Baggenstoss and Weigel 2006; Markovitz et al. 1959; Stoolmiller and Dorfman 1969; Tlapak-Simmons et al. 2004). PmHS2 also is inhibited by UDP. The UDP inhibition is influenced by the nucleotide sugar concentration, and high substrate concentrations reduce the UDP inhibition. We observed that the specific activity of PmHS2 remained constant during the incubation period from 2 to 12 h, despite the decrease of available nucleotide sugars and the subsequent increase in UDP. This showed that the UDP concentration present in the polymerization assay has to reach a certain concentration limit which seems to be above 4 mM in order to be an inhibitor of PmHS2 activity.
To summarize, we observed that purified PmHS2 is able to synthesize relatively long heparosan polymers with an Mw up to 130 kDa. In the presence of 5 mM of each UDP-sugar, a maximal specific activity of 0.145 nmol/µg/min was found. We showed evidence that a change in the initial nucleotide sugar concentrations influenced the final product molecular weight and polydispersity and confirmed that PmHS2 is a nonprocessive enzyme. Future studies on heparosan synthase will allow us to develop an in vitro production system in which heparosan polymer chain length can be controlled.
Acknowledgments
The authors thank Marinella van Leeuwen for her help to isolate from Pasteurella multocida the heparosan synthase gene pmhssB and Ben van den Broek for his precious help for the HPSEC sample analysis. This project is carried out in close collaboration with Schering–Plough and financially supported by the Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (www.b-basic.nl) through B-Basic, a public–private NWO-ACTS program (Advanced Chemical Technologies for Sustainability).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Baggenstoss B, Weigel PH. Size exclusion chromatography-multiangle laser light scattering analysis of hyaluronan size distributions made by membrane-bound hyaluronan synthase. Anal Biochem. 2006;352:243–251. doi: 10.1016/j.ab.2006.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli R, Porro F, Tarsia P. The heparins and cancer: review of clinical trials and biological properties. Vasc Med. 2004;9:205–213. doi: 10.1191/1358863x04vm566ra. [DOI] [PubMed] [Google Scholar]
- Chen M, Bridges A, Liu J. Determination of the substrate specificities of N-acetyl-D-glucosaminyltransferase. Biochemistry. 2006;45:12358–12365. doi: 10.1021/bi060844g. [DOI] [PubMed] [Google Scholar]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol. 2003;328:307–317. doi: 10.1016/S0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, White CL. Identification and molecular cloning of a heparosan synthase from Pasteurella multocida Type D. J Biol Chem. 2002;277:7209–7213. doi: 10.1074/jbc.M112130200. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, White CL. Identification of a distinct, cryptic heparosan synthase from Pasteurella multocida Types A, D, and F. J Bacteriol. 2004;186:8529–8532. doi: 10.1128/JB.186.24.8529-8532.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis PL, Gunay NS, Toida T, W-j M, Linhardt RJ. Identification of the capsular polysaccharides of Type D and F Pasteurella multocida as unmodified heparin and chondroitin, respectively. Carbohydr Res. 2002;337:1547. doi: 10.1016/S0008-6215(02)00219-7. [DOI] [PubMed] [Google Scholar]
- Gosselin S, Alhussaini M, Streiff MB, Takabayashi K, Palcic MM. A continuous spectrophotometric assay for glycosyltransferases. Anal Biochem. 1994;220:92–97. doi: 10.1006/abio.1994.1303. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Cook NJ, Gottfridson E, Lind T, Lidholt K, Roberts IS. Characterization of the glycosyltransferase enzyme from the Escherichia coli K5 capsule gene cluster and identification and characterization of the glucuronyl active site. J Biol Chem. 1998;273:11752–11757. doi: 10.1074/jbc.273.19.11752. [DOI] [PubMed] [Google Scholar]
- Hodson N, Griffiths G, Cook N, Pourhossein M, Gottfridson E, Lind T, Lidholt K, Roberts IS. Identification that KfiA, a protein essential for the biosynthesis of the Escherichia coli K5 capsular polysaccharide, is an alpha-UDP-GlcNAc glycosyltransferase. The formation of a membrane-associated K5 biosynthetic complex requires KfiA, KfiB, and KfiC. J Biol Chem. 2000;275:27311–27315. doi: 10.1074/jbc.M004426200. [DOI] [PubMed] [Google Scholar]
- Jackson RL, Busch SJ, Cardin AD. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- Jing W, DeAngelis PL. Synchronized chemoenzymatic synthesis of monodisperse hyaluronan polymers. J Biol Chem. 2004;279:42345–42349. doi: 10.1074/jbc.M402744200. [DOI] [PubMed] [Google Scholar]
- Kane TA, White CL, DeAngelis PL. Functional characterization of PmHS1, a Pasteurella multocida heparosan synthase. J Biol Chem. 2006;281:33192–33197. doi: 10.1074/jbc.M606897200. [DOI] [PubMed] [Google Scholar]
- Krupa JC, Shaya D, Chi L, Linhardt RJ, Cygler M, Withers SG, Mort JS. Quantitative continuous assay for hyaluronan synthase. Anal Biochem. 2007;361:218–225. doi: 10.1016/j.ab.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AYY. The effects of low molecular weight heparins on venous thromboembolism and survival in patients with cancer. Thromb Res. 2007;120:S121–S127. doi: 10.1016/S0049-3848(07)70140-8. [DOI] [PubMed] [Google Scholar]
- Lee HG, Cowman MK. An agarose gel electrophoretic method for analysis of hyaluronan molecular weight distribution. Anal Biochem. 1994;219:278. doi: 10.1006/abio.1994.1267. [DOI] [PubMed] [Google Scholar]
- Markovitz A, Cifonelli JA, Dorfman A. The biosynthesis of hyaluronic acid by group A Streptococcus. VI. Biosynthesis from uridine nucleotides in cell-free extracts. J Biol Chem. 1959;234:2343–2350. [PubMed] [Google Scholar]
- May BJ, Zhang Q, Li LL, Paustian ML, Whittam TS, Kapur V. Complete genomic sequence of Pasteurella multocida, Pm70. Proc Natl Acad Sci U.S.A. 2001;98:3460–3465. doi: 10.1073/pnas.051634598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrby K. Low-molecular-weight heparins and angiogenesis. APMIS. 2006;114:79–102. doi: 10.1111/j.1600-0463.2006.apm_235.x. [DOI] [PubMed] [Google Scholar]
- Pandit KK, Smith JE. Capsular hyaluronic-acid in Pasteurella multocida Type-A and its counterpart in Type-D. Res Vet Sci. 1993;54:20–24. doi: 10.1016/0034-5288(93)90005-z. [DOI] [PubMed] [Google Scholar]
- Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat Prod Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- Rimler RB. Presumptive identification of Pasteurella multocida serogroups A, D and F by capsule depolymerisation with mucopolysaccharidases. Vet Rec. 1994;134:191–192. doi: 10.1136/vr.134.8.191. [DOI] [PubMed] [Google Scholar]
- Sismey-Ragatz AE, Green DE, Otto NJ, Rejzek M, Field RA, DeAngelis PL. Chemoenzymatic synthesis with distinct Pasteurella heparosan synthases: monodisperse polymers and unnatural structures. J Biol Chem. 2007;282:28321–28327. doi: 10.1074/jbc.M701599200. [DOI] [PubMed] [Google Scholar]
- Stoolmiller AC, Dorfman A. The biosynthesis of hyaluronic acid by Streptococcus. J Biol Chem. 1969;244:236–246. [PubMed] [Google Scholar]
- Tlapak-Simmons VL, Baron CA, Weigel PH. Characterization of the purified hyaluronan synthase from Streptococcus equisimilis. Biochemistry. 2004;43:9234–9242. doi: 10.1021/bi049468v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann WF, Schmidt MA, Jann B, Jann K. The structure of the capsular polysaccharide (K5 antigen) of urinary-tract-infective Escherichia coli 010:K5:H4. A polymer similar to desulfo-heparin. Eur J Biochem. 1981;116:359–364. doi: 10.1111/j.1432-1033.1981.tb05343.x. [DOI] [PubMed] [Google Scholar]
- Weigel PH. Functional characteristics and catalytic mechanisms of the bacterial hyaluronan synthases. Iubmb Life. 2002;54:201–211. doi: 10.1080/15216540214931. [DOI] [PubMed] [Google Scholar]